

Abstract
Active vasodilatation (AVD) in human, non-glabrous skin depends on functional cholinergic fibres but not on acetylcholine (ACh). We tested whether AVD is a redundant system in which ACh and vasoactive intestinal polypeptide (VIP) are co-released from cholinergic nerves. (1) We administered VIP by intradermal microdialysis to four discrete areas of skin in the presence of different levels of the VIP receptor antagonist, VIP(10−28), also delivered by microdialysis. Skin blood flow (SkBF) was continuously monitored by laser Doppler flowmetry (LDF). Mean arterial pressure (MAP) was measured non-invasively and cutaneous vascular conductance (CVC) calculated as LDF/MAP. Subjects were supine and wore water-perfused suits to control whole-body skin temperature (Tsk) at 34 °C. Concentrations of 54 μm, 107 μm, or 214 μm VIP(10−28) were perfused via intradermal microdialysis at 2 μl min−1 for approximately 1 h. Then 7.5 μm VIP was added to the perfusate containing VIP(10−28) at the three concentrations or Ringer solution and perfusion was continued for 45-60 min. At the control site, this level of VIP caused approximately the vasodilatation typical of heat stress. All VIP(10−28)-treated sites displayed an attenuated dilatation in response to the VIP. The greatest attenuation was observed at the site that received 214 μm VIP(10−28) (P < 0.01). (2) We used 214 μm VIP(10−28) alone and with the iontophoretically administered muscarinic receptor antagonist atropine (400 μA cm−2, 45 s, 10 mM) in heated subjects to test the roles of VIP and ACh in AVD. Ringer solution and 214 μm VIP(10−28) were each perfused at two sites, one of which in each case was pretreated with atropine. After 1 h of VIP(10−28) treatment, individuals underwent 45−60 min of whole-body heating (Tsk to 38.5 °C). VIP(10−28), alone or in combination with atropine, attenuated the increase in CVC during heat stress, suggesting an important role for VIP in AVD.
Regulation of internal temperature within a very narrow range is essential to maintenance of normal physiological function. Most heat flow between the environment and body occurs via the skin. Thus, a primary function of the skin circulation is to control internal body temperature. Vasoconstriction of the cutaneous vasculature allows conservation of body heat. Vasodilatation and sweating provide the means for dissipation of heat from the body core to the external environment.
Neural (reflex) control of the circulation in non-glabrous skin in humans is mediated though two branches of the sympathetic nervous system (Johnson & Proppe, 1996). The noradrenergic vasoconstrictor system is tonically active and controls the response to cold exposure as well as the subtle modulations in vascular tone under normothermic conditions. Reflex cutaneous vasodilatation in response to body heating can be broken into two parts. Initially, a rapid vasodilatation resulting in about a doubling of skin blood flow (SkBF) is observed due to removal of active vasoconstrictor tone (Roddie et al. 1957b; Johnson & Proppe, 1996). With further heating, and generally beginning coincident with the onset of sweating, there is a rapid and profound active vasodilatation (AVD) (Roddie et al. 1957). This vasodilatation is due to the release of vasodilator transmitters from sympathetic cholinergic nerves (Grant & Holling, 1938; Kellogg et al. 1995). AVD can be responsible for 90 % of the skin vasodilatation observed during increases in internal temperature (Johnson et al. 1986). During normothermic conditions, while a person is at rest, total SkBF is approximately 200-500 ml min−1. However, with full activation of AVD, SkBF can approach 8 l min−1 or about 60 % of the cardiac output (Johnson & Proppe, 1996).
