

Abstract
The present study was aimed at investigating whether, besides its pivotal role in Ca2+-independent contraction of smooth muscle, Rho-kinase is involved in the mechanisms underlying the Ca2+ signal activated by noradrenaline in arteries. In rat aorta and mesenteric artery, the Rho-kinase inhibitor Y-27632 (10 μM) completely relaxed the contraction evoked by noradrenaline (1 μM) and simultaneously inhibited the Ca2+ signal by 54 ± 1 % (mesenteric artery) and 71 ± 15 % (aorta), and the cell membrane depolarisation by 56 ± 11 % (mesenteric artery). A similar effect was observed in arteries contracted by AlF4−, while in KCl-contracted arteries, Y-27632 decreased tension without changing cytosolic Ca2+. The same effects were observed with another inhibitor of Rho-kinase (HA1077) but not with an inhibitor of protein kinase C (Ro-31–8220). Effects of Y-27632 were not prevented by incubating the artery in 25 mM KCl, with K+ channel blockers or with the Ca2+ channel blocker nimodipine. Y-27632 did not affect either the increase in the production of inositol phosphates activated by noradrenaline, or the release of Ca2+ from non-mitochondrial stores evoked by InsP3 in permeabilised aortic cells, or the Ca2+ signals evoked by thapsigargin or caffeine. The capacitative Ca2+ entry activated by thapsigargin was not impaired by Y-27632, but the entry of Ba2+ activated by noradrenaline in the presence of nimodipine was blocked by 10 μM Y-27632. These results indicate that Rho-kinase is involved in noradrenaline activation of a Ca2+ entry distinct from voltage- or store-operated channels in rat arteries.
Cytosolic Ca2+ concentration ([Ca2+]cyt) is the main regulator of contraction in smooth muscle cells. A rise in [Ca2+]cyt leads to the activation of myosin light chain kinase (MLCK). Phosphorylation of myosin light chain (MLC) results in muscle contraction while dephosphorylation promotes smooth muscle relaxation. However, Ca2+-independent contractions have been described, and ascribed to Ca2+ sensitisation of contractile proteins (Kitazawa et al. 1989). The small GTPase Rho and its effector, Rho-kinase, play an important role in the Ca2+-independent regulation of smooth muscle contraction (Hirata et al. 1992). Evidence for the involvement of Rho-kinase is provided by the observations that addition of the catalytic subunit of Rho-kinase to permeabilised vessels results in contraction (Kureishi et al. 1997) and that the Rho-kinase inhibitor Y-27632 inhibits the contraction induced by phenylephrine or GTPγS (Uehata et al. 1997). Rho-kinase-dependent contraction is mediated through the increase in the level of phosphorylated MLC via the inhibition of MLC phosphatase (Fukata et al. 2001).
In vascular smooth muscle, cGMP-evoked relaxation is associated with an increase in smooth muscle myosin light chain phosphatase activity (Wu et al. 1996), and with inhibition of both Rho-dependent Ca2+ sensitisation of the contractile proteins and actin cytoskeleton organisation (Sauzeau et al. 2000). In addition, cGMP has been shown to inhibit the agonist-evoked Ca2+ signal (Ghisdal et al. 2000). However, it is not known whether inhibition of the Ca2+ signal could be related to the inactivation of Rho.
The objective of the present study was to investigate the role of Rho-kinase in the mechanisms underlying the increase in [Ca2+]cyt evoked by noradrenaline in rat aorta and mesenteric artery. α-Adrenergic receptors are known to activate the Gq-phospholipase C (PLC) pathway (Exton, 1994). The increase in [Ca2+]cyt evoked by noradrenaline originates from inositol trisphosphate (InsP3)-evoked Ca2+ release from the sarcoplasmic reticulum (SR) and from extracellular Ca2+ entry through voltage-operated Ca2+ channels (VOCs) and receptor and store-operated non-selective cationic channels (McFadzean & Gibson, 2002). The results indicate that Rho-kinase is involved in the G protein-dependent activation of non-selective cation channel in rat aorta and mesenteric arteries.
METHODS
Male Wistar rats (about 250 g) were used. The rats were anaesthetised with diethyl ether and killed by decapitation. The aorta and the superior mesenteric artery were rapidly removed, immersed in physiological solution (composition (mM): NaCl 122, KCl 5.9, NaHCO3 15, glucose 10, MgCl2 1.25 and CaCl2 1.25, supplemented with indomethacin (10 μM) and gassed with a mixture of 95 % O2-5 % CO2), and carefully cleaned of all fat and connective tissue. All experiments were carried out in accordance with national guidelines.
Measurement of contractile tension and cytosolic Ca2+ concentration
Artery rings were inverted, endothelium was gently removed and the rings were incubated for 3–3.5 h at room temperature in physiological solution (composition as above) containing 5 μM fura-2 acetoxymethyl ester (fura-2-AM) and 0.05 % Cremophor EL, as described (Ghisdal et al. 2000). After the loading period, the rings were mounted between two hooks under a tension of 8 mN (mesenteric artery) or 20 mN (aorta) in a 3 ml cuvette continuously perfused with physiological solution supplemented with NG-nitro-L-arginine (L-NNA, 100 μM) at 37 °C. The cuvette was part of a fluorimeter (CAF, JASCO, Tokyo, Japan) which allowed estimation of the calcium signal. The muscle tone was measured using an isometric force transducer. The artery segment was incubated for 30 min in physiological solution and was thereafter stimulated with 100 mM KCl solution (composition (mM): NaCl 27, KCl 100, NaHCO3 15, glucose 10, MgCl2 1.25, CaCl2 1.25, indomethacin 0.01). After washing and a further 15 min recuperation, the preparation was stimulated as required. When possible, control responses were recorded before incubation of the tissue with the inhibitor and application of a second stimulation. The amplitude of the second response was compared with the response measured before treatment. Control experiments without inhibitor were performed. Ca2+-free solution was prepared from physiological solution without Ca2+ supplemented with EGTA (0.1 mM). The increase in Ca2+ signal was measured with reference to the signal measured immediately before the stimulation, except for increase following re-addition of Ca2+ to a Ca2+-free solution, which was measured with reference to the baseline signal in Ca2+-containing solution. At the end of the experiment, the fura-2-Ca2+ signal was calibrated as previously described (Ghisdal et al. 2000). In some experiments, in particular when Ba2+ was used instead of Ca2+, the Ca2+ signal was expressed as the ratio of fluorescence at 340 and 380 nm (F340/F380), corrected for the autofluorescence measured at 340 and 380 nm after addition of MnCl2 (10 mM). The changes in fluorescence ratio were expressed either as percentages of the basal values or were normalised to the change evoked by the KCl stimulation. The rates of Ca2+ or Ba2+ entry were measured by the average slope of the change in fluorescence ratio calculated during the first minute after the addition of the cation into the perfusion solution (MacLab, AD Instruments Pty Ltd, Castle Hill, Australia).
Simultaneous measurement of contractile tension and membrane potential
Smooth muscle cell membrane potential was recorded in a segment of the superior mesenteric artery, 2 mm length, inverted and mounted in a myograph as described (Ghisdal et al. 2000). A glass microelectrode filled with 1.5 M KCl (resistance 50–80 MΩ) was advanced through the luminal surface of the arterial segment. Potential differences were measured with reference to the earthed bath by means of a Dagan amplifier (Minneapolis, MN, USA).
