

Abstract
Accumulating evidence suggests that extracellular signal-regulated kinases (ERK1/2) play a key role in regulating vascular tone. To test the hypotheses that ERK1/2 modulate cerebral artery agonist-induced contraction, and that this changes with developmental age, we measured both total and phosphorylated ERK1/2 in adult and fetal ovine cerebral arteries. In middle cerebral arteries (MCA) we also examined tension and [Ca2+]i responses to phenylephrine (PHE), in the absence and presence of the ERK1/2 inhibitor U-0126 and the mitogen-activated protein kinase kinase (MAPKK or MEK) inhibitor PD-98059. In the fetus, but not adult, U-0126 potentiated PHE-induced contraction. In both age groups, inhibition by U-0126, but not PD-98059, decreased the PHE-induced [Ca2+]i increase; in fact for adult, this eliminated any significant [Ca2+]i increase. In turn in the adult, but not fetus, protein kinase C (PKC) inhibition by staurosporine (3 × 10−8 M) prior to ERK1/2 inhibition by U-0126 (10−5 M) prevented this elimination of [Ca2+]i increase. In adult and fetal cerebral arteries basal total ERK1/2 levels were similar. However, in fetal arteries the basal phosphorylated ERK1/2 levels were significantly less than in adult. In fetal, but not adult, cerebral arteries, 10−6–10−4m PHE increased ERK1/2 phosphorylation in a concentration- and time-dependent manner. The ERK1/2 inhibitor U-0126, but not the MEK inhibitor PD-98059, lowered basal activated ERK1/2 levels in vessels of both age groups. These results suggest that basal levels of phosphorylated ERK1/2 play an important role in suppressing Ca2+ sensitivity, perhaps by PKC inhibition. The developmental increase in cerebral artery basal phosphorylated ERK levels from fetus to adult, suggests a transition in the regulation of contraction from Ca2+ sensitivity in the fetal arteries to Ca2+ concentration in the adult vessels.
In mature vascular smooth muscle (VSM) cells the main contraction pathway is via Ca2+-calmodulin-mediated phosphorylation of myosin light chain (Khalil & Morgan, 1993). During the past decade, accumulating evidence suggests that in vascular smooth muscle cells, key components of the MAPK cascade (MAPK kinase (MEK), and its substrates, the extracellular signal-regulated kinases ERK1 and ERK2 (ERK1/2) of 44 kD (p44) and 42 kD (p42), respectively; Boulton & Cobb, 1991; Blenis, 1993) play a role in modulating contraction/relaxation, in addition to their role in signal transduction from plasma membrane receptors to nuclear transcriptional events (Cobb et al. 1991; Adam, 1996; Dessy et al. 1998). Although the relationship of ERK1/2 activation and cell proliferation and differentiation is well established (Childs et al. 1992; Sugden & Clerk, 1997), relatively less is known of the role of ERKs in VSM contraction and relaxation. Several agonists that produce VSM contraction simultaneously activate ERKs (Khalil & Morgan, 1993; Somlyo & Somlyo, 1994; Adam et al. 1995; Katoch & Moreland, 1995; Watts, 1996; Epstein et al. 1997; Dessy et al. 1998; Ratz, 2001). Thus, while the majority of evidence suggests an important function for ERKs in VSM contraction, others have denied such a role (Nixon et al. 1995; Gorenne et al. 1998).
Cerebral arteries show significant vessel-specific and age-related developmental differences in agonist-induced contraction. For instance, these vessels display major differences in α-adrenergic receptor-mediated activity and inositol 1,4,5-trisphosphate (Ins(1,4,5)P3) responses (Longo et al. 1966), Ins(1,4,5)P3-receptor density (Zhou et al. 1997), calcium channel function (Long et al. 1999; Blood et al. 2002), potassium channel function (Long et al. 2000a), sarcoplasmic reticulum Ca2+ stores (Long et al. 2000b), protein kinase C (PKC) function (Longo et al. 2000) and so forth. Given that ERKs appear to play a major role in the regulation of vascular tone, it seemed reasonable to consider their function in regulating cerebrovascular reactivity, and their temporal activation pattern in response to α-adrenergic agonists. Of interest to us was the idea that the ERKs would play a particularly prominent role in developing VSM, in which one would expect a ‘synthetic’, as opposed to the more mature ‘contractile’, phenotype in the adult (Owens, 1998).
Thus, in the present studies we tested the hypotheses that in cerebral arteries, ERKs become activated (phosphorylated) upon α-adrenergic-stimulated contraction, and that the degree of agonist-induced ERK activation correlates with vascular tension and intracellular Ca2+ concentration, [Ca2+]i. We also tested the hypothesis that in developing fetal cerebral arteries, ERKs play an important role in regulating Ca2+ sensitivity, as compared with the adult.
METHODS
Experimental animals and tissues
For these studies, we used cerebral as well as common carotid arteries (CCA) from near-term fetal (≈140 days) and non-pregnant adult sheep (< 2 years) obtained from Nebeker Ranch (Lancaster, CA, USA). The ewes were anaesthetized and killed with 100 mg kg−1 intravenous sodium pentobarbital, following which we obtained anterior, middle and posterior cerebral arteries, or in the case of tension and [Ca2+]i measurements, the main branch middle cerebral artery (MCA). We have shown that this method of killing has no significant effect on vessel reactivity, as compared to use of other anaesthetic agents (Pearce et al. 1991). All surgical and experimental procedures were performed within the regulations of the Animal Welfare Act, and the National Institutes of Health's Guide for the Care and Use of Laboratory Animals was strictly adhered to, as was The Guiding Principles in the Care and Use of Animals approved by the Council of the American Physiological Society, and this work followed by the Animal Care and Use Committee of Loma Linda University.
Studies were performed in isolated vessels cleaned of adipose and connective tissue, as previously described (Longo et al. 1996). For each study ‘n’ was the number of vessels from 4 or 5 animals. To avoid the complications of endothelial-mediated effects, we removed the endothelium by carefully inserting a small wire three times (Longo et al. 1996). The vessels were used immediately for the experiments. In arteries used for responses to phenylephrine (PHE) and ERK1/2 inhibitors, we added the inhibitor for 20 min prior to administration of agonist. We used the common carotid arteries to work out details of the several assays, and as a basis of comparison for the results in the cerebral arteries. Unless otherwise noted, all chemicals were obtained from Sigma Chemical Co. (St Louis, MO, USA).
Immunoblotting of ERK1/2
Fetal and adult sheep cerebral arteries were first isolated, cleaned in the Krebs buffer and then rapidly frozen in liquid nitrogen. Frozen samples were homogenized with a tissue grinder. Homogenized samples then were incubated for 10 min in the lysing buffer (mm): 20 Hepes-KOH, 1 EDTA, 1 dithiothreitol, 1 phenylmethylsulfonyl fluoride, 10 KCl, 1.5 MgCl2, 10 μg ml−1 leupeptin, 2 μg ml−1 aprotinin, pH 7.5. Nuclei and debris were pelleted by centrifugation at 1000 g for 20 min. The whole cell lysate was then stored at −80 °C.