Although cutaneous AVD was first identified seven decades ago (Lewis & Pickering, 1931), the neural mechanism(s) remains unclear. The typical temporal coincidence of sweating and AVD led to the proposal that sudomotor function is responsible for both events (Fox & Hilton, 1958) but such a sudomotor-vasomotor link remains unproven (Johnson & Proppe, 1996). Postsynaptic muscarinic receptor blockade with atropine blocks the sweating response, but only slightly inhibits AVD (Roddie et al. 1957a; Kellogg et al. 1995). At first glance, the lack of blockade of AVD by atropine might be considered as convincing evidence of a noncholinergic origin of AVD. However, Kellogg et al. (1995) found strong evidence that AVD is mediated through cholinergic nerves, but not entirely by acetylcholine (ACh). This was shown through the use of intradermal botulinum toxin, which acts presynaptically to abolish the release of all transmitters selectively from cholinergic nerves (Valtorta & Arslan, 1993). Treatment with botulinum toxin abolished both the sweating and the AVD responses to heat stress (Kellogg et al. 1995). These findings led to the conclusion that AVD is dependent on functional cholinergic nerves, but is not reliant on ACh, and hence a result of cholinergic nerve co-transmission.
In considering co-transmission, one logical choice for a co-transmitter, as proposed by Hökfelt, et al. (1980), is the basic 28-amino acid peptide, vasoactive intestinal peptide (VIP) (Mutt et al. 1974). VIP is a known vasodilator and is often co-localized with ACh in cholinergic nerve terminals (Lundberg, 1981; Henning & Sawmiller, 2001). Additionally, VIP is believed to be the transmitter responsible for atropine-resistant vasodilatation in exocrine glandular tissue (Lundberg, 1981) and is found in neurons associated with the vasculature of human skin and sweat glands (Hartschuh et al. 1984; Vaalasti et al. 1985).
Savage, et al. (1990) caused heat stress in patients with cystic fibrosis (CF), a population with very low levels of VIP in postganglionic neurons (Heinz-Erian et al. 1985) and found that those patients exhibited normal AVD in response to hyperthermia. They therefore concluded that VIP was probably not the vasodilator responsible for AVD (Savage et al. 1990). As an alternative, it is our proposal that a redundant system, in which ACh and VIP are co-released from cholinergic nerves, is responsible for the vasodilatation in response to hyperthermia. In the absence of one neurotransmitter, the other is sufficient to elicit a response not obviously different from that of the intact system. Such a mechanism would explain not only the atropine-resistant vasodilatation in healthy subjects, but also the apparently normal AVD in CF patients.
METHODS
The Institutional Review Board (IRB) of the University of Texas Health Science Center at San Antonio approved all experiments. All procedures conformed to the Declaration of Helsinki. Voluntary, and written informed consent was given by all subjects prior to participation. All subjects were nonsmokers and took no medications (24 h) or caffeine (12 h) prior to the start of the experiment.
To test our proposal several steps had to be completed. We needed to deliver VIP into the skin in sufficient quantity to elicit a vasodilatation similar to that seen during whole-body heating (Savage et al. 1990; Kellogg, et al. 1993; Shastry, et al. 2000). In a series of preliminary studies, we identified 7.5 μm VIP, delivered via intracutaneous microdialysis, as causing that rate and extent of vasodilatation. Next, it was necessary to select an appropriate concentration of a VIP antagonist to obtain a maximum blockade of the VIP-induced vasodilatation but with minimal confounding effects. That was the aim of protocol 1 (see below). The antagonist we selected was a fragment of the endogenous peptide, VIP(10−28). This fragment contains the C-terminus necessary for binding to the VIP receptor (Chakder & Rattan, 1993) yet lacks the N-terminus responsible for stimulation adenylate cyclase activity (Turner et al. 1986). Finally, in protocol 2, we tested for roles of ACh and VIP in the reflex response to body heating by antagonizing the effects of these neurotransmitters, individually and together, using atropine and VIP(10−28).
