

Abstract
Nerve growth factor (NGF) causes a rapid sensitisation of nociceptive sensory neurones to painful thermal stimuli owing to an action on the heat and capsaicin receptor TRPV1 (formerly known as VR1). We have developed a new technique to study this rapid sensitisation of TRPV1 by monitoring the effects of NGF on the increase in intracellular calcium concentration ([Ca2+]i) following exposure to capsaicin. Brief applications of capsaicin caused a rise in [Ca2+]i, and NGF was found to enhance this rise in 37 % of capsaicin-responsive neurones within 2 min. Pathways responsible for transducing the sensitisation of TRPV1 by TrkA, the NGF receptor, were characterised by observing the effects of inhibitors of key members of NGF-activated second messenger signalling cascades. Specific inhibitors of the ras/MEK (mitogen-activated protein and extracellular signal-regulated kinases) pathway and of phospholipase C did not abolish the NGF-induced sensitisation, but wortmannin, a specific inhibitor of phosphatidylinositol-3-kinase (PI3K), totally abolished the effect of NGF. Pharmacological blockade of protein kinase C (PKC) or calcium-calmodulin-dependent protein kinase II (CaMK II) activation also prevented NGF-induced sensitisation, while blockade of protein kinase A (PKA) was without effect. These data indicate that the crucial early pathway activated by NGF involves PI3K, while PKC and CaMK II are also involved, probably at subsequent stages of the NGF-activated signalling pathway.
Nerve growth factor (NGF) has been well characterised as essential for the growth and development of sensory neurones. An involvement of NGF as an important extracellular signalling molecule in enhancing the sensation of pain has, however, only more recently been described. Injection of NGF induces both thermal and mechanical hyperalgesia in the adult rat (Lewin et al. 1993) and causes hypersensitivity to noxious heat and mechanical stimuli in humans (Petty et al. 1994). NGF appears to signal an important component of physiological inflammation, as removal of endogenous NGF by the injection of NGF-specific antibodies largely reverses both the thermal and the mechanical hyperalgesia caused by injection of complete Freund's adjuvant (CFA; Woolf et al. 1994; McMahon et al. 1995). Lewin et al. (1993) showed that sensitisation to noxious thermal stimuli developed within minutes of an injection of NGF into the hind paw of a rat, and is therefore far too rapid to involve upregulation of gene transcription. This rapid sensitisation subsequently developed into a thermal and mechanical hyperalgesia lasting for days, and there is a general consensus that changes in expression of proteins involved in nociception are important in maintaining long-term hyperalgesia (Lewin & Mendell, 1993; Lee et al. 2002; Bron et al. 2003) The sensitisation caused by NGF is mediated by the TrkA receptor, because in p75NTR-null mice NGF can still induce thermal hyperalgesia, indicating that p75NTR is unlikely to be essential for NGF-mediated sensitisation to noxious thermal stimuli (Bergmann et al. 1998).
The rapid sensitisation to noxious thermal stimuli observed by Lewin et al. (1993) was shown to result from a direct action of NGF on peripheral nociceptors (Shu & Mendell 1999, 2001). In these experiments capsaicin, the active ingredient of chilli peppers, was used in place of thermal stimuli to activate the heat and capsaicin receptor, TRPV1 (vanilloid receptor 1, initially called VR1; Caterina et al. 1997), and an enhancement of the membrane current gated by a brief capsaicin application was observed within 10 min of NGF application. TRPV1 is the only ion channel gated by capsaicin, but it is not the only mechanism by which noxious heat is detected, as TRPV1-/- mice are insensitive to capsaicin but respond to noxious heat (Davis et al. 2000; Caterina et al. 2000). The use of capsaicin as a surrogate for noxious heat in these experiments therefore demonstrates that NGF-activated second messenger signalling cascades cause a direct sensitisation of TRPV1.
Activation of TrkA receptors recruits many signalling molecules that can bind to the intracellular phosphorylated tyrosine residues within TrkA by means of Src homology (SH2) domains. Three proteins in particular have been identified based on their specific binding to phosphorylated Trk receptors: Shc, which activates the ras/MEK pathway; phospholipase C gamma-1 (PLCγ1), which cleaves phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5-P2) to inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG); and phosphatidylinositol-3-kinase (PI3K), which 3-phosphorylates PtdIns-4,5-P2 (Vetter et al. 1991; Soltoff et al. 1992; Raffioni & Bradshaw, 1992; Obermeier et al. 1993; Dikic et al. 1995). In the present study we investigated the role of each of these three pathways in TRPV1 sensitisation by the use of specific inhibitors.
The end point of a putative sensitisation pathway may be phosphorylation of TRPV1 itself. The amino acid sequence of TRPV1 contains potential phosphorylation sites for many different serine/threonine kinases, most notably protein kinase C (PKC), protein kinase A (PKA) and calcium-calmodulin-dependent protein kinase II (CaMK II). Of these, PKC and PKA have been shown to enhance TRPV1-mediated responses (Hingtgen et al. 1995; Cesare & McNaughton, 1996; Lopshire & Nicol, 1998; Cesare et al. 1999; Vellani et al. 2001; Bhave et al. 2002; Numazaki et al. 2002). We therefore investigated the effects of kinase inhibitors on TRPV1 sensitisation.
There is some disagreement over the pathways intervening between the activation of TrkA by NGF and sensitisation of TRPV1. Chuang et al. (2001) suggested a system of regulation similar to that used for some other TRP channels, i.e. that binding of NGF to TrkA activates PLCγ, leading to breakdown of PtdIns-4,5-P2 and the relief of TRPV1 from constitutive inhibition by PtdIns-4,5-P2. Shu & Mendell (2001) proposed instead that phosphorylation of TRPV1 by PKA was involved, as inhibition of PKA reduced the amplitude of sensitisation of TRPV1 caused by NGF, whilst inhibition of mitogen-activated protein kinases (MAPKs) or PKC caused no significant alterations in the sensitising effects of NGF on TRPV1. In a related study Aley et al. (2001) found that sensitisation by adrenergic receptor activators was partly mediated by MEK, which is also activated by NGF.
In the present study we set out to identify the intracellular signalling pathways that mediate NGF-evoked sensitisation of TRPV1 in mouse dorsal root ganglion (DRG) neurones. A new technique was developed to study rapid thermal sensitisation in large numbers of neurones by monitoring the effects of NGF on the response of sensory neurones to capsaicin. Isolated, cultured sensory neurones were loaded with a Ca2+-sensitive dye and imaged using confocal microscopy. Brief applications of capsaicin caused a rise in [Ca2+]i resulting from activation of TRPV1, and the effects of NGF on the amplitude of these capsaicin-induced Ca2+ increases could then be monitored. NGF was shown to sensitise the response to capsaicin in 37 % of capsaicin-responsive neurones. The intracellular signalling pathway responsible for transducing the sensitisation of TRPV1 by NGF was then characterised by observing the effects of inhibitors of key members of the NGF-activated second messenger signalling cascades. Of the three pathways known to be activated by TrkA, inhibition of PI3K completely abolished sensitisation, while inhibition of PLC or the ras/MEK pathway was without effect on the numbers of neurones sensitised, though there was some reduction in the amplitude of the sensitisation. Inhibition of PKC or CaMK II also abolished sensitisation, suggesting an involvement of these kinases further down the pathway leading to sensitisation.