A 10 % polyacrylamide gel was loaded with 15–20 μg protein mixed with an equal volume of 2 × electrophoresis sample buffer per lane. Protein concentration was determined by the Bradford method (Bradford, 1976). The samples were boiled for 5 min before loading and electrophoresed at 90 V for 2 h. A Mini Trans-Blot Electrophoretic Transfer Cell system (Bio-Rad Laboratories, Hercules, CA, USA) was used to transfer proteins to a nitrocellulose membrane at 100 V for 1 h. Blocking for non-specific binding was performed by incubating the membrane for 1 h in blotting solution (5 % non-fat milk in 1 × Tris-buffered saline (TBS) with 0.1 % Tween-20 (TTBS)) at room temperature (22 °C). We then performed overnight incubation of 1:1000 diluted primary antibody (rabbit anti-phosphorylated ERK1/2) or 1:2000 dilution (rabbit anti-total ERK1/2; Cell Signaling Technology, Beverly, MA, USA) in blotting solution at 4 °C. Then, we washed the membrane three times with TTBS. Horseradish peroxidase (HRP)-conjugated secondary antibody was used to incubate the membrane for 2 h at 1:2000 dilution in TTBS at the room temperature. Following incubation with the secondary antibody, the membrane was washed three times in TTBS, 5 min each, and one time in TBS. The membrane was then incubated in chemiluminescence luminol reagent (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 min, and the protein band was detected by using Hyperfilm (Amersham Life Science, Inc., Arlington Heights, IL, USA). We quantified the bands by densitometry (Alpha Innotech Corp., San Leandro, CA, USA). In an attempt to maintain the immunoblotting labelling conditions constant, we used the same titre of polyclonal ERK1/2 antibodies and the same protein concentration in all samples. We chose the polyclonal antibody titres and protein concentrations on the basis of preliminary experiments to determine optimal conditions on the linear portion of the titration curve. α-Actin was used as an internal control for uniform protein loading. In a separate study we have shown that this protein is expressed to an equal amount from preterm fetus to adult (Y. Zhao, W. Long, W. J. Pearce & L. D. Longo, unpublished observations). For immunoblot concentration-response or time course studies, cerebral arteries (i.e. anterior, middle and posterior) from two brains were pooled to obtain enough protein for a single assay.
ERK activity
Nuclei and debris from fetal and adult sheep cerebral arteries were pelleted by centrifugation at 1000 g for 20 min. The supernatant was assayed for ERK1/2 activity with an assay kit (Amersham Corp., Piscataway, NJ, USA), that measures the incorporation of [γ-32P]ATP into a synthetic peptide (KRELVEPLTPAGEAPNQALLR) as a highly specific MAPK substrate for p42/p44 ERKs (Eguchi et al. 1996). The reaction was carried out with the cell lysate (≈1 μg of protein) in 75 mm Hepes buffer, pH 7.4, containing 1.2 mm MgCl2, 2 mm substrate peptide, 1.2 mm ATP and 1 μCi of [γ-32P]ATP for 30 min at 30 °C. The resultant solution was applied to a phosphocellulose membrane and extensively washed in 75 mm orthophosphoric acid and then in H2O. We measured the radioactivity trapped on the membrane by liquid scintillation counting. Activity was expressed as picomoles per minute per milligram of protein.
Measurement of [Ca2+]i and isometric tension
We cut the fetal or adult middle cerebral arteries into rings of 2 mm in length, mounted them on two tungsten wires (0.13 mm diameter; A-M Systems, Carlsborg, WA, USA), attaching one wire to an isometric force transducer (Kent Scientific, Litchfield, CT, USA), and the other to a post attached to a micrometer used to vary resting tension in a 5 ml tissue bath mounted on a Jasco CAF-110 Intracellular Ca2+ Analyzer (Jasco Inc., Easton, MD, USA). The tension value, along with vessel inside diameter, wall thickness, length and potassium-induced force, enabled calculation of force per unit cross-sectional area, as previously described (Pearce et al. 1991). To load the VSM cells with the acetoxymethyl ester of fura-2 (fura-2/AM; Molecular Probes, Eugene, OR, USA), a fluorescent Ca2+ indicator, MCA rings were equilibrated at 25 °C for 40 min in an oxygenated standard Krebs solution containing (mm): 122NaCl, 25.6 NaHCO3, 5.56 dextrose, 5.17 KCl, 2.49 MgSO4 1.60 CaCl2, 1.18 KH2PO4 and 0.027 EDTA. The bath chambers were bubbled with 95 % O2-5 % CO2. Following this, for the measurement by fura-2 fluorescence, we re-equilibrated the vessel at 38 °C for 30 min, as we have previously described (Long et al. 1999). As we have noted, although some investigators may prefer the transformation of fluorescence to intracellular Ca2+ concentration, in tissues such as cerebral arteries the presentation of the ratio is less ambiguous. During all contractility experiments, we continuously digitized, normalized and recorded contractile tensions and the fluorescence ratio (F340/380) using an on-line computer. For all vessels, we evaluated the contractile response for tension and fluorescence ratio by measuring the maximum peak height and expressing it as Emax or percentage of Kmax (maximal effect, a measure of ‘efficacy’) and calculated pD2 (the negative logarithm of the half-maximal concentration for PHE and an index of tissue ‘sensitivity’ or ‘potency’) (Long et al. 1999). In the presence of fura-2, neither K+- nor agonist-induced tensions were significantly different from those contractions in the absence of the dye (data not shown).
In arteries used for response to the ERK1/2 inhibitor U-0126 or the MEK inhibitor PD-98059 following the PHE dose response, we administered U-0126 (10−5 M) or PD-98059 (3 × 10−5 M) (Promega, Madison, WI, USA) for 20 min and repeated the PHE dose response.
Statistics
We tested the null hypothesis by use of one-way ANOVA. We used the Student-Newman-Keuls post hoc test to determine differences between age groups. We used Student's paired t test to compare two groups. We analysed concentration-response curves by computer-assisted non-linear regression to fit the data by use of GraphPad Prism (GraphPad Software, San Diego, CA, USA). A value of P < 0.05 was considered statistically significant; n = the number of arteries tested and the number of animals from which arteries were obtained.
RESULTS
Basal levels of total and activated ERKs in adult and fetal cerebral arteries
To determine the relative abundance of both total and activated, i.e. phosphorylated, ERKs in adult and fetal cerebral arteries, we first measured these by Western immunoblot under basal conditions in which the vessels were neither stretched nor stimulated. As seen in Fig. 1A, while basal levels of total ERKs 1 and 2 (p44 and p42) were similar in the cerebral arteries of both age groups, the levels of phosphorylated ERKs in the fetal vessels were significantly less than that of the adult. α-Actin levels are shown as an internal control. Figure 1B shows the densitometric analysis for activated p44 and p42 levels in the two age groups, with the value for both isoforms in fetal arteries being 0.73 ± 0.10 that of adult (P < 0.05; n = 5).
Figure 1. Basal, control levels of total and phosphorylated p42/p44 extracellular signal-regulated kinases (ERK1/2) in adult and fetal cerebral arteries.

In fetal cerebral arteries the levels of phosphorylated ERK1/2 were significantly less than that of the adult vessels, while the levels of total ERKs were similar in the two age groups. A, adult and fetal cerebral artery levels of phosphorylated ERK1/2 (upper panel), total ERKs (middle panel) and α-actin band for internal control (lower panel). B, densitometric analysis of activated p44 and p42 bands in adult and fetal cerebral arteries normalized to α-actin, with fetal levels for both ERK1 and 2 ∼0.73 ± 0.10 that of the adult. Data are means ± s.e.m. for 8 separate experiments; *P < 0.05.