Intra-dermal microdialysis was the technique we chose for delivery of both VIP and its antagonist because it allows local and continuous delivery of pharmacological agents to the cutaneous interstitium at high concentrations without confounding systemic effects. All pharmacological agents were sterilized using a 13 mm Acrodisc syringe filter with 2 μm membrane (Pall Gelman Laboratory, Ann Arbor, MI, USA) prior to administration to the subjects. Each microdialysis probe was constructed of a microdialysis membrane with an 18 kDa cut-off (Spectrum, Laguna Hills CA, USA), 1 cm in length and 200 μm in diameter, and polyimide tubing with a 0.0049 inch internal diameter. The probe was reinforced with 0.0015 inch diameter coated stainless steel wire (Kellogg et al. 1999). While the microdialysis membrane has a nominal 18 kDa molecular mass cut-off, in practice, passage of molecules larger than 5 kDa is significantly limited (Huang et al. 1996). Pilot studies confirmed that this membrane was adequate for passage of VIP and VIP(10−28), with molecular masses of 3.3 kDa and 2.3 kDa, respectively.
Subjects reported to the lab for microdialysis probe insertion approximately 2.5 h before the start of data collection. Prior to microdialysis probe insertion, cold packs were applied to the ventral surface of the forearm as a temporary anaesthetic. The probes were then fed through the lumen of a 25-gauge needle. Needles were inserted intra-dermally into the arm such that entry and exit points were approximately 2.5 cm apart. Probes were aligned such that the microdialysis membranes were centred within the dermis. The needles were then removed leaving the probe in place. Approximately 2 h after probe placement, subjects were prepared for the experiments. This allowed ample time for the trauma to the skin to subside before the start of data collection (Anderson et al. 1994).
After microdialysis probe insertion, the subjects dressed in a tube-lined suit and rested in the supine position. The suit covered the entire body with the exception of the head, hands, feet, and areas of arm instrumented with microdialysis/blood flow probes. Water, of varying temperatures, was perfused through the tubes to control whole-body skin temperature (Tsk). Tsk was assessed by the weighted average of six thermocouples placed at predetermined locations on the abdomen, lower back, upper chest, thigh, calf, and upper back (Taylor et al. 1989).
SkBF was measured by laser Doppler flowmetry (LDF) (Saumet et al. 1988; Johnson, 1990; Öberg, 1990). The LDF probe was placed directly over the microdialysis probe, ensuring the centre of the probe was aligned with the centre of the microdialysis membrane. Mean arterial pressure (MAP) and heart rate (HR) were measured continuously and non-invasively by photoplethysmography at the finger (Finapres, Ohmeda, Engelwood, CO, USA) (Friedman et al. 1990). Cutaneous vascular conductance (CVC) was calculated as a ratio of the LDF measurement to MAP.
At the end of each protocol, 25 mM of sodium nitroprusside (SNP), a nitric oxide (NO) donor, was given via the microdialysis probes to all treatment sites to elicit a maximal vasodilatation of the skin vessels (Kellogg et al. 1999). This allowed CVC to be expressed as a percentage of the maximal CVC for each site (Kellogg et al. 1993).
Protocol 1: identification of an appropriate concentration for the antagonist, VIP(10−28), to obtain an optimal blockade of a 7.5 μm VIP-induced vasodilatation
Eight subjects (six males and two females, between the ages of 19 and 35 years) participated in this protocol. Each subject was instrumented with four microdialysis probes as described above. Water was perfused through the tubes of the suit to maintain Tsk at 34 °C throughout this protocol. At this temperature subjects are typically neither warm nor cold and tonic cutaneous vasoconstrictor activity is minimal (Johnson & Proppe, 1996). Sterile Ringer solution was perfused (2 μl min−1) at all four sites for a baseline period of 5 −15 min or until steady-state blood flow was observed. Subsequently, sites received either 54, 107 or 214 μm VIP(10−28) or Ringer solution at the control site (Fig. 1). This phase of the experiment lasted approximately 1 h, until a steady-state blood flow was observed. Then, a combination of antagonist (VIP(10−28)) at unchanged concentration and agonist (7.5 μm VIP, identified in preliminary studies) was administered at the three sites previously infused with antagonist alone while the control site received 7.5 μm VIP alone. This portion of the experiment lasted about 50 min. Finally, 25 mM SNP, a NO donor, was given at all sites to elicit a maximum vasodilatation (Kellogg et al. 1999).
Figure 1. Protocol 1: blockade of 7.5 μm VIP-induced vasodilation.