METHODS
Primary culture of neonatal DRG neurones
Neonatal mice (2–5 days old, C57BL/6J strain) were killed by cervical dislocation and then decapitated, the spinal cord was removed, and approximately 40–45 DRGs were removed and transferred into 2 ml phosphate-buffered saline (PBS, Life Technologies). Nerve trunks and connective tissue were dissected away and the DRGs were transferred into 2 ml Dulbecco's Modified Eagle's Medium (DMEM, Life Technologies) supplemented with 0.25 % collagenase (type IV, Worthington) for 1 h in a humidified incubator (37 °C, 5 % CO2), then washed with 20 ml medium A (DMEM containing 10 % fetal bovine serum (FBS); Life Technologies) and resuspended in 2 ml medium A. DRGs were triturated through a 23 gauge needle followed by a 25 gauge needle to produce a suspension of single cells. Cells were concentrated by centrifugation at 1000 r.p.m. (110 g) for 10 min and the resulting pellet was resuspended in 1.2 ml medium B (DMEM containing 10 % FBS, 1 % penicillin-streptomycin solution (5000 u ml−1, Life Technologies), 1 % L-glutamine (20 mm, Life Technologies), 100 ng ml−1 NGF (Promega) and 10 μm cytosine arabinoside (Sigma)).
Glass coverslips (BDH, 13 mm diameter, thickness 0) coated with 10 μg ml−1 poly-L-lysine (Sigma) and 5 μg ml−1 laminin (Stratec) were placed in four-well plates (Costar) and washed in medium B. Neuronal suspension (100 μl) was then slowly added to 1.2 ml medium B in each well. Neurones were incubated for 48 h at 37 °C in a humidified incubator gassed with 5 % CO2 in air. After 48 h in culture, NGF was withdrawn by washing and replacing medium B with medium C (DMEM containing 1 % penicillin-streptomycin, 1 % L-glutamine, 10 % N-2 supplement (Life Technologies) and 500 ng ml−1 anti-NGF antibody (mAb 27/21, Boehringer Mannheim). Neurones were maintained in medium C for 24 h with all experiments performed on the third day of culture. Neonatal neurones survive NGF withdrawal well provided they have an initial period of exposure to NGF and there was no evidence of neuronal death in the 24 h period of culture without NGF.
Imaging of intracellular calcium
Neurones were loaded with the acetoxymethyl ester of the calcium-sensitive fluorophore fluo-4 (5 μm in DMEM for 15 min at 37 °C). Coverslips were then mounted in an imaging chamber and continuously perfused with Hank's balanced salt solution (HBSS (mm): 140 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes (all Sigma), 5 D-glucose, pH 7.4). Cells were imaged using a BioRad MRC-600 confocal microscope fitted with a × 20 objective lens, NA 0.75. Fluo-4 was excited at 488 nm and images were captured every 3 s using the TCSM time course software package (BioRad). Fluorescence from individual neurones was monitored as a function of time by outlining a region near to but within the cell boundary, and measuring total fluorescence signal from this region.
Neurones were distinguished from non-neuronal cells by the increase in [Ca2+]i which is caused by application of KCl (25 mm) in HBSS (15 s application at the beginning of the experiment). Capsaicin (Calbiochem, typically 500 nm in HBSS) was then applied for 15 s every 4 min. The amplitude of the Ca2+ signal in response to capsaicin declined substantially over the first few applications because of Ca2+-dependent tachyphylaxis (see e.g. Vellani et al. 2001). NGF (Promega, 100 ng ml−1 in HBSS, 2 min) was applied between the fifth and sixth applications of capsaicin, when the response to capsaicin had largely stabilised. The protocol is shown in Fig. 1A and was modified where indicated.
Figure 1. Enhancement of the capsaicin-induced Ca2+ increase by nerve growth factor (NGF).
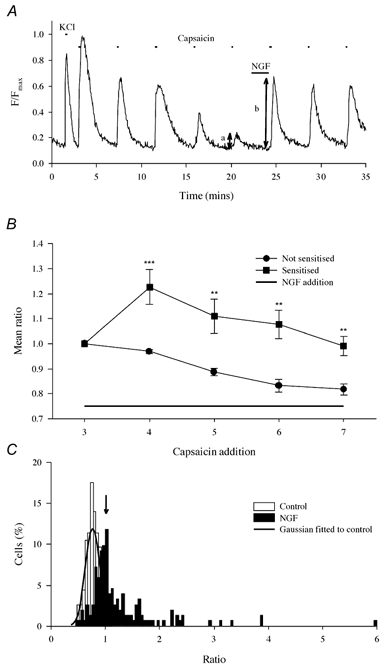
A, trace illustrating a typical experimental procedure. Ca2+ increases shown in a single neurone from a coverslip containing (typically) 10–30 neurones. Neurones loaded with the Ca2+-sensitive fluorophore fluo-4 as detailed in Methods. The Ca2+ increase observed on application of KCl (25 mm, 15 s first exposure) distinguished neuronal from non-neuronal cells. Subsequent applications of capsaicin (500 nm, 15 s) caused activation of TRPV1, which was potentiated by exposure to NGF (100 ng ml−1, 2 min). NGF did not itself cause an increase in [Ca2+]i in any experiment. The ratio b/a of the Ca2+ increases before and after exposure to NGF was used as an index of enhancement. B, NGF causes a long-lasting enhancement. Neurones (ncell = 32) were separated into two groups: those in which an enhancement was (▪) or was not (•) observed in the first exposure following NGF addition (lower bar). Bars give ± s.e.m. Significance levels (two-tailed t test) are: **P < 10−2; ***P < 10−3. (NB in this experiment a continuous NGF exposure followed the third capsaicin application while in all others a 2 min exposure was given between the fifth and sixth capsaicin applications). C, collected ratio values obtained from experiments as in A. Open bars give ratios obtained without exposure to NGF (ncell = 112, nexp = 8). The distribution was well fitted by a Gaussian function with mean of 0.76, s.d. of 0.134 and upper 95 % two-tailed confidence limit of 1.023 (arrow). Filled bars give ratios following 2 min exposure to NGF (100 ng ml−1; ncell = 152, nexp = 26). Following NGF exposure 38.74 ± 5.61 % of ratios exceeded the 95 % confidence limit, and the mean of these ratio values was 1.81 ± 0.14.
Maximal dye fluorescence (Fmax) was measured at the end of the experiment following elevation of [Ca2+]i to a high level by application of the Ca2+ ionophore ionomycin (10 μm) in a high Ca2+, high K+ solution (mm: 30 CaCl2, 125 KCl, 10 Hepes, pH 7.4). Background fluorescence from instrumental sources and from intrinsic cell fluorescence was then measured by lysing cells with water and was subtracted from the fluorescence signal to obtain the Ca2+-dependent fluorescence F, which was then expressed as F/Fmax.
All experiments were carried out at room temperature (18–20 °C).
Data analysis
Amplitudes of Ca2+ increases, ΔF/Fmax, caused by stimulation of neurones with capsaicin were measured by subtracting the ‘baseline’F/Fmax (mean for 30 s prior to capsaicin addition) from the peak F/Fmax achieved on exposure to capsaicin. Possible sensitising actions of, for example, NGF on the capsaicin-induced Ca2+ increase were characterised by taking the ratio of ΔF/Fmax after the sixth to that after the fifth application of capsaicin (b and a in Fig. 1, respectively). In the absence of any treatment the distribution of these ratios was well fitted by a normal distribution (see Fig. 1C), and from the mean and standard deviation of this control distribution a criterion level for denoting a neurone as exhibiting sensitisation in response to NGF application was set at 1.96 s.d. above the mean (the 5 % two-tailed confidence level). The mean percentage of cells exhibiting sensitisation in response to NGF for each experimental condition was compared statistically by one-way analysis of variance (ANOVA) with Bonferroni's post hoc test. Pairwise comparisons were carried out using Student's t test, using SPSS for Windows.