Determination of inositol phosphates
The artery segments were incubated in modified physiological solution (mM: NaCl 118, KCl 4.7, CaCl2 1.25, MgCl2 1.25, KH2PO4 1.2, EDTA 0.5, NaHCO3 25, Hepes 3.3, glucose 10, Tris-HCl 20, pH 7.4) supplemented with indomethacin (10 μM) and L-NNA (100 μM) at 37 ° C for 1 h. Subsequently, artery segments were incubated for 4 h at 37 °C in fresh buffer containing 20–25 μCi ml−1 of [3H]myo-inositol. At the end of the incubation, [3H]inositol-labelled artery segments were washed in buffer for 10 min and then transferred to physiological solution containing 10 mM LiCl plus noradrenaline (0.1–1 μM) and Y-27632 (10 μM) as required. Incubation was carried out for 30 min and stopped by rapid freezing of the tissue samples. Inositol phosphates were determined as described (Ghisdal et al. 2000). Data were normalised to the protein content of each sample.
45Ca2+ fluxes in β-escin permeabilised A7r5 cells
A7r5 cells were used between the 15th and the 20th passage after receipt from ECACC (European Collection of Cell Cultures, UK). The cells were cultured at 37 °C in DMEM medium (Gibco, catalogue no. 41965–039) supplemented with 2 mM glutamine and 10 % fetal bovine serum, 50 u ml−1 penicillin and 50 μg ml−1 streptomycin. The cells were seeded in 12-well dishes. Permeabilisation was carried out by replacing the culture medium with 2 ml of permeabilisation medium containing 120 mM KCl, 30 mM imidazole/HCl (pH 6.8), 2 mM MgCl2, 1 mM ATP, 1 mM EGTA, 30 μM β-escin, at 22 °C. The permeabilisation medium was removed after 30 min and the cells were washed twice with the same but β-escin-free medium. 45Ca2+ fluxes were measured as described by Missiaen et al. 1992. 45Ca2+ uptake was accomplished by incubation of the cells in 2 ml of loading solution containing 120 mM KCl, 30 mM imidazole/HCl (pH 6.8), 5 mM MgCl2, 5 mM ATP, 0.44 mM EGTA, 10 mM NaN3, 150 nM free Ca2+ (20 μCi ml−145Ca2+) at 22 °C for 50 min. The cells were then washed three times in an efflux medium containing 120 mM KCl, 30 mM imidazole/ HCl (pH 6.8), 2 mM MgCl2, 1 mM ATP, 1 mM EGTA, 5 mM NaN3, 2 μM thapsigargin. A 1 ml volume of this medium was then added to the cells and replaced every 2 min. At the end of the experiment the 45Ca2+ remaining in the stores was released by incubation with 1 ml of a 1 % SDS solution for 45 min. Ca2+ release was expressed as the amount of Ca2+ leaving the stores in 2 min divided by the total store content at that time.
Drugs
Y-27632 ((+)-(R)-trans-4-(1-aminoethyl)-N-(4-pyridyl) cyclohexanecarboxamide dihydrochloride monohydrate) was a gift from Mitsubishi Pharma Corporation (Osaka, Japan). Fura-2 acetoxymethylester (fura-2-AM), HA1077 and Ro-31–8220 were from Calbiochem (EuroBiochem, Bierges, Belgium). [3H]myo-inositol was from NEN Life Science Products (Zaventem, Belgium), 45Ca2+ was from Amersham Biosciences (Little Chalfont, UK). All other compounds were obtained from Sigma. AlF4− was produced by the combination of 5 mM sodium fluoride (NaF) and 30 μM aluminum chloride (AlCl3).
Statistical analysis
Quantified data are presented as means ±s.e.m. The noradrenaline concentration producing 50 % of the maximal effect (EC50) and the concentration of Y-27632 producing 50 % inhibition of the noradrenaline responses (IC50) were calculated by non-linear curve fitting (Prism, GraphPad). Log values were used for statistical analysis. Comparisons were made by Student's t test. Differences with P values smaller than 0.05 were considered significant.
RESULTS
Effect of the Rho-kinase inhibitor Y-27632 on contraction and the Ca2+ signal evoked by noradrenaline, AlF4− and KCl
Figure 1 shows the effect of Y-27632 (10 μM) on the Ca2+ signal and contraction in mesenteric artery. Contraction evoked by noradrenaline was associated with a rapid increase in the cytosolic Ca2+ concentration ([Ca2+]cyt) followed by a sustained component which was maintained for at least 10 min. Y-27632 (10 μM) produced a simultaneous decrease in contraction and in [Ca2+]cyt (Fig. 1A). After 1–2 min, the contraction was completely relaxed and the Ca2+ signal was depressed by 54 ± 1 % (n = 5). Noradrenaline concentration-effect curves for contractile tension and Ca2+ signal were established in control tissues and after 15 min pre-incubation of artery rings with 10 μM Y-27632. The curves are shown in Fig. 2A and B. Y-27632 markedly depressed the contraction curve and produced a significant rightward shift of the Ca2+ signal curve: the noradrenaline pD2 value (-logEC50) for the Ca2+ signal was decreased from 7.25 ± 0.16 in untreated arteries (n = 5) to 5.87 ± 0.44 in Y-27632-treated arteries (n = 5) (P < 0.05).
Figure 1. Representative recordings of the effect of Y-27632 on the Ca2+ signal and the contraction evoked by noradrenaline (A), AlF4− (B) or 100 mM KCl (C) in rat mesenteric artery rings.
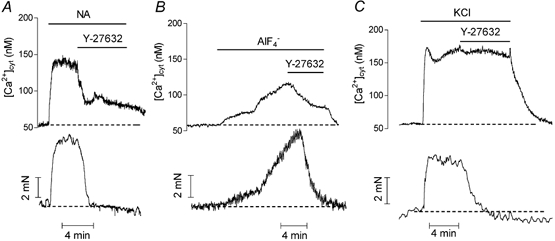
[Ca2+]cyt (upper trace) and contraction (lower trace) were measured simultaneously in fura-2-loaded arteries. Traces A, B and C are from different arteries. Noradrenaline (NA, 1 μM), AlF4− (30 μM), KCl (100 mM) and Y-27632 (10 μM) were applied as indicated.
Figure 2. Effect of Y-27632 on noradrenaline concentration-response curves in fura-2-loaded mesenteric artery (A and B) and aorta (C and D).
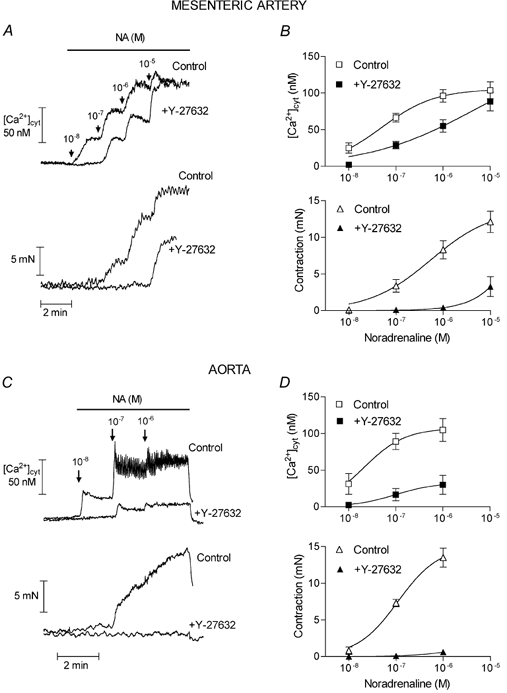
[Ca2+]cyt (upper traces and graphs) and contraction (lower traces and graphs) were simultaneously measured in control artery rings or in artery rings pre-incubated for 10 min in the presence of 10 μM Y-27632. A and C, typical traces obtained in untreated and Y-27632-treated segments from the same artery are superimposed. B and D, mean values from 4–5 different arteries. Vertical bars represent the s.e.m. values.
