

Abstract
The α7 nicotinic acetylcholine receptor (nAChR) has been implicated widely in behavioural functions and dysfunctions related to the hippocampus, but the detailed mechanisms by which this receptor contributes to these behavioural processes have yet to be elucidated. In the present study, sustained application (5 min) of nicotine significantly lowered the threshold for synaptic plasticity, and thus a long-lasting potentiation was induced by a stimulus that would normally evoke only a short-term potentiation. This effect appeared to be mediated by α7 nAChRs, as it was inhibited by the α7 nAChR-specific antagonist α-bungarotoxin (100 nm), but not by mecamylamine (50 μm) or dihydro-β-erythroidine (DHβE; 1 μm) at concentrations known to be selective for non-α7 nAChRs. Further pharmacological dissection revealed that the effect was also abolished by the NMDA receptor antagonist, D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5; 50 μm). This blockade, however, unmasked a slowly developing nicotine-induced potentiation of field excitatory postsynaptic potential that appeared to be dependent on both α7 nAChR activation and non-α7 nAChR desensitisation. This secondary effect of nicotine was blocked by a combination of picrotoxin (50 μm) and saclofen (100 μm), and thus appeared to be mediated via GABAergic interneurons. The important implication of this study was that the sustained application of α7 nAChR agonists could modulate the conditions for synaptic plasticity through multiple transduction pathways, and not simply the inactivation of α7 nAChRs. These α7-nAChR-dependent mechanisms could reconcile the discrepancies between the previously reported behavioural versus electrophysiological effects of nicotine in the hippocampus.
The α7 nicotinic acetylcholine receptor (nAChR) plays an important role in mediating the functions of learning and memory (Levin et al. 2002), and in the cognitive-enhancing properties of nicotine (Rezvani & Levin, 2001). Interest in this receptor has escalated recently due to its potential as a powerful therapeutic target in Alzheimer's disease (AD): α7 nAChR agonists may not only help to alleviate the behavioural symptoms of AD (Papke et al. 2000), but also act to inhibit the process of neurodegeneration (Wang et al. 2000; Kihara et al. 2001). The hippocampus is both integral to memory function and a primary site of neurodegeneration in AD, and accordingly this region shows high levels of α7 nAChR expression (Seguela et al. 1993; Breese et al. 1997). More specifically, the α7 nAChRs have principally been localised to GABAergic interneurons in the hippocampus (Freedman et al. 1993) where they mediate rapidly desensitising nicotinic currents (Jones & Yakel, 1997; Frazier et al. 1998), which may form the basis of fast cholinergic transmission (Alkondon et al. 1998; Frazier et al. 1998; Jones et al. 1999). The α7 nAChR also appears to be located presynaptically, and has been shown to enhance the release of both GABA and glutamate from hippocampal neurons (Gray et al. 1996; Alkondon et al. 1997; Radcliffe & Dani, 1998). However, despite the increasing knowledge concerning α7 nAChRs within the cellular circuitry of the hippocampus, most electrophysiological studies have failed to find pronounced effects of α7 nAChR agonists on hippocampal population activity or plasticity.
The α7 nAChR displays a distinctive range of features, including high Ca2+ permeability, high single-channel conductance and rapid desensitisation upon the application of agonists (Seguela et al. 1993; Sudweeks & Yakel, 2000). The rapid desensitisation kinetics of the α7 nAChR may explain the paucity of effects of α7 nAChR activation on hippocampal network responses. Indeed, recent emphasis has been placed on the precise location and timing of α7 nAChR activation (Ji et al. 2001; Buhler & Dunwiddie, 2002), which is of undoubted importance in endogenous cholinergic signalling. However, it is not clear how this conceptual framework can explain why the systemic administration or local infusion of α7 nAChR agonists modulates hippocampal memory processing in vivo (Rezvani & Levin, 2001; Levin et al. 2002).
The aim of the study presented here was to attempt to resolve this discrepancy by exploring the effects of sustained α7 nAChR activation in vitro. The only effect already observed is very subtle: the application of α7 nAChR agonists can lower the threshold for long-term potentiation (LTP) so that it can be induced by stimuli that would normally elicit only a short-term potentiation (STP, lasting ≈10–20 min; Fujii et al. 1999). Therefore, the effects of nicotine (5 min application) on STP were examined and tested for sensitivity to the α7 nAChR antagonist, α-bungarotoxin. In the CA1 region of the hippocampus, both STP and LTP are dependent on NMDA receptors (Schulz & Fitzgibbons, 1997). In order to explore whether any nicotinic augmentation of STP was due to increased NMDA receptor activation, the effects of nicotine were examined in the presence of the NMDA receptor antagonist, D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5). Finally, since the α7 nAChRs are widely expressed on GABAergic interneurons in the hippocampus (Freedman et al. 1993; Frazier et al. 1998), any observed effects of nicotine could result from modulation of GABAergic transmission (Ji & Dani, 2000; Buhler & Dunwiddie, 2002). To test this hypothesis, experiments were performed in the presence of antagonists for GABAA and GABAB receptors: picrotoxin and saclofen, respectively.