Immunocytochemistry
Cultures of DRG neurones (control or exposed to stimuli such as NGF or bradykinin) were fixed for 10 min at room temperature (4 % w/v formaldehyde, 4 % w/v sucrose dissolved in 50 % PBS:50 % distilled water) and were then washed three times in PBS before permeabilisation in Triton X-100 (0.2 % in PBS, 10 min at 4 °C) and incubation with primary antibody (overnight at 4 °C in TTBS: 0.1 M Tris HCl, 0.9 % NaCl w/v, 0.3 % Triton X-100 v/v, pH 7.4) in the presence of 10 % goat serum (Sigma) to reduce non-specific binding. Primary antibodies (polyclonal) were used at the following concentrations: anti-PKCδ (Olivier & Parker, 1994), 12.5 μg ml−1; anti-PKCε (Schaap et al. 1989), 15 μg ml−1; anti-PKCζ (Upstate Biotechnologies), 12 μg ml−1; anti-CaMK II (Transduction Laboratories), 0.5 μg ml−1. Coverslips were then washed three times in PBS followed by exposure to secondary antibody (anti-rabbit Alexa Fluo 488, Molecular Probes, 4 μg ml−1, 2 h at room temperature) and mounted with Moviol (Calbiochem). Neurones were visualised using a BioRad MicroRadiance confocal microscope. Specificity of antibodies was checked by pre-incubation with the peptide used to raise the antibody (at × 20 antibody concentration); in all cases neurone-specific labelling was completely blocked.
RESULTS
NGF-induced sensitisation of TRPV1
Neonatal mouse neurones were used in the present study as they are readily cultured and a high proportion respond to capsaicin. We found that 83 % of neonatal mouse DRG neurones cultured for 3 days as described in Methods responded to 500 nm capsaicin with an increase in Ca2+-dependent fluorescence of ΔF/Fmax > 0.05. To investigate the possibility that the remaining 17 % of neurones were capsaicin-sensitive but that the responses were below the threshold of detection, we exposed cells to phorbol myristate acetate (PMA, 1 μm), which enhances the response of TRPV1 in DRG neurones by activating PKC (Cesare & McNaughton, 1996; Vellani et al. 2001). Treatment of cultures with PMA substantially enhanced the mean amplitude of the Ca2+ increase elicited by capsaicin, but caused no alteration in the proportion of neurones that responded to capsaicin with a detectable Ca2+ signal (data not shown).
NGF caused a sensitisation of the response to capsaicin which was large in some neurones (Fig. 1A) and absent in others. The variability in the degree of sensitisation is likely to reflect the variable expression of the TrkA receptor for NGF in the population of DRG neurones (Molliver & Snider, 1997). The sensitisation caused by NGF was both rapid (< 2 min, Fig. 1A) and long-lasting, as sensitisation at a level significantly above control remained for at least 16 min in the continued presence of NGF (Fig. 1B).
Repeated application of capsaicin caused progressive desensitisation of the Ca2+ increase, an effect that has been shown to depend on Ca2+ entry through capsaicin-gated channels (Docherty et al. 1996; Vellani et al. 2001). We therefore allowed desensitisation to reach a quasi-steady state by applying capsaicin five times before exposing the neurone to NGF (see protocol in Fig. 1A; Fig. 1B is the only experiment in which an earlier application protocol was adopted). Sensitisation was measured by calculating the ratio between the Ca2+ increase observed on the sixth capsaicin application, after exposure to NGF, to that observed on the fifth application, immediately before exposure to NGF.
In order to determine the degree of sensitisation and the proportion of neurones sensitised by NGF we first studied the variability of the Ca2+ signal with no NGF added. The ratios produced by dividing the sixth by the fifth capsaicin-evoked Ca2+ increase in 112 cells (ncell = 112) from eight separate experiments (nexp = 8) were found to be normally distributed with a mean of 0.76, s.d. of 0.134 and upper 95 % two-tailed confidence limit of 1.023 (Fig. 1C). The mean percentage of ‘false positives’ with ratios above 1.023 was 1.88 ± 1.24 % (all values are means ± s.e.m.).
Upon addition of NGF for 2 min between the fifth and sixth pulses of capsaicin, the ratio values shifted to the right to a variable extent (Fig. 1C). A simple and readily calculated index of sensitisation was the percentage of neurones for which the ratio exceeded the 5 % two-tailed confidence limit (arrow in Fig. 1C). Sensitisation on this definition occurred in 38.74 ± 5.61 % of neurones. Because 1.88 % of these ratio values were false positives (see above), ≈37 % of DRG neurones in culture were sensitised by NGF. A second useful index of sensitisation is the mean enhancement of the ratio in these sensitised neurones, which was 1.81 ± 0.14.
Sources of Ca2+ contributing to NGF-induced sensitisation
Activation of TRPV1 causes a direct influx of Ca2+ though the capsaicin-gated ion channel, but it also depolarises the neurone, which may supplement the Ca2+ entry by allowing entry through voltage-gated Ca2+ channels (VGCCs). Release from intracellular stores may also enhance the Ca2+ signal. An effect of NGF on these processes could enhance the Ca2+ signal without the need to invoke a direct effect on TRPV1. In addition, an NGF-dependent change in the threshold of other voltage-sensitive ion channels (Zhang et al. 2002), or indeed an NGF-dependent modulation of any process which alters neuronal excitability, could also lead to enhanced Ca2+ influx.
To test whether or not the observed sensitisation was attributable to an indirect effect of NGF, the depolarising effects of TRPV1 activation were mimicked by applying pulses of 25 mm KCl, which caused a Ca2+ signal of comparable magnitude to that elicited by 500 nm capsaicin, and NGF was applied between the fifth and sixth KCl applications (Fig. 2A). In parallel experiments the ratio values were calculated in control experiments without the addition of NGF. NGF did not cause a significant difference between the mean ratio values, which were 0.870 ± 0.097 for control experiments and 0.869 ± 0.078 with NGF (P = 0.82, not significant, two-tailed t test). The percentage of neurones above the upper 95 % two-tailed confidence limit (1.13 for exposures to KCl) was also not significantly different (0 % in control and 2.42 ± 1.32 % following NGF; see histogram in Fig. 2D). These data show that NGF did not sensitise the depolarisation-induced Ca2+ influx, and therefore that the effect of NGF on the Ca2+ responses evoked by capsaicin is not due to changes in neuronal excitability.
Figure 2. NGF enhances Ca2+ entry through TRPV1 but not via other routes.