In order to exclude the possibility that Y-27632 could inhibit noradrenaline-evoked responses by interacting with the adrenergic receptor, arteries were stimulated with AlF4−, which activates G proteins directly (Boonen & De Mey, 1990). As illustrated in Fig. 1B, 10 μM Y-27632 inhibited the Ca2+ signal evoked by AlF4− by 63 ± 5.4 % (n = 4), while contraction was abolished. The effect of Y-27632 was also investigated in artery contracted by 100 mM KCl. KCl contractions were evoked in the presence of phentolamine (1 μM) to inhibit possible effects of noradrenaline, which could be released from nerve terminals. In contrast with stimulations involving G protein activation, in KCl-contracted artery Y-27632 decreased the contractile tension but did not significantly affect the Ca2+ signal (Fig. 1C). An investigation of the effects of Y-27632 in rat aorta gave similar results except that the noradrenaline-evoked Ca2+ signal was more sensitive to the inhibitory effect of Y-27632 in the aorta than in the mesenteric artery (in the aorta, the Ca2+ signal in response to 1 μM noradrenaline was inhibited by 71 ± 15 % with 10 μM Y-27632, n = 5). This was confirmed by the marked depression of the noradrenaline concentration-response curves in the aorta in the presence of the Rho-kinase inhibitor (Fig. 2C and D). In order to address the possibility that the effects of Y-27632 could be mediated via other kinases than the Rho-kinase and particularly via protein kinase C (PKC), we tested the effects of another inhibitor of Rho-kinase, HA1077 (Uehata et al. 1997; Sward et al. 2000), and of a selective inhibitor of PKC, Ro-31–8220, in aortic segments. The effects of HA1077 were similar to the effects of Y-27632 although HA1077 was slightly less potent than Y-27632: 10 μM HA1077 relaxed the contraction and inhibited the Ca2+ signal evoked by noradrenaline (1 μM) in aorta by 67 ± 10 % and 37 ± 10 % (n = 4), respectively (Fig. 3A). The increase in [Ca2+]cyt evoked by 100 mM KCl solution was not significantly affected (the Ca2+ signal in the presence of 10 μM HA1077 was 91 ± 4 % (n = 3, P > 0.05) of the Ca2+ signal in the absence of the inhibitor) but the contraction was inhibited by 84 ± 4 % (n = 3). The inhibitor of PKC, Ro-31–8220 (3 μM), did not affect the responses to noradrenaline and did not prevent the effects of Y-27632 (Fig. 3B).
Figure 3. Representative recordings of the effect of HA1077 and Ro-31–8220 on Ca2+ signal (upper traces) and contraction (lower traces) evoked by noradrenaline in fura-2-loaded aorta.
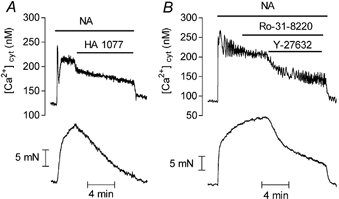
Noradrenaline (NA, 1 μM), HA1077 (10 μM), Ro-31–8220 (3 μM) and Y-27632 (10 μM) were applied as indicated.
The relation between the concentration of Y-27632 and its inhibitory effect on contraction and on the Ca2+ signal evoked by a sub-maximal concentration of noradrenaline (1 μM) in mesenteric artery is shown in Fig. 4. As observed in Fig. 1 and Fig. 2, part of the Ca2+ signal was resistant to Y-27632 (maximum inhibition was 54 ± 0.9 %, n = 3), while contraction was inhibited by 91 ± 4 % (n = 3). However, the pA2 (-logIC50) values for the effects of Y-27632 on the Ca2+ signal and on contraction were not different (6.29 ± 0.10 and 6.28 ± 0.03, respectively). The maximum effect was obtained at concentrations of Y-27632 higher than 3 μM. A concentration of 10 μM, which was maximally effective, was used in all following experiments.
Figure 4. Concentration-inhibition curves for effects of Y-27632 on the Ca2+ signal and the contraction evoked by noradrenaline in mesenteric artery.
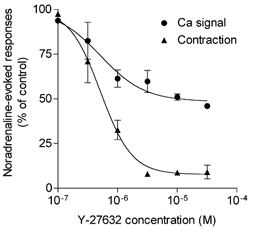
Fura-2-loaded arteries were incubated with Y-27632 (10 μM) for 15 min before stimulation with 1 μM noradrenaline. Responses were expressed as a percentage of the control responses recorded before the incubation with Y-27632. Each point is the mean value of 4–5 determinations. Vertical bars represent the s.e.m. values.
Effect of Y-27632 on noradrenaline-evoked depolarisation
Noradrenaline-evoked contraction of mesenteric artery is accompanied by a depolarisation of vascular smooth muscle cells (Mulvany et al. 1982; Ghisdal et al. 2000). We investigated whether, in addition to its effect on the Ca2+ signal, Rho-kinase inactivation affected noradrenaline-evoked depolarisation in the mesenteric artery. The mean resting membrane potential of mesenteric artery smooth muscle cells was −46.2 ± 0.7 mV (n = 14). Noradrenaline (1 μM) depolarised smooth muscle cells to −35.8 ± 1.6 mV and simultaneously produced a contraction of 4.8 ± 0.8 mN (n = 8). In most cells, rhythmic oscillations of the membrane potential of 5–10 mV of amplitude were superimposed on the tonic depolarisation (Fig. 5). The pre-incubation of arteries with Y-27632 did not change the resting membrane potential (−46.0 ± 1.1 vs. −45.5 ± 1.1 mV, n = 6) but modified the response to noradrenaline. In the presence of the Rho-kinase inhibitor, the depolarisation evoked by noradrenaline presented a peak followed by a stable response (Fig. 5), which was significantly lower than in the control (cells were depolarised by 10.6 ± 1.2 mV (n = 8) and 6.0 ± 1.0 mV (n = 5) in the absence and in the presence of 10 μM Y-27632, respectively, P < 0.05). Simultaneously, the contractile response to noradrenaline was completely abolished (Fig. 5).
Figure 5. Effect of Y-27632 on the depolarisation evoked by noradrenaline in mesenteric artery smooth muscle cells.
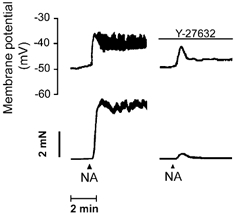
Representative recording of the change in membrane potential (upper trace) and in tension (lower trace) evoked by noradrenaline (NA, 1 μM) in mesenteric artery before and after incubation with Y-27632 (10 μM). Traces are from the same artery segment.
Influence of K+ channel blockers or increased external KCl concentration on the effects of Y-27632
In order to determine whether the Y-27632-evoked inhibition of the Ca2+ signal and the depolarisation in response to noradrenaline that was observed in mesenteric artery could be caused by an interaction of Rho-kinase with K+ channels, we investigated the effect of Rho-kinase inhibition in the presence of a cocktail of the following K+ channel blockers: 1 mM 4-aminopyridine to inhibit voltage-operated K+ channels, 0.1 μM apamin to inhibit small-conductance Ca2+-activated K+ channels, 0.1 μM charybdotoxin to inhibit large-conductance Ca2+-activated K+ channels, and 10 μM glybenclamide to inhibit ATP-dependent K+ channels (Nelson & Quayle, 1995). The results indicated that neither K+ channel blockers nor 25 mM KCl, which change the equilibrium potential of K+ to less negative values, prevented the inhibitory effect of Y-27632 on the Ca2+ signal evoked by noradrenaline: Y-27632 inhibited the noradrenaline-evoked Ca2+ signal by 40 ± 9.7 % (n = 5), 42 ± 7.7 % (n = 5) and 49 ± 1.4 % (n = 5) in the presence of the cocktail of K+ channel blockers, in a solution containing 25 mM KCl and in untreated mesenteric arteries, respectively.