METHODS
Hippocampal slice preparation
Male albino guinea-pigs (200–350 g) were decapitated under halothane anaesthesia, in accordance with UK Home Office guidelines, and 400 μm transverse hippocampal slices were prepared. Slices were maintained at room temperature in artificial cerebrospinal fluid (ACSF), which contained (mm): 126 NaCl, 26 NaHCO3, 10 glucose, 3.7 KCl, 2.4 CaCl2, 1.3 MgSO4 and 1.3 KH2PO4, saturated with 95 % O2-5 % CO2. After incubation for at least 1 h, an individual slice was transferred to a submerged recording chamber and continuously superfused at 32–33 °C with oxygenated ACSF at a rate of 1–2 ml min−1
Electrophysiological recordings and analysis
Field excitatory postsynaptic potentials (fEPSP) were recorded in the stratum radiatum of CA1 in response to stimulation of the Schaffer collateral and/or commissural pathway, using micropipettes filled with ACSF (resistance < 3 MΩ). Electrical stimuli (0.1 ms square pulses) were delivered through a concentric bipolar electrode (FHC, Bowdoinham, ME, USA) controlled via a stimulus isolation unit (Digitimer, Welwyn Garden City, UK), with the stimulus strength adjusted to evoke a half-maximal response (5–20 V). The recording micropipettes were pulled from thin-wall microfibre, 1.2 mm capillary borosilicate tubing using a Flaming and/or Brown-type horizontal puller (Sutter Instruments, San Rafael, CA, USA). Electrical signals were recorded with a NeuroData IR-283 amplifier (NeuroData Instruments, New York, USA), and subjected to further × 20 amplification and low-pass filtering at 1 kHz through in-house electronics. The initial slope of the fEPSP was measured on-line using an Intel Pentium-based computer with Signal software (Cambridge Electronic Design, Cambridge, UK).
STP was induced by a tetanus of 10 pulses at 50 Hz, unless otherwise stated. In STP experiments, a STP tetanus was initially applied in the absence of drugs to establish the inherent plasticity of the slice. If there was no clear STP lasting more than 10 min, the slice was rejected. Otherwise, the responses were allowed to stabilise (> 20 min), and the effect of drug treatment was assessed on a second STP tetanus given immediately before washout of the drug(s).
With the exception of those experiments conducted in the presence of picrotoxin, changes in the fEPSP were normalised to the LTP induced at the end of the experiment by a tetanus of 100 pulses at 100 Hz. For the presentation of the time course of experiments, fEPSP values were averaged over 1 or 2 min (three or six responses, respectively). For statistical comparison between drug groups, fEPSP values were averaged over the 5 min (15 responses) preceding the next experimental condition. Data are presented as the mean ± s.e.m. Comparisons for differences in the means were assessed by one-way ANOVA followed by post hoc unpaired t tests or Student-Newman-Keuls multiple comparison t tests, as appropriate.
Drug application
All drugs were diluted in oxygenated ACSF immediately prior to use, and were applied via the perfusion system. Nicotine, mecamylamine and α-bungarotoxin were purchased from Sigma-Aldrich (Poole, UK). d-AP5, picrotoxin and saclofen were purchased from Tocris Cookson (Bristol, UK). The stock solution (× 1000) of picrotoxin was made in ethanol and then aliquoted and frozen at −20 °C. All other drug stock solutions (× 1000) were made in distilled water and then aliquoted and frozen at −20 °C.
RESULTS
Nicotine augments STP via the α7 nAChRs
An initial set of experiments explored the relationship between the number of pulses in a 50 Hz tetanus delivered to the Schaffer collateral and/or commissural pathway, and the degree of potentiation of the fEPSP recorded in the CA1. It was found that 10 pulses at 50 Hz produced a robust STP that returned to near-baseline levels within approximately 15 min, and this tetanus stimulation was used to induce STP in all other experiments. As a reference, the degree of potentiation that could be induced by a 50 Hz tetanus was saturated by 50 stimulation pulses, although this was only ≈75 % of the LTP induced by 100 pulses at 100 Hz.
The application of nicotine (5 nm to 50 μm) 5 min prior to the STP tetanus facilitated the development of a long-lasting potentiation (Fig. 1A). A sigmoidal dose-response curve fitted to the data estimated that the EC50 for the effect of nicotine was ≈96 nm and that the maximum potentiation was ≈48 % of LTP (Fig. 1B). Further experiments to explore the transduction pathway for this effect were conducted using 5 μm nicotine, as this dose produced a near-maximal effect and was less likely to produce non-specific effects than higher concentrations of nicotine.
Figure 1. Nicotine facilitates the induction of a long-lasting potentiation.

A, a typical example of the effect of nicotine application on the change in the response to a STP tetanus (↓). Following the STP tetanus, the fEPSP returned to near-baseline values within 20 min. The application of 5 μm nicotine (5 min) led to the development of a long-lasting potentiation of the fEPSP following the STP tetanus. For analysis, the degree of potentiation was normalised to the LTP induced at the end of the experiment, with a tetanus of 100 Hz for 1 s (↑). Representative recordings of the fEPSP measured for the baseline, control STP, nicotine STP, and LTP periods are shown in inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV. B, dose-response curve for the effect of nicotine (5 nm to 50 μm; n = 3). C, the effects of α7 and non-α7 nAChR antagonists, 100 nmα-bungarotoxin and 50 μm mecamylamine, respectively, on the augmentation of STP produced by 5 μm nicotine (nic). The antagonists were applied for 5 min prior to the application of nicotine, and then both drugs were co-applied for a further 5 min prior to STP induction. To check for effects of the antagonists themselves, experiments were also performed in which the antagonists alone were applied for 10 min prior to STP induction. The graph shows the potentiation of the fEPSP produced both by the pre-drug test STP tetanus and the second STP tetanus following drug application. The potentiation produced by the test STP stimulus was not significantly different between different drug groups, while the statistical analysis of the effects of nAChR antagonists is detailed in D. Error bars have been removed for clarity (○ control, • 5 μm nicotine, ▵ 50 μm mecamylamine, ▴ 5 μm nicotine + 50 μm mecamylamine, ▪ 100 nmα-bungarotoxin, ▪ 5 μm nicotine + 100 nmα-bungarotoxin). D, the nicotinic augmentation of STP was not significantly inhibited by mecamylamine (mec), but was partially inhibited by α-bungarotoxin (α-Btx). The application of α-bungarotoxin itself was able to augment STP, suggesting that the effect of nicotine (nic) was due to both the activation and desensitisation of α7 nAChRs (n = 3–4; **P < 0.01; ***P < 0.001 cf. control; †P < 0.05; ††P < 0.01 cf. 5 μm nicotine).