Effect of ERK antagonists on PHE-induced contraction, [Ca2+]i and phosphorylated ERK levels
To examine the effect of ERK inhibition on phenylephrine-induced tension and [Ca2+]i in adult and fetal cerebral arteries, we first determined the PHE dose-response relations under control conditions. Next, we administered 10−5 M U-0126, and in 20 min repeated the PHE dose- response curve (n = 4). As seen in Fig. 2A for adult MCA, in the presence of U-0126 the PHE dose-response was shifted one-half log unit to the right (pD2 = 4.6 ± 0.1) as compared to control (pD2 = 5.2 ± 0.1; P < 0.05), i.e. sensitivity decreased. (The corresponding Emax values were 1.2 ± 0.2 and 1.1 ± 0.2.) Even more striking in the adult vessel was the effect of U-0126 in totally eliminating the normal PHE-induced [Ca2+]i increase (Fig. 2B); Emax = 0), but with full maximal tension.
Figure 2. Phenylephrine (PHE) dose-response relations for adult and fetal middle cerebral arteries (MCA) in the presence (▵) or absence (•) of the ERK1/2 inhibitor U-0126.
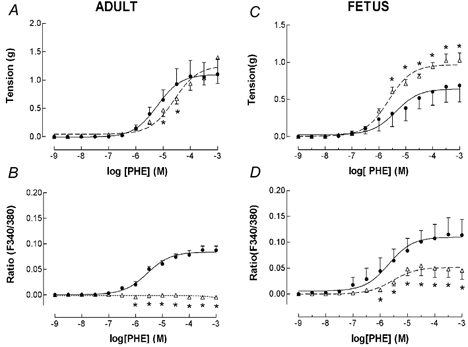
Arterial segments were first contracted with 120 mm K+ to obtain peak tension. After washing and re-equilibration to baseline tension, we induced subsequent contractions using cumulative doses of PHE added in half-log increments (n = 4 each; see Methods for details). A, vascular tension (g) for adult MCA under control conditions (•, continuous line). Also shown is the PHE dose-response following administration of 10−5 M U-0126 (▵, dashed line). Points are means ± s.e.m.; *P < 0.05 vs. control. B, fluorescence ratio for adult MCA in response to PHE under control conditions, and following administration of 10−5 M U-0126 (symbols same as above). C, vascular tension for fetal MCA in response to PHE under control conditions, and in the presence of U-0126 (▵, dashed line). D, fluorescence ratio for fetal MCA under control conditions, and following administration of U-0126 (symbols same as above).
In contrast, in fetal MCA ERK inhibition showed a very different pattern of response. In the presence of 10−5 M U-0126, PHE-induced contraction was augmented with Emax = 1.0 ± 0.1 vs. 0.6 ± 0.1 for control at 10−4m PHE (P < 0.05; Fig. 2C). Also of interest, in the presence of the ERK antagonist, fetal MCA showed a markedly inhibited [Ca2+]i response (Emax = 0.05 ± 0.03 vs. 0.10 ± 0.03 for control; Fig. 2D), although this was not completely eliminated as in the adult vessel.
To compare the effect of the MEK inhibitor PD-98059 with that of the ERK inhibitor U-0126 on tension and [Ca2+]i, we repeated the PHE dose-response relations in the absence or presence of 3 × 10−5 M PD-98059. As seen in Fig. 3, for vessels of both age groups these responses were quite different from those seen with U-0126. In adult cerebral arteries, PD-98059 was without significant effect on either tension (Fig. 3A) or [Ca2+]i (Fig. 3B). In marked contrast to the adult, and differing from the response in the presence of U-0126, administration of PD-98059 resulted in the fetal MCA having a significant decrease in PHE-induced tension (Emax = 0.4 ± 0.1 vs. control value 1.1 ± 0.1; P < 0.05; Fig. 3C). In addition, while the PHE-induced [Ca2+]i response was less than under control conditions (0.04 ± 0.02 vs. control of 0.10 ± 0.02; P < 0.05; Fig. 3D), it was similar to that response seen in the presence of U-0126.
Figure 3. PHE dose-response relations for adult and fetal middle cerebral arteries (MCA) in the presence (▵) or absence (•) of the MEK inhibitor PD-98059.
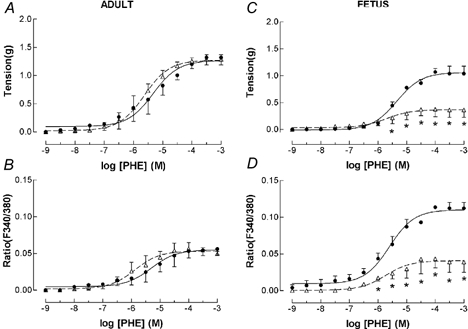
Arterial segments were first contracted with 120 mm K+ to obtain peak tension. After washing and re-equilibration to baseline tension, we induced subsequent contractions using cumulative doses of PHE added in half-log increments (n = 4; see Methods for details). A, vascular tension (g) for adult (•, continuous line) MCA under control conditions. Also shown is the PHE dose-response following administration of 10−5 M PD-98059 (▵, dashed line). Points are means ± s.e.m. B, fluorescence ratio for adult MCA in response to PHE under control conditions, and following administration of 10−5 M PD-98059 (symbols same as above). C, vascular tension for fetal MCA in response to PHE under control conditions (•, continuous line), and in presence of PD-98059 (▵, dashed line). *P < 0.05 vs. control. D, fluorescence ratio for fetal MCA under control conditions, and following administration of PD-98059 (symbols same as above).
To understand more completely the relation of vascular tension to [Ca2+]i in the absence or presence of these antagonists, we plotted these relations in response PHE alone, and in the presence of either 10−5 M U-0126 or 3 × 10−5 M PD-98059. For adult cerebral artery, the relation of PHE-induced tension to fluorescence ratio was reasonably linear, with a slope of 27.1 ± 2.1, r = 0.67. In the presence of U-0126, in contrast, tension increased with no significant change in [Ca2+]i (P < 0.01). In contrast to these results, for adult cerebral arteries in the presence of PD-98059 the slope of the relation of tension to [Ca2+]i was not significantly different (slope = 21.1 ± 1.5, r = 0.59) from that of control PHE-induced curves (slope = 20.2 ± 1.3, r = 0.67). For the fetal cerebral arteries in response to PHE, tension increased somewhat less for a given increase in fluorescence ratio than in the adult (slope = 5.7 ± 0.3, r = 0.72). In view of the significant increase of PHE-induced tension in the presence of U-0126 in the fetal artery (see Fig. 2C), the slope was greater than that for PHE alone (11.8 ± 1.6, r = 0.52, P < 0.05). Again for the fetal cerebral artery, the PHE-induced tension and [Ca2+]i and those in the presence of PD-98059 were not significantly different (slope = 9.9 ± 1.4, r = 0.78, and slope = 4.7 ± 1.5, r = 0.47).
To examine the role of MEK and ERK1/2 inhibitors on PHE-induced (10−5 M) phosphorylated ERK1/2 levels, we examined these by Western immunoblot following the administration of two different inhibitors of ERK phosphorylation. As shown in Fig. 4A (and Table 1) in fetal cerebral arteries, the antagonist U-0126 (10−5 M) of itself resulted in a significant reduction in the basal level of phosphorylated ERK1/2. Also following U-0126 administration, 10−5m PHE stimulation produced no significant increase in phosphorylated ERK1/2 levels above those seen with U-0126 alone (Fig. 4, Table 1). By comparison, the MEK antagonist PD-98059 (3 × 10−5 M) resulted in no significant inhibition of the basal phosphorylated ERK1/2 levels, although it prevented the increase seen in response to 10−5m PHE. Figure 4B shows the densitometric analysis of activated p44 and p42 levels under these conditions (n = 4 each; see Table 1).
Figure 4. In fetal cerebral arteries the ERK inhibitor U-0126 decreased the basal level of phosphorylated ERK1/2, and also inhibited the PHE-induced response.