Effect of Y-27632 on the production of inositol phosphates evoked by noradrenaline
α-Adrenergic receptors are known to be coupled with Gq proteins which activate phospholipase Cβ to increase the production of InsP3 (Exton, 1994). Ca2+ signalling activated by noradrenaline in arteries is dependent on the increase in InsP3 production (Ghisdal et al. 2000). In order to investigate whether the inhibitory effect of Y-27632 on the noradrenaline-dependent Ca2+ signal could be related to an inhibition of the PLC activity, we measured its effect on the accumulation of inositol phosphates produced by noradrenaline in the aorta and the mesenteric artery. Table 1 summarises the results and shows that no significant difference could be observed between untreated arteries and arteries incubated in the presence of Y-27632 (10 μM).
Table 1.
Effect of Y-27632 on the accumulation of inositol phosphates evoked by noradrenaline in aorta and mesenteric artery
| Accumulation of inositol phosphates (% of basal control value) | ||||
|---|---|---|---|---|
| Mesenteric artery (n = 8) | Aorta (n = 5) | |||
| Untreated | +Y-27632 | Untreated | +Y-27632 | |
| Control | 100 | 99 ± 5 | 100 | 105.3 ± 4.5 |
| Noradrenaline (0.1 μm) | 230 ± 21* | 216 ± 16* | 343 ± 18* | 315 ± 22* |
| Noradrenaline (1 μM) | 395 ± 33* | 388 ± 20* | 437 ± 19* | 411 ± 25* |
Artery rings loaded with [3H]myo-inositol were pretreated with or without Y-27632 (10 μm) for 20 min and further incubated with noradrenaline (0.1–1 μm) for 30 min.
P < 0.01 vs. control.
Effect of Y-27632 on the release of Ca2+ from non-mitochondrial stores evoked by InsP3
To address the question of whether Rho-kinase regulates InsP3 receptors, we measured the effect of Y-27632 on InsP3-induced Ca2+ release in permeabilised monolayers of cultured aortic A7r5 cells. This cell line is a well-characterised model system for analysing InsP3-induced Ca2+ release under conditions of unidirectional flux (Missiaen et al. 1992). After permeabilisation of the cells with β-escin and loading of non-mitochondrial stores with 45Ca2+, incubation of the cells in an efflux medium led to a passive efflux of 45Ca2+ out of the stores. The addition of InsP3 to the intracellular medium produced an immediate release of 45Ca2+. As shown in Fig. 6, the increase in fractional release of Ca2+ evoked by 0.1–10 μM InsP3 did not differ between control cells and cells treated with 10 μM Y-27632.
Figure 6. Effect of Y-27632 on InsP3-evoked 45Ca2+ release in permeabilised A7r5 aortic cells.
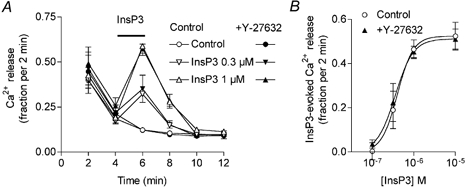
A, 45Ca2+-loaded Ca2+ stores were allowed to release Ca2+ passively in the efflux medium with or without Y-27632 (10 μM). InsP3 (0.3 and 1 μM) was applied for 2 min as indicated by the horizontal bar. Ca2+ release was plotted as fractional release (the amount of Ca2+ leaving the stores in 2 min divided by the total store content at that time). Points are means from four experiments. B, Ca2+ release evoked by a 2 min application of various concentrations of InsP3 in the absence or presence of 10 μM Y-27632.
Effect of Y-27632 on responses to noradrenaline in the presence of the Ca2+ channel blocker nimodipine
In rat aorta and mesenteric artery, the contractions evoked by noradrenaline are dependent on both intracellular and extracellular Ca2+. Part of the entry of Ca2+ produced by noradrenaline is associated with the opening of VOCs (Godfraind & Dieu, 1981; Morel & Godfraind, 1991). In the mesenteric artery, as in the aorta, in the presence of the L-type Ca2+ channel blocker nimodipine (1 μM), noradrenaline produced a transient peak of [Ca2+]cyt followed by a sustained phase which plateaued at about 30–45 % of the response measured in the absence of nimodipine (in the absence and in the presence of nimodipine, noradrenaline increased the Ca2+ signal in mesenteric artery by 114 ± 20 nM, n = 6, and 35 ± 14 nM, n = 5, (P < 0.05), and in aorta by 83 ± 7 nM, n = 8 and 37 ± 4 nM, n = 8 (P < 0.05), respectively) (Fig. 7). Simultaneously, in agreement with a previous report (Godfraind & Dieu, 1981), contraction was less affected by nimodipine in the aorta where it was reduced by 25 %, from 19 ± 0.9 mN (n = 8) to 15 ± 1.2 mN (n = 8, P > 0.05), than in the mesenteric artery, where contraction was inhibited by 57 %, from 7.8 ± 0.8 mN (n = 9) to 3.4 ± 0.8 mN (n = 5, P < 0.05). In the presence of 10 μM Y-27632, the plateau phase of the nimodipine-resistant increase in [Ca2+]cyt was depressed by 67 ± 8 % in the mesenteric artery (n = 3, P < 0.05 compared with the inhibition produced by Y-27632 in the absence of nimodipine) and by 63 ± 5 % in the aorta (n = 7, not significantly different from the effect of Y-27632 in the absence of nimodipine) (Fig. 7).
Figure 7. Inhibition of the responses to noradrenaline by Y-27632 in nimodipine-treated aorta.
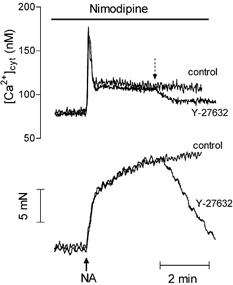
Representative recording of the effect of noradrenaline (1 μM NA) on [Ca2+]cyt (upper trace) and contractile tension (lower trace) in fura-2-loaded aortic segments pretreated with the Ca2+ channel blocker nimodipine (1 μM). Y-27632 (10 μM) or vehicle were added at the time indicated by the dashed arrow. Control and Y-27632 traces were from different segments of the same aorta.
Effect of the Rho-kinase inhibitor Y-27632 on responses to noradrenaline in Ca2+-free solution and after re-addition of Ca2+
The components of the Ca2+ signal evoked by noradrenaline were further investigated in aorta perfused with Ca2+-free solution. Perfusion with Ca2+-free solution produced a small decrease in the basal F340/F380 ratio, which was reversed when Ca2+ was re-added to the perfusion solution (not shown). Noradrenaline applied after 90 s perfusion of the aorta with Ca2+-free solution produced a large but transient increase in Ca2+ signal (peak was 141 ± 23 nM Ca2+, n = 12) associated with a sustained contraction that was depressed compared with the tension developed in Ca2+-containing solution (9.6 ± 0.8 mN, n = 12, and 19 ± 0.9 mN, n = 7, respectively, P < 0.01). The re-addition of Ca2+ to the bathing solution produced a slow increase in [Ca2+]cyt which reached a steady level of 67 ± 6.7 nM above the baseline (n = 12). Figure 8 shows that in aortas pre-incubated with Y-27632, the peak Ca2+ response evoked by noradrenaline in Ca2+-free solution was significantly depressed (40 ± 5 % inhibition at peak, n = 7) (Fig. 9A). The rate of Ca2+ entry evoked by the re-addition of Ca2+ to the solution was decreased by 59 %, from 1.37 ± 0.27 F340/F380 ratio units s−1 to 0.64 ± 0.10 ratio units s−1 (n = 6, P < 0.05), and the steady-state Ca2+ level was inhibited by 75 ± 5 % (n = 6) (Fig. 9B).