To explore which nAChR mediated the effect of nicotine on STP, experiments were conducted in the presence of α7 and non-α7 nAChR antagonists: α-bungarotoxin (100 nm) and mecamylamine (50 μm), respectively. In each experiment, a STP tetanus was applied in the absence of drugs to establish the inherent plasticity of the slice, and once the responses had stabilised (> 20 min), the drug treatments were applied prior to a second STP tetanus (see Fig. 1). To control further for variability between the slices, the degree of potentiation was normalised to LTP induced at the end of the experiment. There were no significant differences in the inherent plasticity of the slices used in the different drug groups (one-way ANOVA; F(5,15) = 0.10, P = 0.99; see Fig. 1C), but there were various effects of nicotine and nAChR antagonists (one-way ANOVA; F(5,15) = 13.02, P < 0.001). The application of 5 μm nicotine prior to the STP tetanus produced a long-lasting potentiation of the fEPSP (44.3 ± 3.6 % of LTP), which was significantly greater than the potentiation produced in control experiments (P < 0.001; Fig. 1D). This effect of nicotine was not significantly inhibited by the non-α7 nAChR antagonist, mecamylamine (Fig. 1D). The application of mecamylamine alone produced a small increase in the potentiation produced by the STP tetanus (16.7 ± 2.6 % of LTP), but this effect was not significantly different from the control (Fig. 2B).
Figure 2. The nicotinic augmentation of STP is mediated via activation and inactivation of α7 nAChRs.

A, the application of the competitive nAChR antagonist, DHβE, at non-selective concentrations (10 μm), completely blocked the nicotinic augmentation of STP. Two top arrows represent the points at which a STP tetanus was given. Bars represent the time course of drug application. Bottom arrow represents the point at which LTP was induced. Representative recordings of the fEPSP measured for the baseline, control STP, STP following nicotine and 10 μm DHβE application, and LTP periods are shown inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV. B, at concentrations of 1 μm, DHβE did not antagonise α7 nAChRs, but effectively blocked the main classes of non-α7 nAChRs. It was found that only 10 μm DHβE, and not 1 μm DHβE, blocked the augmentation of STP produced by nicotine application (n = 3–4; **P < 0.01 cf. 5 μm nicotine; †††P < 0.001 cf. 5 μm nicotine + 1 μm DHβE). C, the bath application of 10 mm choline, a selective α7 nAChR agonist, was able to mimic the potentiation of STP produced by nicotine. Two top arrows represent the points at which a STP tetanus was given. Bars represent the time course of drug application. The bottom arrow represents the point at which LTP was induced. Representative recordings of the fEPSP measured for the baseline, control STP, STP following 10 mm choline application, and LTP periods are shown inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV. D, the effects of choline application were dose-dependent, and while 1 mm choline produced a significant augmentation of STP, only 10 mm choline was sufficient to produce a similar a magnitude of potentiation to that observed with application of 5 μm nicotine (n = 3–4; *P < 0.05; **P < 0.01 cf. control; ††P < 0.01 cf. 1 mm choline).
The facilitatory effect of nicotine was partially inhibited by the irreversible α7 nAChR-selective antagonist, α-bungarotoxin (P < 0.05 cf. 5 μm nicotine; P < 0.01 cf. control; Fig. 1D). The application of α-bungarotoxin alone produced a significant potentiation following the STP tetanus (29.5 ± 5.4 % of LTP; P < 0.01 cf. control). This effect was significantly smaller than the potentiation produced by the application of nicotine (P < 0.05), but was very similar to the effect of nicotine in the presence of α-bungarotoxin (Fig. 1D). This suggests that the effect of nicotine on the response to STP stimulation involves both the activation and inactivation of α7 nAChRs.
The block of nAChRs by mecamylamine is slow and use dependent, and to ensure that a role for non-α7 nAChRs in the observed nicotinic augmentation of STP had not been overlooked, the sensitivity to the broad-spectrum competitive nAChR antagonist dihydro-β-erythroidine (DHβE) was also tested. Low concentrations of DHβE (1 μm), which are ineffective in antagonising α7 nAChR-mediated currents, were found to have no significant effect on the nicotinic augmentation of STP (Fig. 2B). However, the effect of nicotine was completely blocked by the non-selective, 10-fold higher concentration of DHβE (P < 0.01 cf. 5 μm nicotine, P < 0.001 cf. 5 μm nicotine + 1 μm DHβE; Fig. 2A and B), which could competitively antagonise both the activation and desensitisation of α7 nAChRs. The nAChR subtype pharmacology was characterised further by examining the effects of the selective α7 nAChR agonist, choline. It was found that the application of 1 mm choline produced a small, but significant, augmentation of the response to STP stimulation (P < 0.05 cf. control; Fig. 2D). A higher, and less selective concentration of choline (10 mm) produced a significantly larger potentiation (P < 0.01 cf. control, P < 0.01 cf. 1 mm choline; Fig. 2C and D), which was similar to the effect of nicotine on STP. Together these results indicate a primary role for α7 nAChRs in mediating the observed effects of nicotine.
Blockade of NMDA receptors reveals a secondary modulatory effect of nicotine
Application of 50 μmd-AP5 alone completely abolished STP, leaving only a post-tetanic potentiation (PTP) lasting less than 2 min (data not shown). The application of 5 μm nicotine did not rescue STP from the effects of d-AP5, and the fEPSP quickly returned to baseline values following a short PTP (Fig. 3A). However, following the PTP, the fEPSP gradually increased again and a significant long-lasting potentiation developed (47.8 ± 3.6 % of LTP; t = 3.19, d.f. = 4, P < 0.05 cf. 50 μmd-AP5, unpaired two-tailed t test; Fig. 3B).