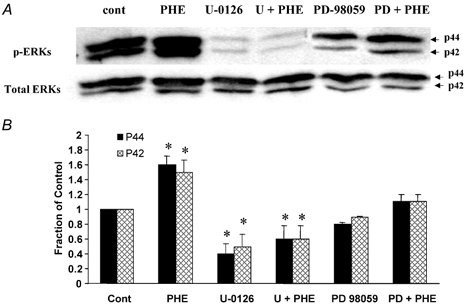
A, Western immunoblots of activated p42/p44 in response to 10−5m PHE ± the ERK inhibitor 10−5 M U-0126, or the MAPK inhibitor 3 × 10−5 M PD-98059. Also shown is the band for total ERKS. B, densitometric analysis of phosphorylated p44 and p42 under these conditions, normalized to the α-actin band (n = 4; *P < 0.05 as compared to control).
Table 1.
Phosphorylated ERK levels in fetal and adult arteries by both Western immunoblot and specific activity
| Fetus | Adult | |||||
|---|---|---|---|---|---|---|
| Densitometry | Activity | Densitometry | Activity | |||
| p44 | p42 | p44 | p42 | |||
| Cerebral Arteries | ||||||
| Control | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| K+ 1.2 × 10−1 | 1.0 | 1.0 | — | 1.0 | 1.0 | — |
| PHE 10−6 M | 1.3 ± 0.2* | 1.7 ± 0.2* | — | 1.0 ± 0.1 | 0.8 ± 0.1 | — |
| 10−5 M | 1.7 ± 0.5* | 1.6 ± 0.2* | 2.4 ± 0.3† | 0.9 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.1 |
| 10−4 M | 2.3 ± 0.4† | 2.8 ± 0.5† | — | 0.9 ± 0.1 | 0.9 ± 0.1 | — |
| U-0126 10−5 M | 0.4 ± 0.1* | 0.5 ± 0.1* | 0.5 ± 0.1 | 0.7 ± 0.4* | 0.6 ± 0.1* | 0.3 ± 0.1 |
| U-0126 + PHE 10−5 M | 0.6 ± 0.2* | 0.6 ± 0.2* | 0.6 ± 0.1 | 0.7 ± 0.1* | 0.6 ± 0.1* | 0.6 ± 0.1 |
| PD-98059 3 × 10−5 M | 0.8 ± 0.1 | 0.9 ± 0.1 | — | 0.8 ± 0.1 | 0.8 ± 0.1 | — |
| PD-98059 + PHE 10−5 M | 1.1 ± 0.1 | 1.1 ± 0.1 | — | 0.8 ± 0.1 | 0.9 ± 0.1 | — |
| Common carotid | ||||||
| control | 1.0 | 1.0 | — | 1.0 | 1.0 | — |
| PHE 10−6 M | 1.6 ± 0.3* | 1.4 ± 0.2 | — | 1.0 ± 0.1 | 1.2 ± 0.2 | — |
| 10−5 M | 2.2 ± 0.3† | 2.0 ± 0.3† | 1.9 ± 0.1† | 1.1 ± 0.2 | 1.2 ± 0.2 | 1.0 ± 0.1 |
| 10−4 M | 2.8 ± 0.4† | 3.4 ± 0.5† | — | 1.1 ± 0.2 | 1.3 ± 0.2 | — |
| U-0126 10 −5 M | 1.1 ± 0.2 | 0.9 ± 0.2 | 0.9 ± 0.1 | 0.6 ± 0.1* | 0.6 ± 0.1* | 0.6 ± 0.1 |
| U-0126 + PHE 10−5 M | 1.1 ± 0.2 | 0.9 ± 0.2 | 0.9 ± 0.1 | 0.6 ± 0.1* | 0.6 ± 0.1* | 0.6 ± 0.1 |
Values for both Western immunoblot densitometry and specific activity are means ± s.e.m. expressed as fraction of control baseline value (see Methods for details). n Values were 4 each for the activity assays and at least 5 each for the immunoblots; K+, potassium; PHE, phenylephrine
P < 0.05
P < 0.01 compared with control.
In cerebral arteries of the adult, although the response of phosphorylated ERK1/2 to 10−5m PHE stimulation was not significant, U-0126, but not PD-98059, resulted in the phosphorylated p42 and p44 levels being decreased significantly below control values (Table 1).
Since one might argue that the levels of phosphorylated ERK1/2, as determined by Western immunoblots, do not accurately reflect ERK1/2 activity, we measured this activity by biochemical assay under several conditions. As shown in Table 1, by this assay in fetal cerebral arteries phosphorylated ERK1/2 activity was about one-half that in the adult vessels. In response to 10−5m PHE, activity in the fetal arteries increased ≈140 % (P < 0.01), while that in the adult vessels increased only ≈10 % (n.s.). Also in agreement with the immunoblots, 10−5 M U-0126 resulted in a significant decrease in basal ERK1/2 activity, as well as inhibition of the PHE-induced increase in arteries of both age groups (Table 1).
PHE concentration-ERK activation response
To examine the dose-response relations for PHE in increasing phosphorylated ERK levels, we exposed adult and fetal cerebral arteries to varying concentrations of PHE, and in 5 min measured the phosphorylated ERK1/2 levels. Figure 5A shows that under baseline conditions, while there was a relative abundance of total ERK1/2 protein in fetal cerebral arteries, the level of phosphorylated ERKs was rather low. In contrast, in response to increasing PHE concentration (10−6–10−4 M), phosphorylated ERK1/2 levels in fetal arteries increased significantly in a dose-dependent manner. Figure 5B shows the densitometric analysis of phosphorylated p44 and p42 at the several concentrations (n = 4 each; see Table 1). As an aside, in fetal common carotid arteries the phosphorylated ERK1/2 responses to PHE were somewhat greater than in the cerebral arteries (Table 1). Such increases in phosphorylated ERKs were not seen with K+ depolarization in either cerebral or common carotid arteries.
Figure 5. PHE concentration-response relations for phosphorylated p42/p44 ERK1/2 in near-term fetal cerebral arteries.

Intact fetal cerebral arteries were stimulated by exposure to 10−6–10−4m PHE for 5 min. A, at 10−6m PHE and above, the immunoblots showed significant increases in activated ERK1/2, as compared to control. Also shown is the band for total ERK1/2. B, densitometric analysis of phosphorylated p44 and p42 normalized to the α-actin band for internal control. Data are means ± s.e.m. for 4 separate experiments; *P < 0.05, †P < 0.01, as compared to control.
By way of contrast to the fetus, Fig. 6 shows the lack of PHE-induced activated ERK responses in adult cerebral arteries. Again in the adult vessels under basal conditions, total ERK1/2 levels were similar to those of the fetus, while phosphorylated ERK1/2 levels were much greater than those in the corresponding fetal vessels. Particularly striking in the adult arteries and in contrast to the response in the fetus, in response to even 10−4m PHE there was no significant increase in phosphorylated ERK1/2 levels above control (Fig. 6A and B).
Figure 6. Lack of PHE-induced increase in phosphorylated p42/p44 ERK1/2 in adult cerebral arteries.
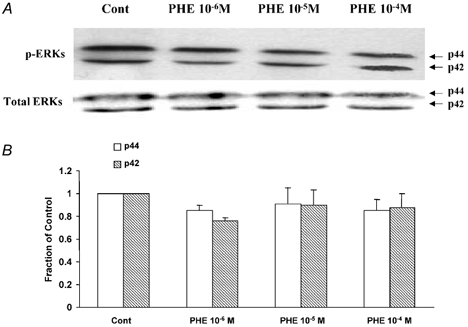
Intact adult cerebral arteries were stimulated by exposure to 10−6–10−4m PHE for 5 min. A, in the adult arteries the immunoblots showed no significant increase in activated ERK1/2 at 10−5-10−4m PHE as compared to control. Also shown is the band for total ERK1/2. B, densitometric analysis of phosphorylated p44 and p42 normalized to the α-actin band (n = 4).