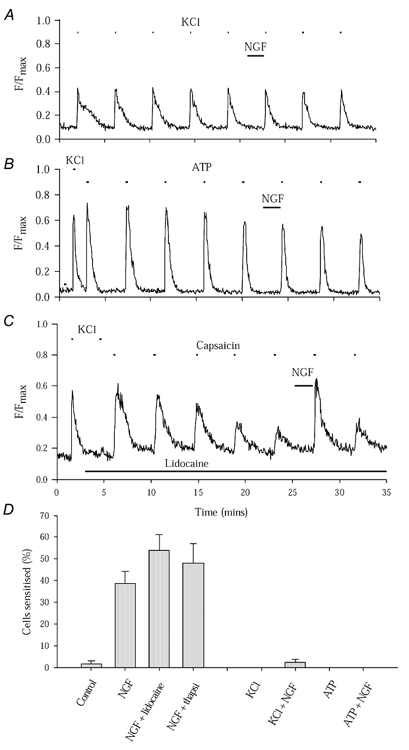
A, NGF has no effect on the Ca2+ increase evoked by KCl (25 mm, 9 s). B, NGF has no effect on the Ca2+ increase evoked by ATP (100 μm, 15 s). Neurones incubated with thapsigargin (10 μm) for 20 min prior to experiments; the Ca2+ increase evoked by thapsigargin lasted 5–7 min, showing that stores were completely emptied with a 20 min pre-exposure. C, enhancement of TRPV1 function by NGF is observed following complete block of voltage-sensitive ion channels by lidocaine (lignocaine; 2 mm, black bar). KCl-evoked Ca2+ increase (see two applications at start) is completely abolished by lidocaine but enhancement of capsaicin-evoked [Ca2+]i increase by NGF is still observed. D, collected results of experiments similar to those in A, B and C, together with experiment similar to C in which thapsigargin (10 μm, applied 20 min prior to experiment) was used to empty subcellular Ca2+ stores. Bars give ± s.e.m. Numbers of cells and separate experiments as follows: control, ncell = 112, nexp = 8; NGF alone, ncell = 152, nexp = 26; NGF + lidocaine, ncell = 60, nexp = 8; NGF + thapsigargin, ncell = 71, nexp = 16; KCl, ncell = 88, nexp = 7; KCl + NGF, ncell = 144, nexp = 7; ATP, ncell = 23, nexp = 9; ATP + NGF, ncell = 63, nexp = 15.
We next tested whether NGF could sensitise Ca2+ signals caused by activation of ATP-gated ion channels. P2X2 and P2X3 ATP-gated channels are present in DRG neurones and ATP released from damaged cells is thought to be an activator of primary sensory neurones (Chen et al. 1995; Hamilton & McMahon, 2000). Neurones were pretreated with thapsigargin (10 μm) to empty Ca2+ stores and so eliminate the possibility that a component of the effects of ATP on [Ca2+]i may result from P2Y-induced release of Ca2+. Neurones were exposed to pulses of 100 μm ATP (Fig. 2B), and TRPV1-expressing cells were selected by testing responsiveness to capsaicin at the end of the experiment. Mean ratio values from experiments with and without exposure to NGF were not significantly different (0.86 ± 0.06 in control experiments and 0.81 ± 0.03 with NGF addition, not significant, P = 0.13). The upper two-tailed 95 % confidence limit from control data was 1.33 and no cells in either group had ratios greater than this (see histogram in Fig. 2D). These data show that NGF does not sensitise responses mediated by P2X receptors in DRG neurones.
We next tested whether inhibition of voltage-gated Na+ channels (VGSCs) or emptying of internal Ca2+ stores changed the proportion of cells sensitised by NGF. Lidocaine (lignocaine; 2 mm throughout the experiment) inhibits all VGSCs in DRGs (Scholz & Vogel, 2000), and we found that the Ca2+ increase in response to application of 25 mm KCl was rapidly and completely abolished (Fig. 2C), but there was no effect on the enhancement by NGF of the capsaicin-evoked Ca2+ signal. As shown in Fig. 2D, the proportion of neurones sensitised by NGF in the presence of lidocaine was 54.03 ± 7.31 %, not significantly different from the proportion sensitised in the absence of lidocaine (P = 0.23). We also emptied intracellular stores using 10 μm thapsigargin, a concentration sufficient to evoke a large Ca2+ signal which returned to baseline within 20 min. After emptying intracellular Ca2+ stores with thapsigargin (20 min pre-application) 48.02 ± 9.01 % of neurones were sensitised by NGF, not significantly different to the proportion in the absence of NGF (P = 0.825). Values with lidocaine and thapsigargin are highly significantly different to the proportion of cells sensitised without NGF application (P = 4.5 × 10−4 and 7 × 10−3, respectively).
These results show that modulation by NGF of VGSCs or VGCCs, or of the release of Ca2+ from intracellular stores, does not mediate the NGF-evoked sensitisation of the Ca2+ signal, and nor was a comparable sensitisation of the Ca2+ signal evoked by P2X ion channel activation observed following NGF application. We conclude that the Ca2+ imaging method outlined above provides a simple and direct means of monitoring direct modulation of TRPV1 following NGF activation, and that the measurement is not contaminated to any significant extent by artefacts which might arise from modulation of other cellular processes by NGF.
Intracellular signalling cascades activated by NGF
NGF-induced autophosphorylation of tyrosine residues in the C-termini of TrkA receptors recruits several intracellular signalling cascades, most notably pathways leading to activation of PLCγ, PI3K and ras (Kaplan & Stephens, 1994; Kaplan & Miller, 2000). The Ca2+ imaging technique described above was used in conjunction with specific inhibitors to block each of these pathways in turn and hence establish their importance in mediating the NGF-induced sensitisation of TRPV1.
Inhibition of phospholipase Cγ
In preliminary experiments we attempted to inhibit PLCγ by applying the widely used inhibitor U73122, which has been shown to abolish agonist-evoked PLC activation (Smith et al. 1990; Broad et al. 1999). U73122 proved to have unacceptable side effects in DRG neurones, however, because at the concentration of 10 μm required to abolish PLC activation, a long-lasting increase in resting [Ca2+]i was observed following capsaicin addition, which reached saturating levels within 3–5 min. This inhibitor was therefore not used further.
An alternative PLC inhibitor is neomycin, which has been used successfully in several systems, including cultured DRG neurones (Eun et al. 2001; Fernandez-Tome et al. 2002). In the presence of neomycin the proportion of neurones sensitised following NGF exposure was 34.6 ± 4.5 %, comparable to the value of 38.74 ± 5.61 % in the absence of neomycin (Fig. 3A and Fig. 4A; difference not significant, P = 0.89, but highly significantly different from neurones without NGF application, P = 1 × 10−3). The mean amplitude of sensitisation in those neurones exceeding the 95 % criterion level was, however, slightly less in the presence of neomycin, with a mean ratio value of 1.34 ± 0.04 (Fig. 4B), significantly less than the value of 1.81 ± 0.14 observed in NGF alone (P = 1.6 × 10−3).
Figure 3. The effects of inhibition of PLC, PI3K, ras, and MEK1 and 2 on NGF-induced sensitisation of TRPV1.
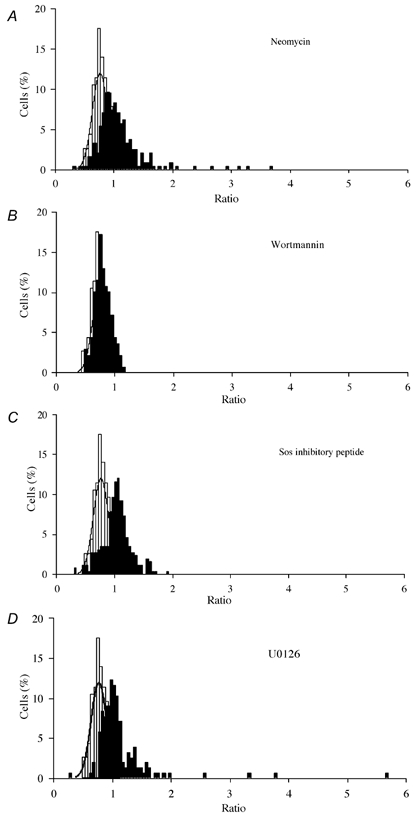
Ratio distributions as in Fig. 1C are shown for NGF (100 ng ml−1) plus the following treatments, compared with control ratio distribution in absence of NGF (fitted with Gaussian function as in Fig. 1C): A, 10 μm neomycin; B, 20 nm wortmannin; C, 10 μm Sos-inhibitory peptide; D, 10 μm U0126. Inhibitors were applied 5 min before experiment and were included in all solutions. Numbers of cells and separate experiments as follows: neomycin, ncell = 240, nexp = 33; wortmannin, ncell = 139, nexp = 20; Sos-inhibitory peptide, ncell = 259, nexp = 20; U0126, ncell = 155, nexp = 8.