Figure 3. The role of NMDA receptors in the nicotinic augmentation of STP.

A, the application of the NMDA receptor antagonist, 50 μmd-AP5, blocked the generation of a gradually decaying STP and left only a PTP lasting less than 2 min. However, when 5 μm nicotine was co-applied with d-AP5, a long-lasting potentiation of the fEPSP gradually developed following the STP tetanus. Two top arrows represent the points at which a STP tetanus was given. Bars represent the time course of drug application. The bottom arrow represents the point at which LTP was induced. Representative recordings of the fEPSP measured for the baseline, control STP, STP following nicotine and d-AP5 application, and LTP periods are shown inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV. B, this long-lasting potentiation was not due to the direct effects of d-AP5, as when d-AP5 was applied alone, there was neither a STP nor a long-lasting potentiation of the fEPSP (n = 3; *P < 0.05 cf. 50 μmd-AP5, unpaired two-tailed t test).
To explore further the synergistic effects of nicotine and d-AP5, these drugs were applied in the absence of STP stimulation, with the effect on the fEPSP still normalised to the LTP induced at the end of the experiment. In the absence of the STP stimulus, the application of 5 μm nicotine produced only a small increase in the fEPSP (5.7 ± 0.02 % of LTP), while the application of 50 μmd-AP5 produced a small depression of the fEPSP (2.8 ± 0.05 % of LTP; Fig. 4A and B). In contrast, the application of nicotine in the presence of d-AP5 produced a slowly developing, long-lasting potentiation of the fEPSP (36.7 ± 0.09 % of LTP; P < 0.01 cf. 5 μm nicotine, P < 0.01 cf. 50 μmd-AP5; Fig. 4A and B). d-AP5 was applied prior to the application of nicotine, with both drugs being washed out at the same time, but the continued presence of d-AP5 did not prevent the long-lasting potentiation produced by nicotine application (data not shown). This gradually developing potentiation was very similar to that produced following STP stimulation in the presence of both nicotine and d-AP5 (see Fig. 3).
Figure 4. The inhibition of NMDA receptors reveals an effect of nicotine in the absence of high-frequency activation.

A, the application of 5 μm nicotine alone had no discernible effect on the fEPSP. It was only when NMDA receptors were inhibited by 50 μmd-AP5, that the application of nicotine produced a slowly developing, long-lasting potentiation of the fEPSP. Bars represent the time course of drug application. Arrow represents the point at which LTP was induced. Representative recordings of the fEPSP measured during baseline, following nicotine application, following nicotine and d-AP5 application, and LTP are shown inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV. B, the potentiation produced by nicotine in the presence of d-AP5 was a synergistic effect, and was not mimicked by either nicotine or d-AP5 alone (n = 3). C, the nicotinic potentiation of the fEPSP in the presence of d-AP5 was not blocked by the non-α7 nAChR antagonist, 50 μm mecamylamine (n = 3). D, The effect of nicotine was partially inhibited by the α7 nAChR antagonist, 100 nmα-bungarotoxin. In contrast to the experiments on the augmentation of STP, α-bungarotoxin had no significant effect when applied alone (n = 3–4). E, following the inhibition of NMDA receptors, the application of 10 μm DHβE, but not 1 μm DHβE, produced a slowly developing, long-lasting potentiation of the fEPSP, in the absence of nicotine application. Bars represent the time course of drug application. The arrow represents the point at which LTP was induced. Representative recordings of the fEPSP measured during baseline, following 1 μm DHβE +d-AP5 application, following 10 μm DHβE +d-AP5 application, and for LTP are shown inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV. F, while 10 μm DHβE appeared to have a synergistic effect with d-AP5, the potentiation produced by nicotine in the presence of d-AP5 was not significantly attenuated by either 1 μm DHβE or 10 μm DHβE. This suggests that 10 μm DHβE, but not 1 μm DHβE, competitively inhibits tonically active nAChRs, which may also be desensitised by nicotine application (n = 3–4: *P < 0.05; **P < 0.01; ***P < 0.001).
The potentiation of the fEPSP induced by nicotine in the presence of d-AP5 was initially characterised by examining the nAChRs that mediated this effect. For all these experiments, 50 μmd-AP5 was applied 5 min prior to the application of nicotine or nAChR antagonists, and the changes in the fEPSP were normalised to the LTP induced at the end of the experiment. The application of 50 μm mecamylamine did not inhibit the nicotine-induced potentiation, and this could not be attributed to any direct effects of mecamylamine (P < 0.01 cf. 5 μm nicotine, P < 0.05 cf. 5 μm nicotine + 50 μm mecamylamine; Fig. 4C). The effect of nicotine was partially inhibited by 100 nmα-bungarotoxin (P < 0.05 cf. 5 μm nicotine), but the residual potentiation was not due to the direct effects of α-bungarotoxin (P < 0.01 cf. 5 μm nicotine, P < 0.05 cf. 5 μm nicotine + 100 nmα-bungarotoxin; Fig. 4D). This suggested that the effect of nicotine was only partially mediated by α7 nAChRs, and further experiments were performed with DHβE in order to elucidate other nAChR subtypes involved. The application of DHβE, at either 1 or 10 μm, was found to attenuate the potentiation produced by nicotine in the presence of d-AP5, but this effect was not significant (Fig. 4F). These mixed results appeared to be due to the dose-dependent effects of DHβE in the absence of nicotine application. While 1 μm DHβE had no synergistic effect with d-AP5 (P < 0.001 cf. 5 μm nicotine, P < 0.05 cf. 5 μm nicotine + 1 μm DHβE), 10 μm DHβE was found to induce a significant potentiation of fEPSP in the presence of d-AP5 (P < 0.01 cf. 1 μm DHβE; Fig. 4E and F). Therefore, α-bungarotoxin- and mecamylamine-insensitive nAChRs appeared to be tonically activated in the slice, with desensitisation of these receptors contributing to the observed d-AP5-dependent nicotinic potentiation.