Time-dependent ERK activation by PHE
To establish the time course of activation of phosphorylated ERKs in response to PHE, we exposed fetal cerebral arteries to 10−5m PHE for time periods of between 1 and 20 min. As shown in Fig. 7A, in response to 10−5m PHE the levels of phosphorylated ERK1/2 (p42 and p44) increased over 2-fold at 5 min, and remained significantly elevated at 20 min. Not shown, the increase at 1 and 2 min was not significant. Also, within 10 min of PHE washout, the phosphorylated ERK levels remained significantly elevated (data not shown). The densitometric values for phosphorylated p44 and p42 at the several time points are shown in Fig. 7B (n = 4 each). For comparison, in fetal common carotid arteries the time course of PHE-induced phosphorylated ERK response was similar to that seen in the cerebral arteries. As noted above, in neither adult cerebral nor common carotid arteries did the phosphorylated ERK1/2 levels increase significantly in response to 10−5m PHE (or higher concentration) at any time up to 20 min (data not shown).
Figure 7. Time course of 10-5m PHE stimulation of phosphorylated ERK1/2 in fetal cerebral arteries.
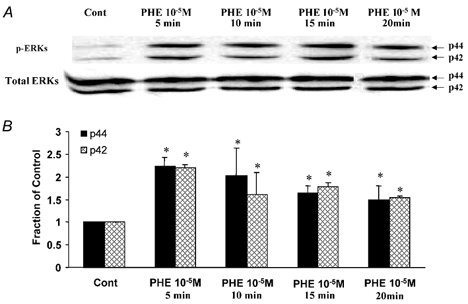
A, Western immunoblot of phosphorylated ERK1/2 at 0, 5, 10, 15 and 20 min. By 5 min the phosphorylated ERK1/2 levels increased significantly, after which they decreased slightly. Levels of total ERKs are also shown. B, densitometric analysis of phosphorylated p44 and p42, normalized to the density of the α-actin band for each respective lane (n = 4; *P < 0.05, as compared to control).
Effect of PKC inhibition on PHE-induced contraction and [Ca2+]i
The effect in adult arteries of ERK inhibition by U-0126 resulting in elimination of the PHE-induced increase in [Ca2+]i, despite only a modest effect on the rise in tension (Fig. 2A and B), was rather mystifying. To explore the mechanistic basis for this phenomenon, and the possible role of PKC in modulating the PHE-induced increase in [Ca2+]i and tension, we examined PHE dose-response relations in the absence or presence of 10−5 M U-0126 following PKC inhibition by staurosporine (3 × 10−8 M). As seen in Fig. 8 for adult MCA, in the presence of staurosporine both tension (Fig. 8A) and [Ca2+]i (Fig. 8B) were significantly inhibited. By way of contrast with staurosporine alone or U-0126 alone (Fig. 2B), in the presence of 10−5 M U-0126 following PKC inhibition, PHE-induced contraction and [Ca2+]i were markedly increased (Fig. 8A and B).
Figure 8. PHE dose-response relations for adult and fetal MCA in presence or absence of the PKC inhibitor staurosporine and the ERK1/2 inhibitor U-0126.
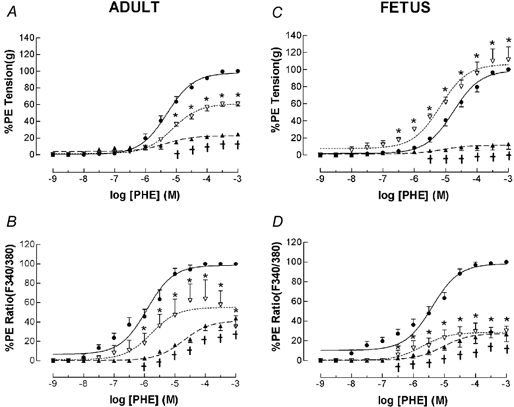
Arterial segments were first contracted with 120 mm K+ to obtain peak tension. After washing and re-equilibration to baseline tension, we induced subsequent contractions using cumulative doses of PHE added in half-log increments (n = 4 each; see Methods for details). A, vascular tension (%PHE max) for adult MCA under control conditions (•, continuous line). Also shown is the PHE dose-response relation following administration of 3 × 10−5 M staurosporine (▴, dashed line; *P < 0.05 vs. control). In addition is the PHE dose-response relation following administration of 3 × 10−8 M staurosporine, followed by 10−5 M U-0126 (▿, dashed line; †P < 0.05 vs. both control and staurosporine alone). Points are means ± s.e.m. B, fluorescence ratio (%PHE max) for adult MCA in response to PHE under control conditions, and following administration of either 3 × 10−8 M staurosporine alone or staurosporine followed by 10−5 M U-0126 (symbols same as above). C, vascular tension for fetal MCA in response to PHE under control conditions, and in the presence of either staurosporine alone (▴) or staurosporine followed by U-0126 (▿). D, fluorescence ratio for fetal MCA under control conditions, and following administration of either 3 × 10−8 M staurosporine alone or staurosporine followed by U-0126 (symbols same as above).
In a similar manner in fetal MCA, staurosporine inhibition of PKC resulted in marked attenuation of both PHE-induced tension (Fig. 8C) and [Ca2+]i (Fig. 8D). In the presence of ERK inhibition by U-0126 following PKC inhibition by staurosporine, PHE-induced tension showed greater sensitivity (pD2 = 5.2 ± 0.1 vs. 4.8 ± 0.1), although the rise in [Ca2+]i was not significantly greater than that with staurosporine alone. (Fig. 8C and D)
DISCUSSION
The role of ERKs in nuclear transcription has been well established in vascular smooth muscle, and many other cell types. In contrast, their function in agonist-mediated contraction-relaxation mechanisms is quite complex and has been less well defined. The present studies offer several important observations regarding the role of ERKs in agonist-induced responses of adult and fetal ovine cerebral arteries. First, the levels of phosphorylated ERKs have functional significance. This is suggested by the effects of both ERK inhibitors, U-0126 and PD-98059, on basal and agonist-induced ERK phosphorylation and cerebral artery tension and [Ca2+]i. In the adult arteries, basal phosphorylated ERK levels were high and did not increase in response to PHE. Also in the adult, U-0126 significantly lowered the basal phosphorylated ERK levels, and resulted in a decreased sensitivity of PHE-induced tension and a total elimination of the PHE-induced [Ca2+]i increase. In contrast, the MEK inhibitor PD-98059 did not affect the basal levels of phosphorylated ERK and had no effect on PHE-induced tension and Ca2+ response. Together with the data on the effect of PKC inhibition on modifying the U-0126-mediated modulation of PHE-induced [Ca2+]i response, this suggests an interaction between ERK and PKC signalling pathways in regulating agonist-mediated [Ca2+]i and tension in adult cerebral arteries. These findings strongly suggest that in the adult vessels, ERKs mediate agonist-induced Ca2+ release but suppress Ca2+ sensitivity, at least in part by modulating PKC activity.