Figure 8. Effect of Y-27632 on the Ca2+ signal and the contraction evoked by noradrenaline in aorta in Ca2+-free solution.
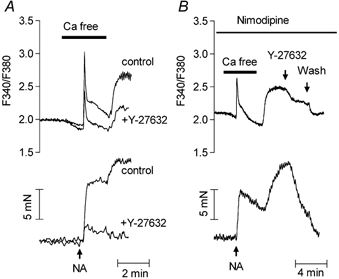
A, representative recording of the effect of pre-incubation of fura-2-loaded aorta with Y-27632 on the Ca2+ signal (upper traces) and contraction (lower traces). Physiological solution was changed to Ca2+-free solution as indicated by the horizontal line. Noradrenaline (NA, 1 μM) was applied as indicated by the arrow. Traces obtained before and after incubation with Y-27632 (10 μM) are superimposed. They were obtained from the same artery ring. B, representative recording of Ca2+ signal (upper trace) and contraction (lower trace) showing the effect of Y-27632 (10 μM) applied during the re-addition of Ca2+ to the perfusion solution in a fura-2-loaded aorta pretreated with nimodipine (1 μM). Noradrenaline (NA, 1 μM) and Y-27632 (10 μM) were applied as indicated.
Figure 9. Effect of Y-27632 on the different components of the Ca2+ signal evoked by noradrenaline in fura-2-loaded aorta in the absence or presence of nimodipine.
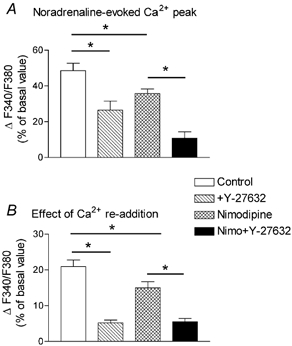
Columns represent the mean values of the increase in F340/F380 ratio measured in Ca2+-free solution at the time of the peak Ca2+ response to noradrenaline (1 μM) (A), or after re-addition of Ca2+ (1.25 mM) to the Ca2+-free solution (B), in untreated aorta, or in aorta pretreated with Y-27632 (10 μM), nimodipine (1 μM), or nimodipine plus Y-27632. The protocol of the experiment was as represented in Fig. 8A. The effect of re-addition of Ca2+ was measured with reference to the baseline fluorescence ratio measured in Ca2+-containing solution. Increases in the F340/F380 ratio were expressed as a percentage of the basal F340/F380 value and are means from 7–13 determinations ±s.e.m.*P < 0.05.
In the presence of nimodipine, the peak Ca2+ signal evoked by noradrenaline in Ca2+-free solution was reduced by 27 ± 8 % and the effect of re-addition of Ca2+ was depressed by 29 ± 10 % (n = 7) compared with untreated arteries. However, the peak response and Ca2+ signal evoked by re-addition of Ca2+ to the Ca2+-free solution were still inhibited by Y-27632 (Fig. 9). In the presence of nimodipine (Fig. 8B), as in its absence (not shown), the application of 10 μM Y-27632 during the plateau phase of the Ca2+ signal evoked by re-addition of Ca2+ to the Ca2+-free solution inhibited the Ca2+ signal with the same potency as that obtained after pre-incubation of the artery with the Rho-kinase inhibitor, suggesting that this effect was not related to the inhibition of the peak of Ca2+.
Effect of the Rho-kinase inhibitor Y-27632 on Ba2+ entry
In order to exclude any intervention of Ca2+ release or storage in the Ca2+ signal and to be free of the activation of Ca2+-dependent feedback mechanisms, we used Ba2+ instead of Ca2+ to monitor the entry of divalent cations activated by noradrenaline (Inoue et al. 2001). Fura-2-loaded aortas were treated with nimodipine to block VOCs and incubated for 5 min in Ca2+-free solution. Addition of Ba2+ (1 mM) to the Ca2+-free solution increased the F340/F380 ratio above the basal value by 18 ± 3 % of the basal signal (n = 6). When arteries were stimulated by 1 μM noradrenaline during incubation in the Ca2+-free solution, the increase in F340/F380 ratio evoked by Ba2+ was significantly higher than in unstimulated aortas (29 ± 1.8 % of the basal ratio, n = 7, P < 0.05 compared with unstimulated aortas) (Fig. 10). Y-27632 did not affect the increase in F340/F380 ratio due to Ba2+ in unstimulated arteries but it significantly decreased the rate and the amplitude of the entry of Ba2+ in noradrenaline-stimulated arteries (Fig. 10). The same inhibition of Ba2+ entry was observed when Y-27632 was applied before noradrenaline (not shown) or after the Ca2+ peak evoked by noradrenaline in Ca2+-free solution (as shown in Fig. 10), indicating that the inhibition was unrelated to the amplitude of the Ca2+ peak evoked by noradrenaline in Ca2+-free solution.
Figure 10. Effect of Y-27632 on Ba2+ entry in fura-2-loaded aorta.
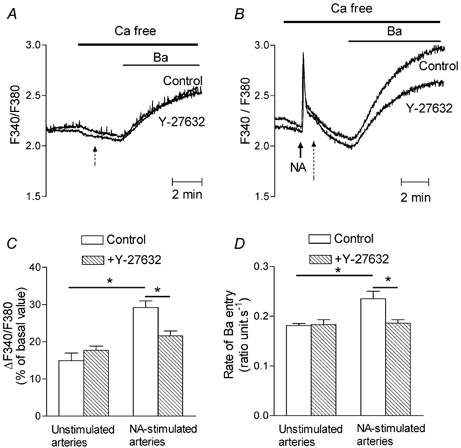
A, superimposed representative recordings of the changes in the F340/F380 ratio evoked by the addition of Ba2+ (1 mM) into nimodipine-containing Ca2+-free perfusion solution. Vehicle (control) or Y-27632 (10 μM) was added at the time indicated by the dashed arrow. Traces were from different aortas. B, superimposed representative recordings of the changes in F340/F380 ratio evoked by the addition of Ba2+ (1 mM) to nimodipine-containing Ca2+-free perfusion solution after stimulation with 1 μM noradrenaline (NA). Vehicle (control) or Y-27632 (10 μM) was added at the time indicated by the dashed arrow. Traces were from different aortas. C, bar graph showing the mean values of the change in the F340/F380 ratio evoked by Ba2+ expressed as a percentage of the basal F340/F380 value in unstimulated aorta (n = 6) and in aorta stimulated by 1 μM noradrenaline (NA, n = 7) in the absence and presence of Y-27632 (10 μM). D, bar graph showing the mean values of the rate of Ba2+ entry expressed as change in F340/F380 ratio s−1 in unstimulated aorta (n = 6) or in aorta stimulated by 1 μM noradrenaline (NA, n = 7) in the absence (□) and presence of Y-27632 (10 μM) (□). *P < 0.05.