GABAergic circuits are involved in the NMDA-receptor-independent effects of nicotine
In the last set of experiments, we attempted to identify the hippocampal circuitry involved in the d-AP5-dependent nicotinic potentiation of synaptic transmission. Although LTP is dependent on the activation of NMDA receptors, it was possible that following NMDA receptor blockade, nicotine application could activate similar downstream mechanisms to those that induce LTP. This hypothesis was tested by examining whether LTP induction could occlude the effects of nicotine. It was found that following LTP induction, the application of nicotine in the presence of d-AP5 still induced a slowly developing potentiation of the fEPSP (166.5 ± 8.0 % of LTP, n = 3; Fig. 5), which was similar to the relative level of potentiation observed at naïve synapses (see Fig. 4A).
Figure 5. The induction of LTP does not occlude d-AP5-dependent nicotinic potentiation.

Following the induction of LTP, the application of 5 μm nicotine in the presence of 50 μmd-AP5 produced a further long-lasting potentiation of the fEPSP. Bars represent the time course of drug application. Arrow represents the point at which LTP was induced. Representative recordings of the fEPSP measured during baseline, following LTP induction, and following nicotine and d-AP5 application are shown inset. Scale bars: horizontal, 5 ms; vertical, 0.5 mV.
Although the effects of nicotine in the presence of d-AP5 appeared to be mediated by multiple nAChRs, we next examined whether this nicotinic modulation might primarily involve the GABAergic circuitry. The inhibition of GABAA receptors led to increased hippocampal excitability, and the development of a pronounced population spike in the stratum radiatum fEPSP and sporadic epileptiform discharges (Fig. 6A). The responses remained stable under these conditions, but it was not feasible to induce LTP at the end of the experiment, and responses were normalised to the pre-drug application period.
Figure 6. The NMDA receptor-independent effect of nicotine is mediated through GABAergic inhibitory circuits.
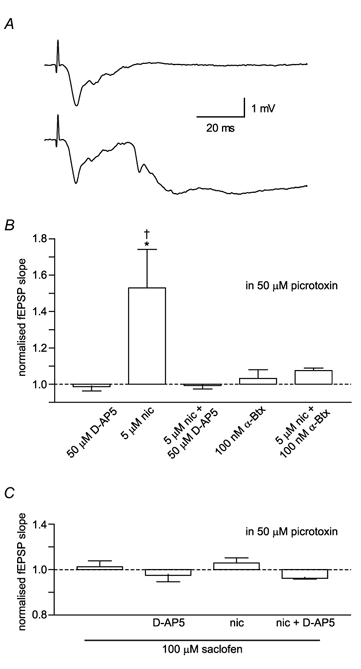
Experiments were conducted in which slices were continually perfused with 50 μm picrotoxin to inhibit GABAA receptors. A, inhibiting GABAA receptors caused the emergence of a population spike in the fEPSP (upper trace) and sporadic epileptiform discharges (lower trace). B, when GABAA receptors were blocked, 5 μm nicotine alone produced a significant potentiation of the fEPSP. This was blocked by 100 nmα-bungarotoxin (n = 3). C, with the additional inhibition of GABAB receptors by 100 μm saclofen, there was no effect of nicotine or synergistic effect between nicotine and d-AP5 (n = 3: *P < 0.05 cf. 5 μm nicotine + 50 μmd-AP5; †P < 0.05 cf. 5 μm nicotine + 100 nmα-bungarotoxin).
In the presence of picrotoxin, the application of nicotine and d-AP5 had no effect (-0.7 ± 1.9 % of pre-drug fEPSP), but nicotine alone was now able to produce a significant increase in the fEPSP (52.9 ± 21.2 % of pre-drug fEPSP; P < 0.05 cf. 5 μm nicotine + 50 μmd-AP5; Fig. 6B). This reversal of effects was not due to the presence of ethanol, in which the picrotoxin was dissolved, as ethanol alone did not inhibit the synergistic effect of nicotine and d-AP5 (39.7 ± 0.1 % of pre-drug; t = 6.27, P < 0.05 cf. 5 μm nicotine, d.f. = 2, unpaired two-tailed t test; data not shown). Indeed, the emergent effect of nicotine in the presence of picrotoxin appeared to result from the increased degree of hippocampal activity, but was still blocked by 100 nmα-bungarotoxin (P < 0.05).
The additional application of 100 μm saclofen, a selective GABAB antagonist, had no effect on the fEPSP (3.6 ± 2.9 % of pre-drug fEPSP). However, the combination of saclofen and picrotoxin blocked both the direct and synergistic effects of nicotine and d-AP5 (one-way ANOVA; F(3,8) = 2.01, P = 0.19; Fig. 6C).