Figure 4. Sensitisation of TRPV1 by NGF following inhibition of PLC, PI3K, ras and MEK.

A, the proportion of neurones sensitised in the presence of 10 μm neomycin, 20 nm wortmannin, 10 μm Sos-inhibitory peptide and 10 μm U0126, to inhibit PLC, PI3K, ras and MEK activation respectively. B, mean ratio values of sensitised neurones. Error bars show means ± s.e.m. Significance levels: *P < 5 %; **P < 1 %; ***P < 0.1 %.
Inhibition of phosphatidylinositol-3 kinase
The contribution of PI3K to NGF-induced sensitisation of TRPV1 was studied by using the potent PI3K inhibitor wortmannin, which at 20 nm is a highly specific inhibitor of PI3K, with actions on other kinases only apparent at much higher concentrations (the lowest being IC50 values of 200 nm for MLCK and 260 nm for SmMLCK, see Davies et al. 2000). In the presence of 20 nm wortmannin the amplitude of the Ca2+ increase caused by activation of TRPV1 by capsaicin was normal, but the sensitising effect of NGF was completely abolished (Fig. 3B). Ratios following NGF application in the presence of wortmannin were normally distributed, with mean ratio 0.78 ± 0.14 (see Fig. 3B), similar to the value of 0.76 ± 0.134 in the absence of NGF. Wortmannin treatment reduced the proportion of capsaicin-responsive neurones that were sensitised by NGF to 5.79 ± 0.58 % (Fig. 4A) compared with 38.74 ± 5.61 % in the absence of wortmannin. The reduction in the proportion of neurones sensitised by NGF in the presence of wortmannin was highly significant when compared with cultures treated with NGF alone (P = 1.2 × 10−3), and the proportion of neurones sensitised by NGF in the presence of wortmannin was not significantly different from the proportion of false positive values in control data (P = 0.120). These data show that PI3K activation is critical for the NGF-activated sensitisation of TRPV1.
Inhibition of Sos-ras binding and MEK activation
Binding of Sos to ras was inhibited by means of a peptide that binds to the SH3 region of Sos (Cussac et al. 1999). The peptide is linked to a 16-residue sequence of the homeodomain of Antennapedia that promotes membrane permeation by a non-receptor-dependent process, causing complete inhibition of Sos-dependent downstream signalling from TrkA. Neurones were exposed to 10 μm Sos-inhibitory peptide (Upstate Biotechnology) for 20 min prior to experimentation, and the peptide was also included throughout. The percentage of neurones sensitised by NGF in the presence of Sos-inhibitory peptide (44.49 ± 5.84 %, Fig. 4A) was comparable to the percentage of neurones sensitised when treated with NGF alone (P = 0.65), but the suppression of some of the largest values of sensitisation meant that the mean ratio value from sensitised cells was 1.21 ± 0.02, compared with the value of 1.81 ± 0.14 from neurones exposed to NGF alone (Fig. 4B; significantly different, P = 6.4 × 10−5).
To further investigate a possible involvement of the ras signalling pathway we used the inhibitor U0126 (10 μm, Calbiochem), a potent and selective inhibitor of the kinases MEK1 (IC50 = 72 nm) and MEK2 (IC50 = 58 nm), which are activated downstream of ras (Davies et al. 2000; Kaplan & Miller, 2000). Figure 3D shows that the proportion of neurones sensitised by NGF when MEK was inhibited was 33.01 ± 2.71 % (Fig. 4A), not significantly different from that observed with NGF alone (P = 0.406) but very significantly higher than control levels of sensitisation (P = 9 × 10−3). The mean amplitude of sensitisation by NGF in the presence of U0126 was 1.41 ± 0.09, significantly different at the 5 % level but not at the 1 % level from that in the presence of NGF alone (P = 0.016, Fig. 4B).
These observations do not support the idea that the ras/MEK pathway is solely responsible for short-term sensitisation, but do suggest a modulatory effect of the ras/MEK pathway on the PI3K-induced signalling cascade, as has been found in other studies (Rodriguez-Viciana et al. 1994).
Involvement of serine/threonine kinases in NGF-induced sensitisation
The amino acid sequence for TRPV1 contains potential phosphorylation sites for many kinases, amongst them PKC, PKA and CaMK II, all of which are involved in many different intracellular signalling pathways and could form part of a second-messenger signalling cascade activated by PI3K and ultimately leading to phosphorylation of TRPV1. Staurosporine is a broad-spectrum kinase inhibitor, which at the concentration used (200 nm) will inhibit PKC, PKA, CamK II and other kinases (Wilkinson & Hallam, 1994). In the presence of staurosporine there was a reduction in the percentage of neurones sensitised by NGF when compared with neurones treated with NGF alone (Fig. 5A), from 38.74 ± 5.61 % to 7.25 ± 2.69 % (difference highly significant, P = 1.9 × 10−5), the latter value not significantly different from the proportion of ‘false positives’ in control data from neurones not treated with NGF (P = 0.107). The mean ratio value for these sensitised neurones is 1.12 ± 0.04 (Fig. 6B), highly significantly different from the mean ratio in NGF alone (P = 1.08 × 10−5).
Figure 5. The effect of inhibition of PKC, PKA and CaMK II on NGF-induced sensitisation of TRPV1.
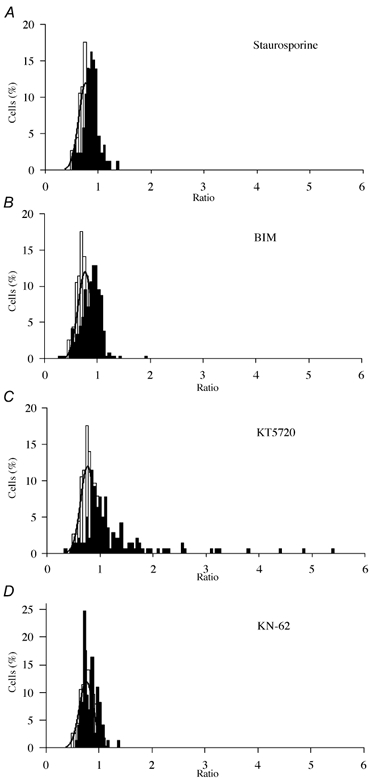
Ratio distributions as in Fig. 1C shown for NGF (100 ng ml−1) plus the following treatments, compared with control ratio distribution in absence of NGF (fitted with Gaussian function as in Fig. 1C): A, 200 nm staurosporine; B, 500 nm BIM; C, 200 nm KT5720; D, 1 μm KN-62. Inhibitors were pre-applied for 5 min and were included in all solutions throughout the experiment. Numbers of cells and separate experiments as follows: staurosporine, ncell = 86, nexp = 7; BIM, ncell = 264, nexp = 15; KT5720, ncell = 141, nexp = 7; KN-62, ncell = 73, nexp = 8.
Figure 6. Sensitisation of TRPV1 by NCF following inhibition of PKC, PKA and CaMK II.