Effect of Y-27632 on agonist-independent responses and on capacitative Ca2+ entry
In order to investigate whether the Ca2+ signal evoked by agonist-independent stimuli was also sensitive to Y-27632, we tested the responses of aorta to caffeine and to thapsigargin. In Ca2+-containing solution, caffeine (10 mM) produced a rapid increase in [Ca2+]cyt which peaked at 148 ± 19 nM Ca2+ (86 ± 17 % of the KCl-evoked response, n = 7) followed by a lower plateau phase (97 ± 12 nM Ca2+, n = 7) associated with a small and transient contraction (2.4 ± 1.1 mN, 23 ± 9 % of the contraction evoked by KCl, n = 7, not shown). The Ca2+ signal evoked by caffeine was not affected by Y-27632 (data not shown). It has been shown that translocation of Rho to the plasma membrane is a necessary step for the activation of Rho-kinase by carbachol, phenylephrine or GTPγS (Fujihara et al. 1997; Gong et al. 1997; Miyazaki et al. 2002). In order to exclude the possibility that the lack of effect of Y-27632 could be due to the non-activation of the Rho A-Rho-kinase pathway in the absence of agonist, the artery was first stimulated with noradrenaline and then caffeine was added to the bathing solution (Fig. 11A and B). In the presence of noradrenaline, caffeine did not produce a further large change in Ca2+ signal: [Ca2+]cyt increased transiently to 158 ± 15 nM and stabilised at 95 ± 18 nM (n = 4). In Y-27632-treated aorta, the Ca2+ signal evoked by noradrenaline alone was depressed by 52 ± 8.3 % (n = 4), as expected from the previous results, while the response to noradrenaline plus caffeine was not impaired (the Ca2+ signal in the presence of Y-27632 was 116 ± 13 % of the response in the absence of Y-27632, n = 4).
Figure 11. Effect of Y-27632 on the Ca2+ signal evoked by caffeine or thapsigargin in fura-2-loaded aorta.
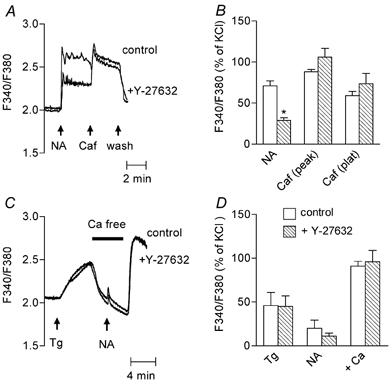
A, representative recording of the effect of pre-incubation of the aorta with Y-27632 (10 μM) on the Ca2+ signal evoked by caffeine. Noradrenaline (NA, 1 μM) was applied 3 min before stimulation with caffeine (Caf, 10 mM). Traces obtained before and after incubation with Y-27632 are superimposed. They were obtained from the same aortic ring. B, bar graph showing mean values of the Ca2+ signal evoked by noradrenaline (NA); caffeine measured at the peak (Caf peak) and the plateau of the response (Caf plat) in control (□) and in Y-27632-treated aorta (□). Data were normalised to the response to 100 mM KCl and are means from 4 determinations ±s.e.m.*P < 0.05, Y-27632-treated vs. untreated aortas. C, representative recording of the effect of Y-27632 on Ca2+ signal evoked by thapsigargin. Thapsigargin (Tg, 1 μM) and noradrenaline (NA, 1 μM) were applied as indicated by the arrows. The physiological solution was changed to a Ca2+-free solution as indicated by the horizontal bar. Traces obtained without and with Y-27632 (10 μM) are superimposed. They were obtained from different aortic rings. D, bar graph showing mean values of Ca2+ signal evoked by thapsigargin (Tg), noradrenaline (NA) measured at the peak of the response in Ca2+-free solution, and after re-admission of Ca2+ (1.25 mM) to the solution (+Ca) in control (□) and in Y-27632-treated aorta (□). Effect of re-addition of Ca2+ was measured with reference to the baseline fluorescence ratio measured in Ca2+-containing solution. Data were normalised to the response to 100 mM KCl and are means from 4 determinations ± S.E.M.
Thapsigargin evokes the release of intracellular Ca2+ stores by blocking the activity of the Ca2+ pump of the SR. All experiments with thapsigargin were performed in the presence of nimodipine in order to observe capacitative Ca2+ entry. In Ca2+-containing solution, the addition of thapsigargin (1 μM) increased [Ca2+]cyt by 56 ± 18 nM (n = 4) (Fig. 11C and D) without producing a marked contraction (not shown). The response to thapsigargin was not modified after incubation with Y-27632. Capacitative Ca2+ entry was estimated from the increase in Ca2+ signal produced by the addition of Ca2+ into the bathing solution after 6 min incubation in Ca2+-free solution in the presence of thapsigargin (Berridge, 1995). Perfusion with Ca2+-free solution caused a slow decrease in the Ca2+ signal. Noradrenaline was applied for 3 min during the incubation of the artery in the Ca2+-free solution, in order to ensure that Rho-kinase was activated (Fig. 11C and D). Under this condition, noradrenaline produced only a small, transient peak in Ca2+ signal, varying between 2 and 30 % of the response to KCl (P < 0.05 compared with the Ca2+ signal measured in the absence of thapsigargin). The re-addition of Ca2+ to the solution produced a large increase in Ca2+ signal above the basal level, which was not affected by Y-27632.
DISCUSSION
The present results indicate that in rat aorta and mesenteric artery, Rho-dependent kinase is involved in the Ca2+ entry activated by noradrenaline or by direct stimulation of G proteins, in addition to its role in the Ca2+ sensitisation of the contractile proteins.
In agreement with previous reports, the Rho-kinase inhibitor Y-27632 inhibited contraction of the aorta and of the mesenteric artery (Uehata et al. 1997). Depression of KCl-evoked contraction confirms that Rho-kinase is activated by depolarisation in rat caudal artery (Mita et al. 2002). Interestingly, the Ca2+ signal evoked by noradrenaline or direct activation of G protein, but not that evoked by high KCl, was significantly inhibited by the Rho-kinase inhibitors Y-27632 and HA1077.
Y-27632 displayed the same potency in inhibiting contraction and the Ca2+ signal activated by noradrenaline. The IC50 value of Y-27632 (500 nM) is in agreement with its reported IC50 value for the inhibition of the Rho-dependent kinases p160ROCK and its isozyme ROCK-II in in vitro assay and for the relaxation of rabbit aorta (Uehata et al. 1997; Davies et al. 2000). Another inhibitor of Rho-kinase, HA1077 (Uehata et al. 1997; Sward et al. 2000), produced the same pattern of effects as Y-27632 and the effects of the two inhibitors were not additive (data not shown). In addition, the PKC inhibitor Ro-31–8220 did not mimic the effect of Y-27632 and did not prevent it. We cannot exclude the possibility that Y-27632 inhibits other kinases or interacts with other targets, but, taken together, these observations suggest that the effects of Y-27632 on contraction and the Ca2+ signal are mediated through its interaction with Rho-dependent kinase.
Ca2+ signalling activated by α-adrenoceptors in arteries is dependent on the activation of PLCβ by Gq proteins (Exton, 1994). PI4 phosphate 5 kinase, which is involved in the synthesis of the PLC substrate, PIP2, has been reported to be a target of Rho-kinase (Oude Weernink et al. 2000). However, prolonged inactivation of Rho protein is required to observe the inhibition of inositol phosphate production in N1E-115 neuroblastoma cells (Zhang et al. 1996) or in mouse fibroblasts (Chong et al. 1994). The present results showed that inhibition of Rho-kinase produced a drop in the noradrenaline-activated Ca2+ signal within a few minutes but did not affect the production of inositol phosphates. Inhibition of the initial rapid increase in InsP3 production evoked by agonists after 5 s of stimulation (Berridge, 1983) cannot be excluded from the present data but would not be consistent with the observation that Y-27632 evenly inhibited the Ca2+ response to noradrenaline 10 min after beginning of stimulation.