Second, fetal cerebral arteries displayed relatively low activated ERK levels under basal conditions, which is counterintuitive given the more active growth and differentiation pathways in these vessels. Nonetheless, these arteries showed significant 1.5- to 2-fold increases in phosphorylated ERK1/2 levels in response to PHE (Figs 4, 5 and 7, and Table 1). In contrast to adult arteries, PD-98059 significantly inhibited PHE-mediated ERK phosphorylation as well as [Ca2+]i increase and tension development. This observation provides further support for the role of phosphorylated ERK in agonist-mediated contractions. Similar to the adult vessels, U-0126, but not PD-98059, significantly decreased basal phosphorylated ERK levels in the fetal arteries. Although the both inhibitors decreased PHE-induced [Ca2+]i increase in the fetal vessels, only U-0126 significantly increased Ca2+ sensitivity. This is consistent with its effect in the adult arteries, and suggests that in cerebral arteries basal levels of phosphorylated ERK play an important role in regulating Ca2+ sensitivity.
Thus, in cerebral arteries of both age groups the ERK pathway appears to play a key role in the regulation of contraction. Nevertheless, the exact nature of that role and its several associated pathways and mechanisms remain an enigma. Here, by the use of both Western immunoblot and specific activity assay, we show for the first time, that although basal total ERK levels were similar in the adult and fetal cerebral arteries, the levels of phosphorylated ERK1/2 were significantly greater in the adult (Fig. 1). The finding that agonist stimulated ERK phosphorylation in fetal arteries but failed to increase phosphorylated ERK levels in the adult suggests that under basal conditions the adult arteries are in a state of maximal ERK stimulation. Given that basal levels of phosphorylated ERK may be important in suppressing Ca2+ sensitivity, the finding of developmental increase in basal phosphorylated ERK from fetal to adult cerebral arteries would suggest a transition in the regulation of contraction from that of Ca2+ sensitivity in the fetal arteries to Ca2+ concentration in the adult.
Role of ERKs in arterial contraction
ERK activation is dependent upon dual phosphorylation of a tyrosine (Try185) and a threonine (Thr187) residue (Alessandrini et al. 1992). In many cell types this activation occurs within 5–10 min of stimulation, and the activity remains elevated for over 20 min (Pang et al. 1993). Studies suggesting that ERK phosphorylation plays a role in modulating arterial contraction include: rabbit femoral artery (Ratz, 2001), rat cerebral arteries (Lagaud et al. 1999), pig carotid artery (Adam et al. 1989, 1995; Katoch & Moreland, 1995), ferret aorta (Khalil & Morgan, 1993; Dessy et al. 1998), rat aorta and mesenteric and tail arteries (Watts, 1996), sheep uterine artery (Xiao & Zhang, 2002) and human peripheral resistance arteries (Touyz et al. 1999). Nonetheless, the exact nature of this role and its associated signalling pathways have remained unclear. In the present study in fetal cerebral arteries, PHE-induced α1-adrenergic-induced contraction was correlated with an increase in ERK1/2 phosphorylation. In contrast, in the adult vessels no association of ERK activation to tension was evident, as in these vessels ERKs appear to be maximally activated under basal conditions. Even in fetal cerebral arteries, although vascular tension increased to near maximum within 1 min of PHE administration, phosphorylated ERK1/2 levels did not increase significantly until ≈5 min (Fig. 7). In addition, 10 min following PHE washout, a time when tension had returned to baseline, phosphorylated ERK1/2 remained significantly elevated (data not shown). Thus, for neither adult nor fetal cerebral arteries was a clear causal relationship demonstrated between ERK1/2 phosphorylation and the increase in vascular tension or [Ca2+]i. In part, this may be compatible with the idea that in the mature (i.e. adult) arteries ERKs play a role in the maintenance of sustained tone, rather than its induction (Takashi & Berk, 1998). Alternatively, tension may be totally independent of phosphorylated ERK levels. Also, K+ depolarization (120 mm) resulted in sustained contraction of both adult and fetal cerebral arteries, without a significant increase in ERK1/2 phosphorylation (Table 1). Additionally in fetal cerebral arteries, the relatively selective inhibitor U-0126 eliminated agonist-induced ERK1/2 phosphorylation (Fig. 4), while at the same time enhancing PHE-induced tension (Fig. 2C).
α1-Adrenergic-mediated contraction and ERKs
In fetal, but not adult, cerebral arteries, the selective α1-AR agonist phenylephrine produced a sustained increase in ERK1/2 phosphorylation, in addition to the usual increases in tension and [Ca2+]i. These results agree with studies in the rabbit femoral artery (Ratz, 2001) and sheep uterine artery (Xiao & Zhang, 2002). α1-Adrenergic receptor stimulation may thus have activated a second cell signalling cascade which, in association with increased [Ca2+]i, resulted in increased ERK1/2 phosphorylation (see Ratz, 2001). The developmental differences in cerebral artery responses of phosphorylated ERK1/2 levels, tension, and [Ca2+]i to PHE may help to explain the lack of response reported by others (Nixon et al. 1995; Gorenne et al. 1998), and the lack of agreement regarding the role of ERKs in vascular reactivity.
ERK and/or MEK inhibition
As noted above, in both adult and fetal cerebral arteries, the relatively selective ERK inhibitor U-0126 reduced both basal phosphorylated ERK1/2 levels, and phosphorylation in response to PHE (Fig. 4 and Table 1). In this regard, U-0126 showed a greater effect than did the MEK inhibitor PD-98059. These data suggest that basal levels of ERKs were active in both adult and fetal non-stimulated cerebral arteries. The differing effects of U-0126 and PD-98059 are not surprising. PD-98059 has been thought to block the phosphorylation of MEK1 by upstream activators, but not efficiently inhibit MEK1 activity once it is phosphorylated (Alessi et al. 1995). In contrast, U-0126 has been reported to be a more selective and potent inhibitor of phospho-MEK1 and constitutively active MEK1 (Favata et al. 1998). More recently, U-0126 also has been reported to inhibit MEK1 phosphorylation by upstream activators (Davies et al. 2000; Ahn et al. 2001). Thus, these compounds may inactivate MEK1 through similar mechanisms, although on the basis of the present (and other) studies, the precise mechanistic differences of their action remain to be resolved.
A particularly striking feature of the present study, was the total elimination by U-0126 of the PHE-induced increase in [Ca2+]i in adult cerebral artery (Fig. 2B), while this response was not significantly affected by PD-98059 (Fig. 3B). The U-0126-associated increase in Ca2+ sensitivity (small decrease in tension in the presence of a greater decrease in [Ca2+]i) emphasizes the role of ERK1/2 in modulating cerebral artery Ca2+ sensitivity and tone. This response pattern (Fig. 2A and B) in adult cerebral arteries is similar to that reported by us in response to PKC stimulation by phorbol 12,13-dibutyrate (PDBu; see Fig. 5A in Longo et al. 2000). A possible reason for the attenuated [Ca2+]i response to PHE in the adult (Fig. 2B) is ERK negative feedback inhibition of PKC under control conditions, with U-0126 blocking this feedback. That such a mechanism is plausible, is suggested by the results of ERK inhibition on PHE-induced responses in the presence of PKC inhibition. As shown in Fig. 8A for adult MCA in the presence of the PKC inhibitor staurosporine (3 × 10−8 M) followed by U-0126 (10−5 M), the PHE-induced increase in [Ca2+]i was significantly attenuated; however, this inhibition was much less than that seen in the presence of U-0126 alone (Fig. 2B).