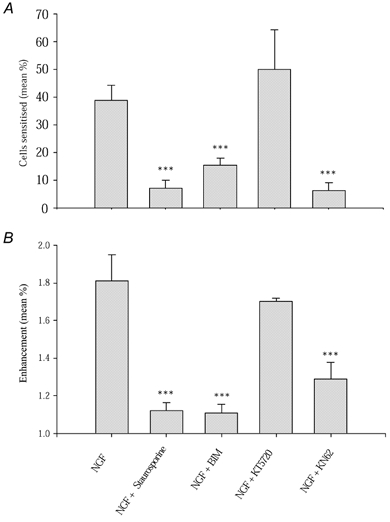
A, the proportion of neurones sensitised by NGF in the presence of 200 nm staurosporine, 500 nm BIM to inhibit PKC, 200 nm KT5720 to inhibit PKA and 1 μm KN-62 to block CaMK II activity. B, mean ratio values of sensitised neurones. Error bars show means ± s.e.m. Significance levels: ***P < 0.1 %.
Specific inhibition of PKC
PKC was inhibited by the use of the specific PKC inhibitor bisindolylmaleimide I (BIM, 500 nm, Calbiochem), which inhibits PKA only at concentrations exceeding 2 μm (Obreja et al. 2002). PKC inhibition largely abolished the NGF-induced sensitisation of TRPV1 (Fig. 5B), but the distribution of ratios was skewed to the right when compared with control, indicating that some modest NGF-induced sensitisation of TRPV1 had occurred. Exposure to BIM reduced the percentage of cells responsive to capsaicin that were sensitised by NGF to 15.33 ± 2.78 % (Fig. 6A), significantly lower than that obtained from neurones treated with NGF alone (38.74 ± 5.61 %, P = 1 × 10−4), but significantly higher than in the absence of NGF under control conditions (1.88 ± 1.24 %, P = 2.62 × 10−4). The mean ratio of enhancement for neurones that were sensitised by NGF in the presence of BIM was 1.11 ± 0.02 (Fig. 6B), lower than with NGF alone (P = 5.8 × 10−6) but above control.
Bradykinin causes sensitisation of TRPV1 by specifically translocating PKCε to the membrane (Cesare et al. 1999), where it phosphorylates serine residues on TRPV1 (Numazaki et al. 2002). In view of the evidence for an involvement of PKC in the NGF-activated signalling pathway leading to sensitisation of TRPV1, we investigated the possibility that PKC isoforms might be translocated to the membrane by NGF. We examined the βI, βII, ε, δ and ζ isoforms of PKC, whose cellular location in the mouse neurones used in the present experiments was similar to that already described for rat DRG neurones (Cesare et al. 1999). Upon exposure to NGF, PKCε was not observed to translocate to the plasma membrane while a clear translocation of PKCε was observed following exposure to bradykinin (Fig. 7). Translocation of the PKC βI, βII, δ and ζ isoforms was also not observed in response to exposure to NGF (data not shown).
Figure 7. Cellular location of PKCε following exposure to bradykinin (left, 1 μm for 30 s) and NGF (100 ng ml−1, 2 min).
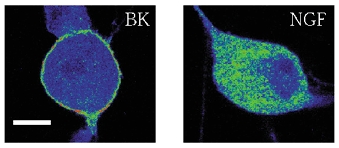
Typical images of neurones fixed and imaged in a confocal microscope as described in Methods. False-colour images with blue indicating low fluorescence and red high fluorescence. Scale bar: 10 μm.
Specific inhibition of PKA
PKA phosphorylates TRPV1 and enhances its response to capsaicin by abolishing desensitisation (Lopshire & Nicol, 1997; Bhave et al. 2002). We tested whether PKA is involved in the sensitisation by NGF of TRPV1 using the specific PKA inhibitor KT5720 (200 nm, Calbiochem; Cai et al. 1999). PKA inhibition did not affect the desensitisation caused by repeated capsaicin addition, and nor, when NGF was applied, did it have any obvious effect on either the proportion of neurones sensitised by NGF or on the amplitude of sensitisation (Fig. 5C). In the presence of KT5720 49.95 ± 14.41 % of neurones were sensitised by NGF (Fig. 6A), not significantly different from NGF alone (P = 0.489), and significantly greater than the proportion in the absence of NGF (P = 5 × 10−3). The mean ratio amplitude in sensitised neurones with KT5720 (1.69 ± 0.11) was also not significantly different from that in the presence of NGF alone (P = 0.52).
Inhibition of CaMK II
We used a CaMKII-specific antibody to determine whether CaMKII is expressed in DRG neurones and therefore could be a possible participant in the signalling pathway leading to modulation of TRPV1. CaMK II was found to be expressed strongly in 56.3 % of cultured mouse DRG neurones (ncell = 1080, nexp = 5, data not shown). We studied the effects of CaMK II inhibition using the specific CaMK II inhibitor, KN-62 (1 μm, Calbiochem), which inhibits CaMK II by binding to the calmodulin binding site (Tokumitsu et al. 1990). We first tested for a possible interaction of KN-62 with PKC by applying the specific PKC activator PMA, which causes a substantial enhancement in current through TRPV1 (Cesare & McNaughton, 1996; Vellani et al. 2001). Inhibition of CaMK II activity by 1 μm KN-62 did not prevent sensitisation of TRPV1 by PMA, showing that KN-62 does not interfere with PKC activation (data not shown). In the presence of the CaMK II inhibitor, however, the sensitising effect of NGF was found to be almost completely abolished (Fig. 5D). With KN-62 6.41 ± 2.71 % of neurones were sensitised by NGF (Fig. 6A), which is significantly less than that with NGF exposure alone (P = 1.5 × 10−4) and not significantly above that in the absence of NGF (P = 0.24). The mean ratio increase of the few sensitised neurones was 1.13 ± 0.06, highly significantly different from the value in NGF (P = 2.8 × 10−5) (Fig. 6B).
DISCUSSION
The Ca2+ imaging technique used in the present study has the advantage that large numbers of neurones can be monitored simultaneously, thus increasing the probability that sensitisation occurring in only a limited population of neurones could be studied in reasonable numbers of cells. NGF was shown to cause a significant enhancement of the response to capsaicin in 37 % of capsaicin-responsive DRG neurones, and the amplitude of the enhancement was up to sixfold. Although it was not directly investigated in the present study, NGF is likely to act via TrkA and not p75NTR receptors because normal sensitisation in response to NGF injection was observed in p75NTR knockout mice (Bergmann et al. 1998).
The possibility was investigated that NGF may act, in part or wholly, through mechanisms other than an enhancement of TRPV1 channel gating. Activation of TRPV1 depolarises the neurones, and some of the Ca2+ increase following exposure to capsaicin may be due to influx through voltage-gated Ca2+ channels or other voltage-dependent Ca2+ entry pathways, which in turn could be modulated by NGF. However, when neurones were directly depolarised with KCl or by activation of ATP-gated ion channels NGF did not enhance the Ca2+ signal, and neither complete block of voltage-gated channels with lidocaine nor emptying intracellular stores with thapsigargin affected the NGF-induced enhancement of TRPV1. The enhancement of the capsaicin-induced Ca2+ signal is therefore due to a direct action of NGF in enhancing Ca2+ influx through TRPV1.