In vascular smooth muscle cell, the increase in cytosolic Ca2+ after noradrenaline stimulation originates from both intracellular stores, mainly located in the SR, and the extracellular compartment, in proportions varying according to the artery. Ca2+ entry occurs through several pathways: VOC, receptor-operated and store-operated Ca2+ channels (McFadzean & Gibson, 2002). Y-27632 inhibited the nimodipine-resistant entry of Ba2+ in noradrenaline-stimulated arteries. Since Ba2+ is not taken up by Ca2+ exchangers and pumps, this result shows unequivocally that Y-27632 inhibits the activation of a cationic channel distinct from the VOC. The observations that Y-27632 affected noradrenaline-evoked Ca2+ and Ba2+ entry similarly when added before or after store emptying, and that InsP3 production and Ca2+ release were not affected, indicate that Rho-kinase might regulate a cationic channel directly. This pathway is distinct from the capacitative Ca2+ entry, or store-operated Ca2+ entry, which was demonstrated by the effect of re-addition of Ca2+ to the medium after store depletion by thapsigargin in Ca2+-free medium (Berridge, 1995). The latter Ca2+ signal was sensitive to low concentrations of Gd3+ (not shown), which is known to be an inhibitor of capacitative Ca2+ entry (Broad et al. 1999), but was not affected by Y-27632. The saturation of the effect of Y-27632 on the fura-2-Ca2+ signal compared with the contractile response probably reflects the selective inhibition of non-selective cationic channels while other Ca2+ sources activated by noradrenaline were unaffected.
In aorta bathed in Ca2+-free solution, noradrenaline produced a large but transient Ca2+ signal, sensitive to thapsigargin but also depressed by nimodipine and by Y-27632. Inhibition of the peak Ca2+ response in Ca2+-free solution by Y-27632 might suggest that Rho-kinase is involved in the mechanisms underlying the release of intracellular Ca2+. This conclusion was not substantiated by the observation that the release of 45Ca2+ from non-mitochondrial stores evoked by InsP3 in β-escin-permeabilised A7r5 aortic cells was not affected by Y-27632. In addition, in freshly isolated cells from rat aorta, Y-27632 (10 μM) did not affect the intracellular Ca2+ release evoked by 50 μM InsP3 monitored by the activation of Ca2+-dependent K+ current (authors' unpublished data). Nevertheless, the limitations of these two observations, the former performed with cultured cells which may not be representative of aorta or mesenteric artery, and the latter using maximal concentrations of InsP3, do not allow us to disregard the possibility that Y-27632 produced a change in InsP3 receptors. The capacity for Ca2+ release of the SR did not appear to be regulated by Rho-kinase since Ca2+ signals evoked by thapsigargin or by caffeine were not impaired by Y-27632. More probably, both Rho-kinase-activated non-selective cationic channels and VOCs contribute to Ca2+ store refilling, as reported for VOCs in several excitable cells (McCarron et al. 2000; Lee et al. 2001).
The non-selective cationic channel regulated by Rho-kinase could be similar to the cationic conductance activated by noradrenaline in rabbit portal vein smooth muscle cells (Byrne & Large, 1988). This conductance is highly permeable to divalent cations (Byrne & Large, 1988; Wang & Large, 1991). Its proposed physiological role is to produce membrane depolarisation with subsequent opening of VOCs and also to allow direct influx of Ca2+ ions. The observation that Y-27632 inhibited noradrenaline-evoked depolarisation in rat mesenteric artery smooth muscle cells is in agreement with a role for this conductance in depolarisation and its regulation by Rho-kinase. It is indeed improbable that the inhibition by Y-27632 of the depolarisation evoked by noradrenaline resulted from an interaction of Rho-kinase with K+ channels, because blockade of most K+ channels with a cocktail of blockers, or a marked increase in the reversal potential of K+ did not prevent the effects of the Rho-kinase inhibitor. The Rho-kinase activated cationic channel does not appear to control resting membrane potential, which was not affected by Y-27632.
Although inhibition of the Rho A-Rho-kinase pathway does not appear to affect the Ca2+ signal in porcine coronary artery (Nobe & Paul, 2001) or in guinea-pig smooth muscle (Lucius et al. 1998), there are several reports suggesting that Rho-kinase could be involved in Ca2+ signalling in rat aorta (Takizawa et al. 1993), and, more recently, in tracheal smooth muscle (Ito et al. 2002) and in cultured human aortic endothelial cells (Yokoyama et al. 2002). There is now compelling evidence for the involvement of the Trp family of channel proteins in the Ca2+ permeable cation channels activated by G-coupled receptors. The mammalian homologue Trp6 has been shown to function as a Ca2+ entry channel independently of store-operated channels (Boulay et al. 1997) and to be an important element of the native current activated by α1-adrenoceptor stimulation in rabbit portal vein (Inoue et al. 2001). In rabbit vena cava, a putative non-selective cationic channel activated by InsP3 channel-mediated Ca2+ release and involved in store refilling may be encoded at least in part by the Trp1 gene (Lee et al. 2002). Further experiments should investigate whether these channel proteins could be a component of the Rho-kinase-regulated channel identified in aorta and mesenteric artery smooth muscle. Differences in the sensitivity of Ca2+ entry to Rho-kinase inhibition between tissues could arise from the expression of different channel proteins.
In conclusion, the present results showed that, in rat aorta and mesenteric artery smooth muscle cells, in addition to its role in Ca2+ sensitisation, Rho-kinase is involved in noradrenaline-evoked activation of Ca2+ entry, distinct from voltage-operated Ca2+ channels or thapsigargin-activated store-operated channels. This Ca2+ entry contributes to depolarisation and to the increase in intracellular [Ca2+].
Acknowledgments
This work was supported by a grant from the Ministère de l'Education et de la Recherche Scientifique (Action Concertée no. 00/05–260) and from the FRSM (grant no. 3.4534.98). The authors thank Mitsubishi Pharma for the gift of the Rho-kinase inhibitor Y-27632, M. C. Hamaide for the assistance in cell culture, and Professor P. Gailly for helpful discussion.