DISCUSSION
Nicotine augments STP via activation and inactivation of the α7 nAChRs
Stimulation of the Schaffer collateral and/or commissural pathway with a tetanus of 10 pulses at 50 Hz was found to produce a STP of the fEPSP in the hippocampal CA1, which returned to baseline levels within about 20 min. Following the application of nicotine for 5 min, this same stimulus induced a long-lasting potentiation of the hippocampal response. This effect of nicotine was not blocked by mecamylamine or low concentrations of DHβE (1 μm), but was inhibited by α-bungarotoxin, suggesting that it was at least partially due to the activation of α7 nAChRs. The irreversible block of α7 nAChRs by α-bungarotoxin itself partially mimicked the effect of nicotine, which suggested that the nicotinic augmentation of STP also involves the desensitisation of α7 nAChRs (Fujii et al. 2000). Therefore, nicotinic augmentation of STP appeared to result from both the activation and inactivation of α7 nAChRs. This hypothesis was supported by the fact that high concentrations of DHβE (10 μm), which competitively antagonises the nicotinic activation, and thus desensitisation, of α7 nAChRs (Hatton & Yang, 2002), completely blocked the effects of nicotine on STP. Furthermore, a selective agonist of the α7 nAChR (Papke et al. 2000), choline, was able to mimic fully the effect of nicotine on STP. The bath application of such high concentrations of choline (1 and 10 mm) may have several non-specific effects on synaptic transmission, particularly on endogenous cholinergic signalling, but all the evidence was consistent with the effects of nicotine on STP being mediated via α7 nAChRs, rather than non-α7 nAChRs. Such an exclusive role for the α7 nAChR is surprising, as many reports have demonstrated the importance of non-α7 nAChRs in modulating synaptic transmission in the rat hippocampus (Fujii et al. 2000; Alkondon & Albuquerque, 2002; Levin et al. 2002). However, very little is known about the role of nAChRs in the guinea-pig hippocampus (Sawada et al. 1994; Nishizaki et al. 2000), or whether there are significant species differences in nAChR function between rodents, but it appears that the nicotinic modulation of synaptic plasticity acts predominantly via α7 nAChRs in this preparation of guinea-pig hippocampus.
As the inactivation of α7 nAChRs was shown to have a facilitatory effect on synaptic plasticity, it is presumed that the STP tetanus stimulation evoked a sufficient degree of ACh release to suppress plasticity under control conditions. As α7 nAChRs mediate excitatory currents (Seguela et al. 1993; Alkondon et al. 1997), it was probable that this effect resulted from the desensitisation of receptors located on GABAergic inhibitory interneurons or their presynaptic terminals. Indeed, while the α7 nAChRs appear to modulate multiple aspects of hippocampal function (Ji & Dani, 2000; Buhler & Dunwiddie, 2002), the α7 subunit is expressed predominantly by GABAergic interneurons (Freedman et al. 1993). Unfortunately, the application of GABA receptor antagonists produces a state of hyperexcitability in the hippocampal CA1 (see Fig. 5A), which leads to epileptiform discharges and the risk of excitotoxic damage following high-frequency afferent stimulation. Therefore, it was not possible to assess directly the extent to which GABAergic circuits mediate the nicotinic augmentation of STP.
As α7 nAChRs show rapid activation and desensitisation kinetics (Seguela et al. 1993; Sudweeks & Yakel, 2000), perhaps a more intriguing aspect of the observed effect was how activation of α7 nAChRs remained significant following sustained application of nicotine (i.e. 5 min of continuous perfusion). Presumably, the activation of α7 nAChRs initiated an intracellular signalling cascade that continued to modulate hippocampal plasticity following the rapid desensitisation of the receptor. Indeed, Ca2+ entry through α7 nAChRs has been shown to activate a number of Ca2+-dependent protein kinases, including mitogen-activated protein kinase (Dineley et al. 2001) and protein kinase A (Dajas-Bailador et al. 2002), and the α7 nAChR may even be directly coupled to the phosphatidylinositol 3-kinase signalling cascade (Kihara et al. 2001). The activation of these intracellular cascades might sensitise the CA1 circuit to high-frequency afferent input in a similar manner to the priming effect of metabotropic glutamate receptor activation on the induction of LTP (Cohen et al. 1998).
A novel effect of nAChR activation revealed by blockade of NMDA receptors
Any attempt to understand the mechanisms by which STP can be converted to a long-lasting potentiation is complicated because the mechanisms of STP itself are poorly resolved. The induction of STP appears to be dependent on the activation of NMDA receptors and intracellular rises in Ca2+, and STP could simply be a reversible form of LTP. In this case, augmentation of STP would simply involve an increase in tetanus activation to suprathreshold levels for LTP, but more downstream mechanisms of STP and LTP expression appear to be independent. In order to gain some insight into the possible downstream targets of α7 nAChR activation, experiments were conducted to explore the role of NMDA receptors in STP and its conversion to a long-lasting potentiation. As expected, the application of the NMDA receptor antagonist d-AP5 prevented the induction of STP, and thus the facilitatory effect of nicotine (Schulz & Fitzgibbons, 1997). However, in the presence of d-AP5, the application of nicotine produced a slowly developing, long-lasting potentiation of the fEPSP, which was independent of high-frequency afferent stimulation. Although this synergistic effect between nicotine and d-AP5 prevented further elucidation of the mechanisms of transition between STP and LTP, it provided the opportunity for some insight into the complex nicotinic signalling pathways modulating synaptic plasticity.
The d-AP5-dependent effects of nicotine did not appear to involve precisely the same mechanisms as the nicotinic augmentation of STP. The effect of nicotine in the presence of d-AP5 was partially inhibited by α-bungarotoxin, but not mecamylamine or low concentrations of DHβE (1 μm), and again appeared to be partially mediated by the α7 nAChR. However, in contrast to experiments examining the nicotinic augmentation of STP, there was no significant effect of α-bungarotoxin itself. Therefore, the nicotinic potentiation in the presence of d-AP5 did not appear to involve desensitisation of the α7 nAChR. This is consistent with the hypothesis that the effect of desensitising α7 nAChRs would only be manifest upon the tetanus-evoked release of ACh. In this case, the residual nicotinic potentiation appeared to be due to the desensitisation of α-bungarotoxin- and mecamylamine-insensitive nAChRs, as the application of 10 μm DHβE without nicotine was able to potentiate synaptic transmission in the presence of d-AP5. Presumably these nAChRs were tonically activated by the basal levels of cholinergic signalling, and therefore had a high sensitivity to ACh. While it is difficult to fit this pharmacological profile to known nAChR subtypes, one possibility is that the desensitisation effect was mediated by α2-containing nAChRs (McQuiston & Madison, 1999; Sudweeks & Yakel, 2000). Human recombinant α2β2 nAChRs expressed in Xenopus oocytes display a relatively high sensitivity to ACh, while being insensitive to mecamylamine and low doses of DHβE (Chavez-Noriega et al. 1997), but the properties of α2-containing nAChRs may be very different in rodents and/or native expression systems (Luetje & Patrick, 1991; McGehee, 1999). Indeed, it is not possible to exclude the involvement of a novel heteromeric α7-containing nAChR (Khiroug et al. 2002) or other uncharacterised subunit combinations (Sudweeks & Yakel, 2000). Although this issue could not be clearly resolved, this set of experiments was important in demonstrating that sustained activation of α7 nAChRs could produce significant long-lasting changes in hippocampal activity.