Sterile Ringer solution was perfused (2 μl min−1) via intradermal microdialysis at all four sites for a baseline period of 5-15 min. Sites then received either VIP(10−28) at the concentrations indicated in the figure, or Ringer solution for approximately 1 h. Then, a combination of VIP(10−28) at the same concentration and 7.5 μm VIP was administered at the three sites previously infused with antagonist alone while the control site received VIP alone. This portion of the experiment lasted about 50 min. Finally, 25 mM sodium nitroprusside (SNP) was given at all sites to elicit a maximal dilatation of the vessels. Tsk was maintained at 34 °C throughout the protocol.
Data analysis
An average CVC was calculated for each 2 min interval and expressed as a percentage of the maximum CVC. The VIP-induced increases in CVC between the steady state achieved after the initial antagonist infusion and the final point of measurement were compared using a one-way ANOVA. A Dunnett's post hoc test was used to compare responses from each treatment site with that from the control sites. Statistical significance was accepted if P < 0.05.
Protocol 2: responses to heat stress in the presence of VIP receptor and cholinergic muscarinic receptor blockade
Seven healthy subjects (four males, three females) participated in this protocol. Four of this group participated in protocol 1. Each subject was instrumented with four intra-dermal microdialysis probes, as described above. Iontophoresis of atropine sulfate (10 mM in propylene glycol), centred over two of the microdialysis sites (sites 2 and 4), produced blockade of muscarinic receptors in 0.6 cm2 areas of skin. The atropine was delivered via 400 μA cm−2 for 45 s, a level shown previously to block completely the vasodilator responses to exogenous ACh (Kellogg et al. 1995). This was done approximately 1 h before the start of data collection.
SkBF measurements were made directly over the centres of the microdialysis membranes. Tsk, blood pressure, and heart rate were measured as in protocol 1. Internal temperature (Tor) was measured by a thermocouple placed in the sublingual sulcus.
This protocol is depicted in Fig. 2. Tsk was maintained at 34 °C in the initial two phases of the protocol. The first phase was a baseline period of 5-15 min, during which sterile Ringer solution was perfused through all four microdialysis probes. In the second phase, 214 μm VIP(10−28) (determined from protocol 1) was perfused through two of the microdialysis membranes (sites 3 and 4). One of those sites had been iontophoretically pretreated with atropine. VIP(10−28) treatment began and continued throughout the periods of normothermia and hyperthermia. Administration of Ringer solution continued at the other two sites, one of which had been pretreated with atropine. Approximately 1 h into the VIP(10−28) treatment, individuals were subjected to whole-body heating by raising the temperature of the water perfusing the suit to elevate Tsk to 38.5 °C. This heat stress lasted 45-60 min. At the end of heating, Tsk was returned to normothermic levels. SNP was then perfused through all four microdialysis membranes to elicit maximal vasodilatation (Kellogg et al. 1999).
Figure 2. Protocol 2: instrumentation of subjects with four microdialysis probes.
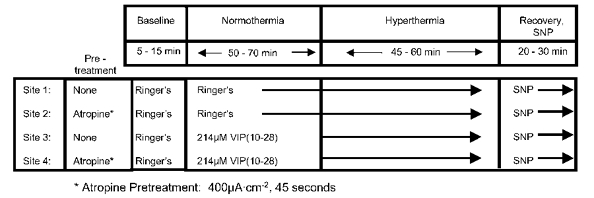
Sites 2 and 4 were pretreated with atropine via iontophoresis. Initially, Tsk was maintained at 34 °C. After a brief baseline period the VIP antagonist, VIP(10−28), was perfused through two of the microdialysis probes (sites 3 and 4), throughout the periods of normothermia and hyperthermia. During VIP(10−28) treatment, individuals were subjected to whole-body heating for 45-60 min, Tsk = 38.5 °C. Tsk was then returned to normothermic levels and sodium nitroprusside (SNP) was perfused through all four microdialysis membranes to elicit maximal vasodilation. Temperature at the sites of blood flow measurement was maintained at 34 °C throughout.