Involvement of intracellular signalling cascades activated by TrkA
Binding of NGF to TrkA receptors activates three main signalling cascades, in which the initial steps involve activation of PLC, PI3K and ras, and we investigated the effect of inhibitors of each of these pathways. The PLC inhibitor U73122 caused an irreversible increase in [Ca2+]i in DRG neurones, so we used instead neomycin, which has also been shown to be an effective inhibitor of PLC. Application of neomycin did not prevent NGF-mediated sensitisation of TRPV1, showing that PLC is unlikely to be involved. In contrast, inhibition of PI3K with wortmannin, which is a specific inhibitor for PI3K at the concentrations used, caused complete abolition of the NGF-induced enhancement of TRPV1 channel gating, highlighting the critical importance of pathways initiated by PI3K. Suppression of ras activation using the Sos inhibitory peptide significantly reduced the amplitude of sensitisation caused by NGF, but did not affect the proportion of neurones in which sensitisation was observed. These results suggest that ras does not cause sensitisation itself but instead enhances the degree to which the PI3K pathway is able to sensitise TRPV1, consistent with the observation that ras interacts with the catalytic domain of PI3K to enhance PI3K activity (Rodriguez-Viciana et al. 1994). The effects of ras are not mediated by downstream activation of MEK1 and 2, because inhibition of these MEKs had no effect on the proportion of neurones sensitised and only a marginally significant effect on the amplitude of sensitisation. This last result agrees with a recent study by Shu & Mendell (2001) who, using the patch-clamp technique, applied the MEK inhibitor PD98059 between the additions of capsaicin, and found no significant reduction in the sensitisation caused by NGF.
In a recent study, Chuang et al. (2001) investigated NGF-induced sensitisation of the response to capsaicin using HEK293 cells transfected with TRPV1 and TrkA receptors. These authors found that activation of PLC was essential for NGF-induced sensitisation of TRPV1 in HEK293 cells, in contrast to the present study where inhibition of PLC with neomycin was found to have no significant effect. They proposed that TRPV1 is tonically inhibited by binding to PtdIns-4,5-P2, and that breakdown of PtdIns-4,5-P2 by PLC removes the inhibition. The disparity between the work of Chuang et al. (2001) and our work may result from the different types of cells in which NGF-evoked sensitisation was studied. In transient expression systems TRPV1 protein is heavily overexpressed by comparison with the physiological expression levels in sensory neurones. TRPV1 has been shown to be more glycosylated in HEK293 cells when compared with DRG neurones (Kedei et al. 2000). TRPV1 has also been shown to form homotetramers in the plasma membrane of DRG neurones, but a range of multimeric states of TRPV1 were found in HEK293 cells. These multimers ranged from monomers to aggregates of protein with molecular weights far greater than the tetrameric form of TRPV1 (Kedei et al. 2000). Finally, TRPV1 was only found in the plasma membrane of DRG neurones whereas it was distributed throughout internal membranes as well as the plasma membrane in transiently transfected HEK293 cells, thus highlighting differences in protein targeting in native and transient expression systems (Jahnel et al. 2001).
Type I PI3Ks are activated by TrkA and preferentially phosphorylate PtdIns-4,5-P2 over PtdIns (Wymann & Pirola, 1998). Phosphorylation of PtdIns-4,5-P2 on the D3 hydroxyl group produces PtdIns-3,4,5-P3, which subsequently recruits PLC to TrkA (Falasca et al. 1998). If the hypothesis proposed by Chuang and colleagues (2001) was correct and PtdIns-4,5-P2 does indeed constitutively inhibit TRPV1, then either PI3K or PLC could remove PtdIns-4,5-P2 from TRPV1. A parsimonious explanation is that differences in signalling cascades between these two cell types could mean that PI3K and PLC can both cause removal of PtdIns-4,5-P2 from TRPV1, by conversion to PtdIns-3,4,5-P3 in DRG neurones and breakdown to DAG and IP3 in HEK293 cells, respectively. This hypothesis would not, however, account for the actions of kinase inhibitors in abolishing NGF-induced sensitisation, as described below.
Involvement of serine/threonine kinases in NGF-induced sensitisation
Consensus sequences for phosphorylation by PKC, PKA and CaMK II exist on intracellular domains of TRPV1, and functionally active sites for PKC and PKA phosphorylation have recently been identified (Bhave et al. 2002; Numazaki et al. 2002). We therefore explored the possibility that activation of kinases by TrkA-recruited pathways may underlie the sensitisation of TRPV1. Application of the broad-spectrum kinase inhibitor staurosporine was found to completely abolish NGF-evoked sensitisation of TRPV1. The specific PKC inhibitor BIM produced a similar but slightly less complete inhibition than that observed using staurosporine, suggesting that PKC is at least partly responsible for NGF-induced sensitisation. Inhibition of PKA with the specific inhibitor KT5720 did not affect sensitisation of TRPV1 by NGF, showing that the action of staurosporine is not due to inhibition of PKA. Immunocytochemical studies were conducted to identify the PKC isoform involved in sensitisation by looking for translocation of PKC within the cell. Both conventional and novel PKCs bind to DAG and phosphatidylserine in membranes following activation (Liu & Heckman, 1998), and translocation of PKC to the membrane is therefore a sensitive indicator of activation (Cesare et al. 1999). Translocation of PKC isoforms βI, βII, ε, δ and ζ was, however, not observed in response to NGF application, suggesting that none of these isoforms is involved. The identity of the PKC isoform which may mediate the sensitisation is therefore presently unclear.
In contrast to the work described here, Shu & Mendell (2001) did not observe an inhibitory effect of BIM (500 nm) on sensitisation by NGF of the capsaicin-induced membrane current in voltage-clamped DRG neurones, but they did report an involvement of PKA. The reasons for the conflicting findings between our work and that of Shu & Mendell (2001) are unclear. There are a number of differences in experimental protocol, of which the most significant may be that capsaicin was repeatedly applied in our experiments, and TRPV1 would have therefore been in a relatively desensitised state, while in the experiments of Shu & Mendell (2001) two applications separated by 10 min were used. A second difference is that neurones in the present study were intact, while the patch clamp technique used by Shu & Mendell (2001) dialyses the intracellular milieu and may interfere with signalling pathways.
Finally, we examined the possibility that CaMK II may be involved in the sensitisation pathway. CaMK II is expressed in 56 % of neurones, and using the specific CaMK II inhibitor KN-62 we found that the NGF-mediated sensitisation of TRPV1 was abolished. These results suggest that both PKC and CaMK II may be involved in sensitisation by NGF. A possible order for the players in the pathway leading to TRPV1 phosphorylation is PI3K → CaMK II → PKC → TRPV1, as sensitisation of TRPV1 by direct activation of PKC using PMA was unaffected by the CaMK II inhibitor.
In conclusion, we have identified the PI3K pathway to be crucial as an initial step in mediating sensitisation of TRPV1 by NGF. The ras pathway may have a modulatory role, but is it is unlikely to be on the direct pathway leading to TRPV1 modulation, as neither ras nor MEK inhibitors abolish the effect of NGF. There is evidence that both PKC and CaMK II are involved downstream of PI3K, but their respective roles, and whether either directly phosphorylates TRPV1, has yet to be established.
Acknowledgments
This work was funded by the Biotechnology and Biological Sciences Research Council.