Another striking effect of U-0126 inhibition was the increase in PHE-induced tension in fetal cerebral artery over that see with PHE alone (Fig. 2C and Fig. 8C). In contrast, following the administration of PD-98059 the fetal PHE-induced tension response was attenuated (Fig. 3C). The [Ca2+]i response in fetal cerebral arteries (Fig. 2D) was also not unlike that we reported in response to PDBu (see Fig. 5C in Longo et al. 2000). Because of a number of significant differences in the role of PKC in adult and fetal cerebral arteries (including a relative lack of PKCε in the fetus; Longo et al. 2000), this may, in part, account for the different responses observed to ERK inhibition in the two age groups. Of interest in fetal MCA, ERK inhibition by 10−5 M U-0126 following inhibition of PKC showed significant increase in tension (Fig. 8C), but not [Ca2+]i (Fig. 8D), similar to that seen with U-0126 alone (Fig. 2C and D). This suggests that in fetal arteries, PKC plays a key role in α1-adrenergic-mediated contraction. Although on the basis of numerous studies it is clear that α-adrenergic-receptor-mediated contraction operates via Ins(1,4,5)P3 and Ca2+, the mechanisms of PKC-mediated contraction and the regulation of Ca2+ sensitivity remain an enigma.
Figure 9 presents in outline these mechanisms, with ERK1/2 negative feedback on PKC and inhibition of Ca2+ sensitivity of myosin light chain phosphorylation. In essence, in adult cerebral arteries, by suppressing Ca2+ sensitivity the activated ERKs help to promote Ca2+-dependent responses. In turn, in fetal vessels with lower levels of phosphorylated ERKs, Ca2+ sensitivity of myosin light chain is enhanced. An important question concerns the mechanism of this ERK-mediated effect on Ca2+ sensitivity. In addition, the role of ERKs on specific α1-AR subtypes and their associated G proteins or phospholipase C responses is of great importance. Quite obviously, further studies are required in this regard.
Figure 9. Proposed signal transduction pathways for Ca2+-dependent (on right) and Ca2+-independent (on left)-mediated contraction in cerebral arteries.
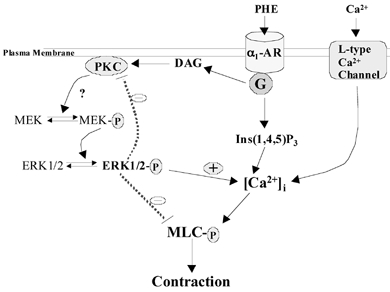
Shown here is α1-adrenergic mediated production of inositol 1,4,5-trisphosphate (Ins(1,4,5)P3) and diacylglycerol (DAG) with activation of protein kinase C (PKC). In turn, this leads to phosphorylation (P) of mitogen-activated protein kinase (MEK) and extracellular signal-regulated kinases (ERK1/2), with a positive role in the regulation of intracellular Ca2+ concentration, and a negative role in the regulation of Ca2+ sensitivity. In addition, activated ERKs demonstrate negative feedback on PKC activation.
Importantly, several studies have reported on non-Ca2+-dependent VSM contraction, mediated by PKC. Jiang & Morgan (1987) first described phorbol ester-induced contraction of both ferret and rat aorta, and differences in [Ca2+]i responses in comparison with PHE. Walsh and coworkers (1994) suggested that PKC may phosphorylate caldesmon and/or calponin to mediate this response. Rembold & Murphy (1998) demonstrated PKC-dependent phosphorylation of myosin can effect such a contractile response in the presence of only a small increase in [Ca2+]i. In rat cerebral arteries Gokina & Osol (1998) also demonstrated PKC's role in increasing myofilament Ca2+ sensitivity. By use of smooth muscle type II knockout mouse, Löhn and colleagues (2002) have shown PKC-induced sustained contraction in cerebral arteries, without an increase in [Ca2+]i. We also have demonstrated such PKC-induced, Ca2+-independent contraction, with important differences between fetus and adult cerebral arteries, possibly related to a relative lack of PKCε in the fetal vessels (Longo et al. 2000).
Perspectives
The present study was designed to examine the role of ERKs in modulating α-adrenergic-induced cerebral artery contraction and [Ca2+]i responses. An additional, the goal was to determine the extent to which these responses differ in the developing fetus, as compared with the adult. The results support the idea that in adult cerebral arteries, by suppressing Ca2+ sensitivity phosphorylated ERKs play a role in mediating the regulation of Ca2+-dependent vascular tone. In contrast, in the developing fetus, the lower basal levels of phosphorylated ERKs appear to play a prominent role in the regulation of contraction by modulating Ca2+ sensitivity. The present studies thus suggest a developmental switch from the regulation of tension by that of Ca2+ sensitivity in the fetus to Ca2+ concentration in the adult. Finally, the studies demonstrate that by regulating Ca2+ sensitivity, in part by PKC-mediated inhibition, ERKs play a key role in the regulation of vascular tone in cerebral arteries of both the adult and fetus. These results fit with our previous studies which demonstrated dependence in fetal cerebral arteries on extracellular Ca2+ (Long et al. 1999; Blood et al. 2002) in association with limited sarcoplasmic reticulum Ca2+ stores (Long et al. 2000a), and differences in function of PKC (Longo et al. 2000). This is in contrast to the adult arteries, which are more dependent upon the classical Ca2+-dependent pathway of agonist-mediated Ins(1,4,5)P3 release, Ins(1,4,5)P3 receptors, sarcoplasmic reticulum Ca2+ stores and [Ca2+]i-induced contraction.
Overall, the present studies reject the first hypothesis posed, that there exists a close relationship between agonist-induced ERK activation and vascular tension and [Ca2+]i. Nonetheless, they demonstrate that in cerebral arteries of both adult and fetus, ERKs play an important role in Ca2+ sensitivity. In addition, the studies illustrate again the complexity of pharmacomechanical coupling in cerebral (and undoubtedly other) vascular smooth muscle, and the significant differences that exist between these mechanisms in the developing fetus, as compared to the adult. We speculate that cross-talk among agonist-mediated MEK, ERK, PKC and related signal transduction mechanisms are even more complex than suggested by this study, and that their change with development is quite important.
Acknowledgments
We thank Brenda Kreutzer for preparing the manuscript. This work was supported by United States Public Health Service Grant HD/HL-03807 to L.D.L.