Data analysis
The CVC for each site, expressed as a percentage of the maximal CVC, was plotted as a function of Tor. The onset of AVD for each subject was identified as the point of dramatic and sustained increase in the slope of this relationship at the control site. This determination was performed by two investigators unware of the origin of the data. In the event of conflict, a third investigator was consulted. Subsequently, the change in CVC, as a percentage of maximum, from the onset of AVD, was calculated for each 2 min period over the next 30 min. These calculations were performed for each site and each subject. The average changes in CVC across all subjects were then expressed as a function of time. A regression analysis of the change in percentage maximum CVC versus time was then performed to compare responses to the treatments. Because the response reached a plateau late in heating (see Results), only the rising portion, as determined from the control site, was used to calculate the rate of change of CVC at each treatment site.
A repeated measures ANOVA with two within factors, VIP blockade and ACh blockade, was performed on the slopes of the normalized CVC vs. time relationships. A Tukey-Kramer post hoc test was used to make pairwise comparisons. A repeated measures ANOVA on the data from the last 2 min of heat stress was performed to compare the net responses to heat stress among treatments. Statistical significance was accepted at P < 0.05.
Thresholds for the onset of active vasodilatation under each experimental treatment were then analysed for each subject. Again, two investigators unaware of the origin of the data made this determination. Since previous evidence (Roddie et al. 1957a; Kellogg et al. 1995) indicated that atropine pretreatment caused a delay in the onset of active vasodilatation (as indicated by the thresholds), a one-tailed t test was used to compare the onset between the control and atropine sites. Statistical significance was accepted if P < 0.05.
RESULTS
In preliminary studies, we found that the responses in CVC to 7.5 μm VIP were similar to those observed during heat stress in both time course and magnitude. Lower concentrations of VIP, while resulting in an increase in CVC, were slow to initiate vasodilatation, produced a slow rate of rise in CVC, and did not cause the magnitude of vasodilatation desired in a reasonable time period. VIP at higher concentrations produced large vasodilator responses which we considered to be too great to be considered as mimicking thermoregulatory vasodilatation and which, further, would be very difficult to antagonize.
Protocol 1
Based upon these preliminary studies, we chose to seek a concentration of VIP(10−28) that was high enough for significant inhibition of a VIP-induced vasodilatation but still low enough to minimize any vasodilator effects of the antagonist. We found early that concentrations over 300 μm caused a prompt, maximal vasodilatation, rendering those sites unsuitable for testing antagonist efficacy. We therefore focused on three concentrations of VIP(10−28): 54 μm, 107 μm, and 214 μm.
Figure 3 shows the average of the responses of the eight subjects during the last 38 min of treatment with the combination of VIP (7.5 μm) and VIP(10−28) at those concentrations at three sites and VIP (7.5 μm) alone at the control sites. Treatment with VIP(10−28) attenuated a 7.5 μm VIP-induced vasodilatation (P < 0.0071). Responses at the sites treated with 107 μm and 214 μm VIP(10−28) differed significantly from those at the control sites (P < 0.05, and P < 0.01, respectively). The vasodilatation caused by 7.5 μm VIP was attenuated by 54 % in the presence of 214 μm VIP(10−28). Concentrations of 54 μm and 107 μm VIP(10−28) attenuated the VIP-induced vasodilatation by 38 % and 40 %, respectively. These results, indicated that 214 μm VIP(10−28) would provide a significant blockade of a VIP-induced vasodilatation without an unacceptable effect on baseline SkBF.
Figure 3. Results from eight subjects for protocol 1.