Mechanisms of d-AP5-dependent α7 nAChR modulation of synaptic transmission
As many forms of synaptic potentiation are dependent on NMDA receptors (Schulz & Fitzgibbons, 1997; Malenka & Nicoll, 1999), it was surprising that the inhibition of NMDA receptors revealed a long-lasting excitatory effect of nicotine on hippocampal activity. In an attempt to understand the mechanisms underlying this unusual form of synaptic plasticity, we first examined whether nicotine might be activating similar downstream pathways as LTP induction. It was shown that the prior induction of LTP did not occlude the d-AP5-dependent nicotinic potentiation, suggesting that the underlying molecular pathways are independent. An alternative hypothesis was that the d-AP5-dependent nicotinic potentiation resulted from long-term changes in inhibition, which would be consistent with the reported role for α7 nAChRs in modulating the activity of local GABAergic interneurons (Freedman et al. 1993; Ji & Dani, 2000; Buhler & Dunwiddie, 2002). Indeed, we found that blocking GABAA receptors with picrotoxin abolished the effect of nicotine in the presence of d-AP5, although, in the absence of fast inhibition, nicotine application alone was capable of inducing an α-bungarotoxin-sensitive potentiation of synaptic transmission. However, the effects of nicotine were completely blocked by a combination of GABAA and GABAB antagonists, suggesting that nicotine acts via GABAergic interneurons.
If the application of nicotine does produce long-term changes in GABAergic inhibition, why are these effects only observed following the blockade of NMDA receptors? The most probable scenario is that activation of nAChRs produces equal but opposite effects on NMDA receptor-dependent and NMDA receptor-independent pathways controlling GABAergic transmission. Therefore, nicotine would have no net effect on hippocampal population activity in control conditions, but would produce long-lasting changes when this balance is disturbed through the blockade of NMDA receptors. Such an equilibrium of nicotinic effects has been demonstrated in a recent report in which nicotine was found to increase glutamate release at autaptic synapses onto hippocampal pyramidal neurons, while inhibiting postsynaptic NMDA currents, and thus had very little overall effect on excitatory postsynaptic currents (Fisher & Dani, 2000). Such complementary effects of α7 nAChR activation on synaptic glutamate currents could also occur at inhibitory interneurons.
One possible model to explain the d-AP5-depedendent effects of nicotine involves activation of α7 nAChRs, increasing glutamate release onto GABAergic interneurons while downregulating postsynaptic AMPA currents, thus establishing an equilibrium between increased NMDA currents and decreased AMPA currents at the postsynaptic interneuron. Following the blockade of NMDA receptors, α7 nAChR activation would decrease the excitatory drive to the interneurons and lead to disinhibition of the pyramidal cells. Such a model could also explain why nicotine application alone induces a potentiation following GABAA receptor inhibition: enhanced glutamate release would be insignificant or saturated in the reverberating excitation patterns observed in the disinhibited slice, thus altering the balance of nicotinic effects in favour of the downregulation of AMPA receptors. This decreased drive to the inhibitory interneurons would reduce the GABAB-mediated inhibition of pyramidal neurons, and therefore lead to potentiation. Why would this effect be blocked by d-AP5? One possibility is that the effect of reducing GABAB-mediated inhibition is to increase the NMDA receptor-mediated activation of pyramidal neurons. Such a hypothesis is highly speculative, but it demonstrates the counter-intuitive principle that inhibition of NMDA receptors could unmask excitatory effects. The precise mechanisms involved cannot be elucidated from this study, and plausible alternative hypotheses could invoke a role for interneurons that selectively target other interneurons (Freund & Buzsaki, 1996), permitting different mechanisms for the disinhibition of pyramidal neurons (Fujii et al. 2000; Ji et al. 2001; Buhler & Dunwiddie, 2002). Indeed, as nAChR activation has been associated with several different intracellular signalling cascades (Dineley et al. 2001; Kihara et al. 2001; Dajas-Bailador et al. 2002), it is possible to imagine many pathways by which blocking NMDA receptors would enable nAChR activation to decrease the GABAergic inhibition of hippocampal CA1 pyramidal neurons.
Conclusion
The fact that both the activation and desensitisation of the α7 nAChR can control hippocampal activity, and that some effects of α7 nAChR agonism are only revealed following inhibition of NMDA receptors, suggests that α7 nAChRs are involved in several different signalling pathways. Indeed, the existence of multiple transduction pathways for α7 nAChR activation is supported by a large body of data (Fujii et al. 1999; Ji et al. 2001; Kihara et al. 2001; Dajas-Bailador et al. 2002). The most surprising finding was that these pathways could be activated by 5 min of continuous application of nicotine. The α7 nAChRs show fast activation and desensitisation kinetics (Seguela et al. 1993), and it has been presumed that only fast local application of nicotinic agonists could reveal the function of these receptors (Ji et al. 2001). Indeed, it may be the case that α7 nAChRs mediate fast cholinergic transmission in the hippocampus in vivo (Alkondon et al. 1998), and that fast application more closely mimics this scenario. However, the application of nicotinic agonists over periods of minutes has pronounced effects on hippocampal learning and memory in vivo, which are partly mediated by the α7 nAChRs (Rezvani & Levin, 2001; Levin et al. 2002). The observed effects of sustained nicotine application on hippocampal activity and plasticity may reconcile the discrepancy between the previously reported behavioural and electrophysiological effects of nicotine. Understanding such aspects of nicotinic signalling could prove invaluable in the development of α7 nAChR agonists as both memory-enhancing drugs and therapeutic agents in AD.