REFERENCES
- Adam LP. Mitogen-activated protein kinase. In: Bárány M, editor. Biochemistry of Smooth Muscle Contraction. San Diego, CA USA: Academic Press; 1996. pp. 167–177. [Google Scholar]
- Adam LP, Franklin MT, Raff GJ, Hathaway DR. Activation of mitogen-activated protein kinase in porcine carotid arteries. Circ Res. 1995;76:183–190. doi: 10.1161/01.res.76.2.183. [DOI] [PubMed] [Google Scholar]
- Adam LP, Haeberl R, Hathaway DR. Phosphorylation of caldesmon in arterial smooth muscle. J Biol Chem. 1989;264:7698–7703. [PubMed] [Google Scholar]
- Ahn NG, Nahreini TS, Tolwinski ND, Resing KA. Pharmacologic inhibitors of MKK1 and MKK2. Methods Enzymol. 2001;332:417–431. doi: 10.1016/s0076-6879(01)32219-x. [DOI] [PubMed] [Google Scholar]
- Alessandrini A, Crews CM, Erikson RL. Phorbol ester stimulates a protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product. Proc Natl Acad Sci U S A. 1992;89:8200–8204. doi: 10.1073/pnas.89.17.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci U S A. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood AB, Zhao Y, Long W, Zhang L, Longo LD. L-type Ca2+ channels in fetal and adult ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2002;282:R131–138. doi: 10.1152/ajpregu.00318.2001. [DOI] [PubMed] [Google Scholar]
- Boulton T, Cobb M. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 1991;2:357–371. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Childs TJ, Watson MH, Sanghera JS, Campbell DL, Pelech SL, Mak AS. Phosphorylation of smooth muscle caldesmon by mitogen-activated protein (MAP) kinase and expression of MAP kinases in differentiated smooth muscle. J Biol Chem. 1992;267:22853–22859. [PubMed] [Google Scholar]
- Cobb MH, Robbins DJ, Boulton TG. ERKs, extracellular signal-regulated MAP-2 kinases. Curr Opin Cell Biol. 1991;3:1025–1032. doi: 10.1016/0955-0674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoceptor stimulation. Am J Physiol. 1998;275:C1081–1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Matsumoto T, Motley ED, Utsunomiya H, Inagami T. Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells. J Biol Chem. 1996;271:14169–14175. doi: 10.1074/jbc.271.24.14169. [DOI] [PubMed] [Google Scholar]
- Epstein AM, Throckmorton D, Brophy CM. Mitogen-activated protein kinase activation: An alternate signaling pathway for sustained vascular smooth muscle contraction. J Vasc Surg. 1997;26:327–332. doi: 10.1016/s0741-5214(97)70196-4. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Gokina NI, Osol G. Temperature and protein kinase C modulate myofilament Ca2+ sensitivity in pressurized rat cerebral arteries. Am J Physiol. 1998;274:H1920–1927. doi: 10.1152/ajpheart.1998.274.6.H1920. [DOI] [PubMed] [Google Scholar]
- Gorenne I, Su X, Moreland RS. Inhibition of p42 and p44 MAP kinase does not alter smooth muscle contraction in swine carotid artery. Am J Physiol. 1998;275:H131–138. doi: 10.1152/ajpheart.1998.275.1.H131. [DOI] [PubMed] [Google Scholar]
- Jiang MJ, Morgan KG. Intracellular calcium levels in phorbol ester-induced contractions of vascular muscle. Am J Physiol. 1987;253:H1365–1371. doi: 10.1152/ajpheart.1987.253.6.H1365. [DOI] [PubMed] [Google Scholar]
- Katoch SS, Morland RS. Agonist and membrane depolarization induced activation of MAP kinase in the swine carotid artery. Am J Physiol. 1995;269:H222–229. doi: 10.1152/ajpheart.1995.269.1.H222. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle cell activation. Am J Physiol. 1993;265:C406–411. doi: 10.1152/ajpcell.1993.265.2.C406. [DOI] [PubMed] [Google Scholar]
- Lagaud GJL, Lam E, Lui A, van Breemen C, Laher I. Nonspecific inhibition of myogenic tone by PD98059, a MEK1 inhibitor, in rat middle cerebral arteries. Biochem Biophys Res Commun. 1999;257:523–527. doi: 10.1006/bbrc.1999.0350. [DOI] [PubMed] [Google Scholar]
- Löhn M, Kämpf D, Gui-Xuan C, Haller H, Luft FC, Gollasch M. Regulation of arterial tone by smooth muscle myosin type II. Am J Physiol. 2002;283:C1383–1389. doi: 10.1152/ajpcell.01369.2000. [DOI] [PubMed] [Google Scholar]
- Long W, Zhang L, Longo LD. Cerebral artery sarcoplasmic reticulum Ca2+ stores and contractility: changes with development. Am J Physiol Regul Integr Comp Physiol. 2000a;279:R860–873. doi: 10.1152/ajpregu.2000.279.3.R860. [DOI] [PubMed] [Google Scholar]
- Long W, Zhang L, Longo LD. Cerebral artery KATP and KCa channel activity and contractility: Changes with development. Am J Physiol Regul Integr Comp Physiol. 2000b;279:R2004–2014. doi: 10.1152/ajpregu.2000.279.6.R2004. [DOI] [PubMed] [Google Scholar]
- Long W, Zhao Y, Zhang L, Longo LD. Role of Ca2+ channels in NE-induced increase in [Ca2+]i and tension in fetal and adult cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 1999;277:R286–294. doi: 10.1152/ajpregu.1999.277.1.R286. [DOI] [PubMed] [Google Scholar]
- Longo LD, Ueno N, Zhao Y, Pearce WJ, Zhang L. Developmental changes in α1-adrenergic receptors, IP3 responses, and NE-induced contraction in cerebral arteries. Am J Physiol. 1996;271:H2313–2319. doi: 10.1152/ajpheart.1996.271.6.H2313. [DOI] [PubMed] [Google Scholar]
- Longo LD, Zhao Y, Long W, Miguel C, Windemuth RS, Cantwell AM, Nanyonga AT, Saito T, Zhang L. Dual role of PKC in modulating pharmacomechanical coupling in fetal and adult cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1419–1429. doi: 10.1152/ajpregu.2000.279.4.R1419. [DOI] [PubMed] [Google Scholar]
- Nixon GF, Iizuka K, Haystead CM, HaysteaD TA, Somlyo AP, Somlyo AV. Phosphorylation of caldesmon by mitogen-activated protein kinase with no effect on Ca2+ sensitivity in rabbit smooth muscle. J Physiol. 1995;48:283–289. doi: 10.1113/jphysiol.1995.sp020879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol Scand. 1998;164:623–635. doi: 10.1111/j.1365-201x.1998.tb10706.x. [DOI] [PubMed] [Google Scholar]
- Pang L, Decker S, Saltiel A. Bombesin and epidermal growth factor stimulate the mitogen-activated protein kinase through different pathways in Swiss 3T3 cells. Biochem J. 1993;289:283–287. doi: 10.1042/bj2890283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce WJ, Hull AD, Long DM, Longo LD. Developmental changes in ovine cerebral artery composition and reactivity. Am J Physiol. 1991;361:R458–465. doi: 10.1152/ajpregu.1991.261.2.R458. [DOI] [PubMed] [Google Scholar]
- Ratz PH. Regulation of ERK phosphorylation in differentiated arterial muscle of rabbits. Am J Physiol Heart Circ Physiol. 2001;281:H114–123. doi: 10.1152/ajpheart.2001.281.1.H114. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Murphy RA. [Ca2+]-dependent myosin phosphorylation in phorbol diester stimulated smooth muscle contraction. Am J Physiol. 1988;255:C719–723. doi: 10.1152/ajpcell.1988.255.6.C719. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal. 1997;9:337–351. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]
- Touyz RM, He G, Deng L-Y, Schiffrin EL. Role of extracellular signal-regulated kinases in angiotensin II-stimulated contraction of smooth muscle cells from human resistance arteries. Circulation. 1999;99:392–399. doi: 10.1161/01.cir.99.3.392. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Andrea JE, Allen BG, Clement-Chomienne O, Collins EM, Morgan KG. Smooth muscle protein kinase C. Can J Physiol Pharmacol. 1994;72:1392–1399. doi: 10.1139/y94-201. [DOI] [PubMed] [Google Scholar]
- Watts SW. Serotonin activates the mitogen-activated protein kinase pathway in vascular smooth muscle: use of the mitogen-activated protein kinase kinase inhibitor PD-098059. J Pharmacol Exp Ther. 1996;279:1541–1550. [PubMed] [Google Scholar]
- Xiao D, Zhang L. ERK MAP kinases regulate smooth muscle contraction in ovine uterine artery: effect of pregnancy. Am J Physiol Heart Circ Physiol. 2002;282:H292–300. doi: 10.1152/ajpheart.2002.282.1.H292. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zhao Y, Nijland R, Zhang L, Longo LD. Ins(1,4,5)P3 receptors in cerebral arteries: Changes with development and high altitude hypoxia. Am J Physiol. 1997;272:R1954–1959. doi: 10.1152/ajpregu.1997.272.6.R1954. [DOI] [PubMed] [Google Scholar]