REFERENCES
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitisation by extracellular signal-regulated kinases. J Neurosci. 2001;21:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann I, Reiter R, Toyka KV, Koltzenburg M. Nerve growth factor evokes hyperalgesia in mice lacking the low-affinity neurotrophin receptor p75. Neurosci Lett. 1998;255:87–90. doi: 10.1016/s0304-3940(98)00713-7. [DOI] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent protein kinase regulates desensitisation of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Broad LM, Cannon TR, Taylor CW. A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanism in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. J Physiol. 1999;517:121–134. doi: 10.1111/j.1469-7793.1999.0121z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R, Klesse LJ, Shah K, Parada LF, Winter J. Activation of Ras is necessary and sufficient for upregulation of vanilloid receptor type 1 in sensory neurons by neurotrophic factors. Mol Cell Neurosci. 2003;22:118–132. doi: 10.1016/s1044-7431(02)00022-2. [DOI] [PubMed] [Google Scholar]
- Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton PA. A novel heat-activated current in nociceptive neurons, and its sensitization by bradykinin. Proc Natl Acad Sci U S A. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN. A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995;377:428–431. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Cussac D, Vidal M, Leprince C, Liu WQ, Cornille F, Tiraboschi G, Roques BP, Garbay C. A Sos-derived peptidimer blocks the Ras signaling pathway by binding both Grb2 SH3 domains and displays antiproliferative activity. FASEB J. 1999;13:31–38. doi: 10.1096/fasebj.13.1.31. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Dikic I, Batzer AG, Blaikie P, Obermeier A, Ullrich A, Schlessinger J, Margolis B. Shc binding to nerve growth factor receptor is mediated by the phosphotyrosine interaction domain. J Biol Chem. 1995;270:15125–15129. doi: 10.1074/jbc.270.25.15125. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S, Boddeke HWGM. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- Eun SY, Jung SJ, Park YK, Kwak J, Kim SJ, Kim J. Effects of capsaicin on Ca(2+) release from the intracellular Ca(2+) stores in the dorsal root ganglion cells of adult rats. Biochem Biophys Res Commun. 2001;285:1114–1120. doi: 10.1006/bbrc.2001.5272. [DOI] [PubMed] [Google Scholar]
- Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998;17:414–422. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Tome MC, Speziale EH, Sterin-Speziale NB. Phospholipase C inhibitors and prostaglandins differentially regulate phosphatidylcholine synthesis in rat renal papilla. Evidence of compartmental regulation of CTP:phosphocholine cytidylyltransferase and CDP-choline:1, 2-diacylglycerol cholinephosphotransferase. Biochim Biophys Acta. 2002;1583:185–194. doi: 10.1016/s1388-1981(02)00208-1. [DOI] [PubMed] [Google Scholar]
- Hamilton SG, McMahon SB. ATP as a peripheral mediator of pain. J Auton Nerv Syst. 2000;81:187–194. doi: 10.1016/s0165-1838(00)00137-5. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnel R, Dreger M, Gillen C, Bender O, Kurreck J, Hucho F. Biochemical characterization of the vanilloid receptor 1 expressed in a dorsal root ganglia derived cell line. Eur J Biochem. 2001;268:5489–5496. doi: 10.1046/j.1432-1033.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- Kedei N, Szabo T, Lile JD, Treanor JJ, Olah Z, Iadarola MJ, Blumberg PM. Analysis of the native quaternary structure of vanilloid receptor 1. J Biol Chem. 2000;276:28613–28619. doi: 10.1074/jbc.M103272200. [DOI] [PubMed] [Google Scholar]
- Lee Y-J, Zachrisson O, Tonge DA, McNaughton PA. Upregulation of bradykinin b2 receptor expression by neurotrophic factors and nerve injury in mouse sensory neurons. Mol Cell Neurosci. 2002;19:186–200. doi: 10.1006/mcne.2001.1073. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Nerve growth factor and nociception. Trends Neurosci. 1993;16:353–359. doi: 10.1016/0166-2236(93)90092-z. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, Mendell LM. Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J Neurosci. 1993;13:2136–2148. doi: 10.1523/JNEUROSCI.13-05-02136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. Activation and recovery of the PGE2-mediated sensitization of the capsaicin response in rat sensory neurons. J Neurophysiol. 1997;78:3154–3164. doi: 10.1152/jn.1997.78.6.3154. [DOI] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: Whole-cell and single-channel studies. J Neurosci. 1998;18:6081–6092. doi: 10.1523/JNEUROSCI.18-16-06081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Molliver DC, Snider WD. Nerve growth factor receptor TrkA is down-regulated during postnatal development by a subset of dorsal root ganglion neurons. J Comp Neurol. 1997;381:428–438. doi: 10.1002/(sici)1096-9861(19970519)381:4<428::aid-cne3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Obermeier A, Halfter H, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Tyrosine 785 is a major determinant of Trk-substrate interaction. EMBO J. 1993;12:933–941. doi: 10.1002/j.1460-2075.1993.tb05734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obreja O, Rathee PK, Lips KS, Distler C, Kress M. IL-1 beta potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002;16:1497–1503. doi: 10.1096/fj.02-0101com. [DOI] [PubMed] [Google Scholar]
- Olivier AR, Parker PJ. Bombesin, platelet-derived growth factor, and diacylglycerol induce selective membrane association and down-regulation of protein kinase C isotypes in Swiss 3T3 cells. J Biol Chem. 1994;269:2758–2763. [PubMed] [Google Scholar]
- Petty BG, Cornblath DR, Adornato BT, Chaudhry V, Flexner C, Wachsman M, Sinicropi D, Burton LE, Peroutka SJ. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol. 1994;36:244–246. doi: 10.1002/ana.410360221. [DOI] [PubMed] [Google Scholar]
- Raffioni S, Bradshaw RA. Activation of phosphatidylinositol 3-kinase by epidermal growth factor, basic fibroblast growth factor, and nerve growth factor in PC12 pheochromocytoma cells. Proc Natl Acad Sci U S A. 1992;89:9121–9125. doi: 10.1073/pnas.89.19.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Schaap D, Parker PJ, Bristol A, Kriz R, Knopf J. Unique substrate specificity and regulatory properties of PKC-epsilon-a rationale for diversity. FEBS Lett. 1989;243:351–357. doi: 10.1016/0014-5793(89)80160-7. [DOI] [PubMed] [Google Scholar]
- Scholz A, Vogel W. Tetrodotoxin-resistant action potentials in dorsal root ganglion neurons are blocked by local anesthetics. Pain. 2000;89:47–52. doi: 10.1016/S0304-3959(00)00345-6. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett. 1999;274:159–162. doi: 10.1016/s0304-3940(99)00701-6. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM. Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol. 2001;86:2931–2938. doi: 10.1152/jn.2001.86.6.2931. [DOI] [PubMed] [Google Scholar]
- Smith RJ, Sam LM, Justen JM, Bundy GL, Bala GA, Bleasdale JE. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J Pharmacol Exp Ther. 1990;253:688–697. [PubMed] [Google Scholar]
- Soltoff SP, Rabin SL, Cantley LC, Kaplan DR. Nerve growth factor promotes the activation of phosphatidylinositol 3-kinase and its association with the trk tyrosine kinase. J Biol Chem. 1992;267:17472–17477. [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62,1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor, VR1, by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter ML, Martin-Zanca D, Parada LF, Bishop JM, Kaplan DR. Nerve growth factor rapidly stimulates tyrosine phosphorylation of phospholipase C-gamma 1 by a kinase activity associated with the product of the trk protooncogene. Proc Natl Acad Sci U S A. 1991;88:5650–5654. doi: 10.1073/pnas.88.13.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson SE, Hallam TJ. Protein kinase C: Is its pivotal role in cellular activation over- stated. Trends Pharmacol Sci. 1994;15:53–57. doi: 10.1016/0165-6147(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na(+) current and delayed rectifier K(+) current in rat sensory neurons. J Physiol. 2002;544:385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]