Acknowledgments
This study was supported by Synaptica. E. O. M was supported by the Schorstein Research Fellowship, Oxford University Medical School. We would like to thank Dr Chris Price for his very helpful comments on the manuscript.
REFERENCES
- Alkondon M, Albuquerque EX. A non-alpha7 nicotinic acetylcholine receptor modulates excitatory input to hippocampal CA1 interneurons. J Neurophys. 2002;87:1651–1654. doi: 10.1152/jn.00708.2001. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Albuquerque EX. alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998;810:257–263. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther. 1997;283:1396–1411. [PubMed] [Google Scholar]
- Breese CR, Adams C, Logel J, Drebing C, Rollins Y, Barnhart M, Sullivan B, Demasters BK, Freedman R, Leonard S. Comparison of the regional expression of nicotinic acetylcholine receptor alpha7 mRNA and [125I]-alpha-bungarotoxin binding in human postmortem brain. J Comp Neurol. 1997;387:385–398. doi: 10.1002/(sici)1096-9861(19971027)387:3<385::aid-cne5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Buhler AV, Dunwiddie TV. Alpha7 nicotinic acetylcholine receptors on GABAergic interneurons evoke dendritic and somatic inhibition of hippocampal neurons. J Neurophysiol. 2002;87:548–557. doi: 10.1152/jn.00316.2001. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h-alpha2/beta2, h-alpha2/beta4, h-alpha3/beta2, h-alpha3/beta4, h-alpha4/beta2, h-alpha4/beta4 and h-alpha7 expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- Cohen AS, Raymond CR, Abraham WC. Priming of long-term potentiation induced by activation of metabotropic glutamate receptors coupled to phospholipase C. Hippocampus. 1998;8:160–170. doi: 10.1002/(SICI)1098-1063(1998)8:2<160::AID-HIPO8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha 7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha 7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JL, Dani JA. Nicotinic receptors on hippocampal cultures can increase synaptic glutamate currents while decreasing the NMDA-receptor component. Neuropharmacology. 2000;39:2756–2769. doi: 10.1016/s0028-3908(00)00102-7. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Wetmore C, Stromberg I, Leonard S, Olson L. Alpha-bungarotoxin binding to hippocampal interneurons: immunocytochemical characterization and effects on growth factor expression. J Neurosci. 1993;13:1965–1975. doi: 10.1523/JNEUROSCI.13-05-01965.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Sumikawa K. Inactivation of alpha7 ACh receptors and activation of non-alpha7 ACh receptors both contribute to long term potentiation induction in the hippocampal CA1 region. Neurosci Lett. 2000;286:134–138. doi: 10.1016/s0304-3940(00)01076-4. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Hatton GI, Yang QZ. Synaptic potentials mediated by alpha 7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci. 2002;22:29–37. doi: 10.1523/JNEUROSCI.22-01-00029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol. 2000;83:2682–2690. doi: 10.1152/jn.2000.83.5.2682. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, Yakel JL. Rat nicotinic ACh receptor alpha 7 and beta 2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol. 2002;540:425–434. doi: 10.1113/jphysiol.2001.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- Levin ED, Bradley A, Addy N, Sigurani N. Hippocampal alpha 7 and alpha 4 beta 2 nicotinic receptors and working memory. Neuroscience. 2002;109:757–765. doi: 10.1016/s0306-4522(01)00538-3. [DOI] [PubMed] [Google Scholar]
- Luetje CW, Patrick J. Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci. 1991;11:837–45. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS. Molecular diversity of neuronal nicotinic acetylcholine receptors. Ann N Y Acad Sci. 1999;868:565–577. doi: 10.1111/j.1749-6632.1999.tb11330.x. [DOI] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. J Neurosci. 1999;8:2887–2896. doi: 10.1523/JNEUROSCI.19-08-02887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Neuroscience – long-term potentiation – a decade of progress. Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Nomura T, Matuoka T, Kondoh T, Enikolopo G, Sumikawa K, Watabe S, Shiotani T, Yoshii M. The antidementia drug nefiracetam facilitates hippocampal synaptic transmission by functionally targeting presynaptic nicotinic ACh receptors. Brain Res Mol Brain Res. 2000;14:53–62. doi: 10.1016/s0169-328x(00)00117-0. [DOI] [PubMed] [Google Scholar]
- Papke RL, Meyer E, Nutter T, Uteshev VV. alpha 7 receptor-selective agonists and modes of alpha 7 receptor activation. Eur J Pharmacol. 2000;393:179–195. doi: 10.1016/s0014-2999(00)00009-1. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci. 1998;18:7075–7083. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol Psychiatry. 2001;49:258–267. doi: 10.1016/s0006-3223(00)01094-5. [DOI] [PubMed] [Google Scholar]
- Sawada S, Ohno-Shosaku T, Yamamoto C. Augmenting action of nicotine on population spikes in the dentate gyrus of the guinea pig. Neurosci Res. 1994;20:317–322. doi: 10.1016/0168-0102(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Fitzgibbons JC. Differing mechanisms of expression for short- and long-term potentiation. J Neurophysiol. 1997;78:321–334. doi: 10.1152/jn.1997.78.1.321. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudweeks SN, Yakel JL. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol. 2000;527:515–528. doi: 10.1111/j.1469-7793.2000.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. β-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]