REFERENCES
- Berridge MJ. Rapid accumulation of inositol trisphosphate reveals that agonists hydrolyse polyphosphoinositides instead of phosphatidylinositol. Biochem J. 1983;212:849–858. doi: 10.1042/bj2120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonen HC, De Mey JG. G-proteins are involved in contractile responses of isolated mesenteric resistance arteries to agonists. Naunyn Schmiedebergs Arch Pharmacol. 1990;342:462–468. doi: 10.1007/BF00169465. [DOI] [PubMed] [Google Scholar]
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem. 1997;272:29672–29680. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- Broad LM, Cannon TR, Taylor CW. A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanism in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. J Physiol. 1999;517:121–134. doi: 10.1111/j.1469-7793.1999.0121z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne NG, Large WA. Membrane ionic mechanisms activated by noradrenaline in cells isolated from the rabbit portal vein. J Physiol. 1988;404:557–573. doi: 10.1113/jphysiol.1988.sp017306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong LD, Traynor-Kaplan A, Bokoch GM, Schwartz MA. The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell. 1994;79:507–513. doi: 10.1016/0092-8674(94)90259-3. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exton JH. Phosphoinositide phospholipases and G proteins in hormone action. Ann Rev Physiol. 1994;56:349–369. doi: 10.1146/annurev.ph.56.030194.002025. [DOI] [PubMed] [Google Scholar]
- Fujihara H, Walker LA, Gong MC, Lemichez E, Boquet P, Somlyo AV, Somlyo AP. Inhibition of RhoA translocation and calcium sensitization by in vivo ADP-ribosylation with the chimeric toxin DC3B. Mol Biol Cell. 1997;8:2437–2447. doi: 10.1091/mbc.8.12.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Amano M, Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci. 2001;22:32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- Ghisdal P, Gomez JP, Morel N. Action of a NO donor on the excitation-contraction pathway activated by noradrenaline in rat superior mesenteric artery. J Physiol. 2000;522:83–96. doi: 10.1111/j.1469-7793.2000.t01-3-00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfraind T, Dieu D. The inhibition by flunarizine of the norepinephrine-evoked contraction and calcium influx in rat aorta and mesenteric arteries. J Pharmacol Exp Ther. 1981;217:510–515. [PubMed] [Google Scholar]
- Gong MC, Fujihara H, Somlyo AV, Somlyo AP. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J Biol Chem. 1997;272:10704–10709. doi: 10.1074/jbc.272.16.10704. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kikuchi A, Sasaki T, Kuroda S, Kaibuchi K, Matsuura Y, Seki H, Saida K, Takai Y. Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. J Biol Chem. 1992;267:8719–8722. [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Ito S, Kume H, Yamaki K, Katoh H, Honjo H, Kodama I, Hayashi H. Regulation of capacitative and noncapacitative receptor-operated Ca2+ entry by rho-kinase in tracheal smooth muscle. Am J Respir Cell Mol Biol. 2002;26:491–498. doi: 10.1165/ajrcmb.26.4.4701. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ J Biol Chem. 1989;264:5339–5342. [PubMed] [Google Scholar]
- Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1997;272:12257–12260. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- Lee CH, Poburko D, Sahota P, Sandhu J, Ruehlmann DO, Van Breemen C. The mechanism of phenylephrine-mediated [Ca2+]i oscillations underlying tonic contraction in the rabbit inferior vena cava. J Physiol. 2001;534:641–650. doi: 10.1111/j.1469-7793.2001.t01-1-00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-H, Rahimian R, Szado T, Sandhu J, Poburko D, Behra T, Chan L, Van Breemen C. Sequential opening of IP3-sensitive Ca2+ channels and SOC during alpha -adrenergic activation of rabbit vena cava. Am J Physiol Heart Circ Physiol. 2002;282:H1768–1777. doi: 10.1152/ajpheart.00637.2001. [DOI] [PubMed] [Google Scholar]
- Lucius C, Arner A, Steusloff A, Troschka M, Hofmann F, Aktories K, Pfitzer G. Clostridium difficile toxin B inhibits carbachol-induced force and myosin light chain phosphorylation in guinea-pig smooth muscle: role of Rho proteins. J Physiol. 1998;506:83–93. doi: 10.1111/j.1469-7793.1998.083bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron JG, Flynn ER, Bradley KN, Muir TC. Two Ca2+ entry pathways mediate InsP3-sensitive store refilling in guinea-pig colonic smooth muscle. J Physiol. 2000;525:113–124. doi: 10.1111/j.1469-7793.2000.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadzean I, Gibson A. The developing relationship between receptor-operated and store-operated calcium channels in smooth muscle. Br J Pharmacol. 2002;135:1–13. doi: 10.1038/sj.bjp.0704468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaen L, De Smedt H, Droogmans G, Casteels R. Ca2+ release induced by inositol 1,4,5-trisphosphate is a steady-state phenomenon controlled by luminal Ca2+ in permeabilized cells. Nature. 1992;357:599–602. doi: 10.1038/357599a0. [DOI] [PubMed] [Google Scholar]
- Mita M, Yanagihara H, Hishinuma S, Saito M, Walsh MP. Membrane depolarization-induced contraction of rat caudal arterial smooth muscle involves Rho-associated kinase. Biochem J. 2002;364:431–440. doi: 10.1042/BJ20020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Yano T, Schmidt DJ, Tokui T, Shibata M, Lifshitz LM, Kimura S, Tuft RA, Ikebe M. Rho-dependent agonist-induced spatio-temporal change in myosin phosphorylation in smooth muscle cells. J Biol Chem. 2002;277:725–734. doi: 10.1074/jbc.M108568200. [DOI] [PubMed] [Google Scholar]
- Morel N, Godfraind T. Characterization in rat aorta of the binding sites responsible for blockade of noradrenaline-evoked calcium entry by nisoldipine. Br J Pharmacol. 1991;102:467–477. doi: 10.1111/j.1476-5381.1991.tb12196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Nilsson H, Flatman JA. Role of membrane potential in the response of rat small mesenteric arteries to exogenous noradrenaline stimulation. J Physiol. 1982;332:363–373. doi: 10.1113/jphysiol.1982.sp014418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Nobe K, Paul RJ. Distinct pathways of Ca2+ sensitization in porcine coronary artery: effects of Rho-related kinase and protein kinase C inhibition on force and intracellular Ca2+ Circ Res. 2001;88:1283–1290. doi: 10.1161/hh1201.092035. [DOI] [PubMed] [Google Scholar]
- Oude Weernick PA, Schulte P, Guo Y, Wetzel J, Amano M, Kaibuchi K, Haverland S, Voss M, Schmidt M, Mayr GW, Jakobs KH. Stimulation of phosphatidylinositol-4-phosphate 5-kinase by Rho-kinase. J Biol Chem. 2000;275:10168–10174. doi: 10.1074/jbc.275.14.10168. [DOI] [PubMed] [Google Scholar]
- Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P, Loirand G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2000;275:21722–21729. doi: 10.1074/jbc.M000753200. [DOI] [PubMed] [Google Scholar]
- Sward K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol. 2000;522:33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa S, Hori M, Ozaki H, Karaki H. Effects of isoquinoline derivatives, HA1077 and H-7, on cytosolic Ca2+ level and contraction in vascular smooth muscle. Eur J Pharmacol. 1993;250:431–437. doi: 10.1016/0014-2999(93)90030-l. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Wang Q, Large WA. Noradrenaline-evoked cation conductance recorded with the nystatin whole-cell method in rabbit portal vein cells. J Physiol. 1991;435:21–39. doi: 10.1113/jphysiol.1991.sp018496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Somlyo AV, Somlyo AP. Cyclic GMP-dependent stimulation reverses G-protein-coupled inhibition of smooth muscle myosin light chain phosphate. Biochem Biophys Res Commun. 1996;220:658–663. doi: 10.1006/bbrc.1996.0460. [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Ishibashi T, Ohkawara H, Kimura J, Matsuoka I, Sakamoto T, Nagata K, Sugimoto K, Sakurada S, Maruyama Y. HMG-CoA reductase inhibitors suppress intracellular calcium mobilization and membrane current induced by lysophosphatidylcholine in endothelial cells. Circulation. 2002;105:962–967. doi: 10.1161/hc0802.104457. [DOI] [PubMed] [Google Scholar]
- Zhang C, Schmidt M, Von Eichei-Streiber C, Jakobs KH. Inhibition by toxin B of inositol phosphate formation induced by G protein-coupled and tyrosine kinase receptors in N1E-115 neuroblastoma cells: involvement of Rho proteins. Mol Pharmacol. 1996;50:864–869. [PubMed] [Google Scholar]