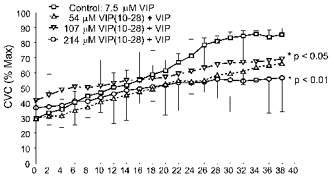
Data are from the final 38 min of combination VIP + VIP(10−28) at three sites and VIP alone at the control sites. Treatment with VIP(10−28) attenuated the 7.5 μm VIP-induced vasodilation (P < 0.007). Dunnett's post hoc test showed responses at the control sites differed significantly from those at sites treated with 107 μm and 214 μm VIP(10−28) (P < 0.05 and P < 0.01, respectively). The greatest attenuation was detected at the sites treated with 214 μm VIP(10−28). Alternating pairs (i.e. control and 214 μm VIP(10−28) vs. 54 μm and 107 μm VIP(10−28)) of s.e.m. error bars are depicted for visual clarity.
Protocol 2
Figure 4 shows data from protocol 2 from a representative subject. Note that skin temperature was elevated over a short period of time, with internal temperature slowly rising thereafter. In all subjects, internal temperature reached a threshold, after which skin blood flow rose at a greater rate. The data beyond this threshold, as shown in Fig. 5, were considered to be reflective of active vasodilatation. Overall, HR rose modestly (P < 0.05) and MAP was not significantly different from the end of the baseline period to the end of heat stress (P > 0.05). Complete data sets for the several antagonist combinations were obtained from six subjects. In one other subject, data from the atropine site were not useable due to probe malfunction.
Figure 4. Typical responses to body heating in protocol 2.
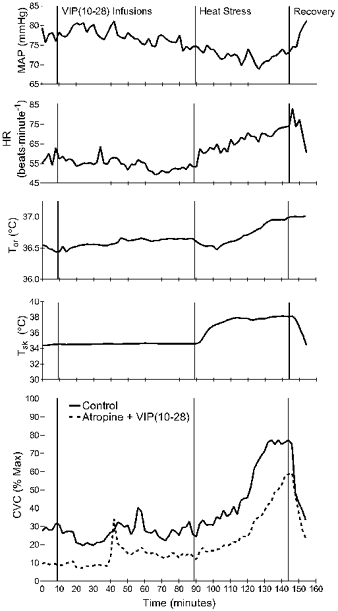
Data are from a single subject. Shown are blood pressure (MAP), heart rate (HR), skin temperature (Tsk), oral temperature (Tor) and cutaneous vascular conductance (CVC) from a control site (continuous line) and from a site treated with atropine and the VIP receptor antagonist VIP(10−28) (dashed line). Atropine pretreatment took place 1 h earlier. Intradermal infusion of VIP(10−28) by microdialysis began after 10 min of baseline data collection. After 1 h, body heating commenced by raising the skin temperature to 38.5 °C. This was not accompanied by local warming of the areas at which blood flow was measured.
Figure 5. Results from protocol 2.
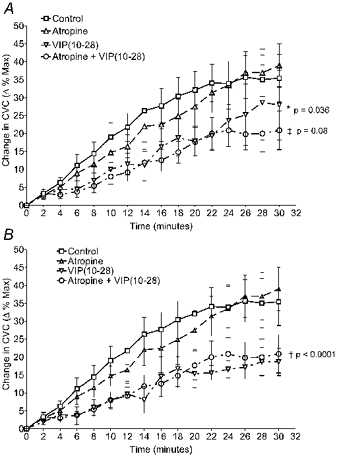
Sites were treated with atropine, VIP(10−28), both antagonists, or Ringer solution only. These are the vasodilator responses, expressed as changes relative to maximal (Δ% Max), from the onset of AVD at the control site to the end of heating. A, repeated measures ANOVA on the magnitude of the change in CVC during the last 2 min of heat stress did not reveal a statistically significant difference among treatment sites (‡P = 0.08). However, one subject displayed a dramatic, unexplained vasodilatation during the last 7 min of heat stress at the VIP(10−28)-treatment site only. B, the average response with these data excluded. VIP(10−28) reduced the vasodilator response significantly (†P < 0.0001). As with the sensitivity, atropine did not significantly affect the magnitude of the change in CVC.
The thresholds for vasodilatation were not significantly different between control and atropine-treated sites (P = 0.13). Originally, we had planned to conduct pairwise comparisons of the onset of AVD at all treatment sites. However, the reduced slopes of the responses at sites treated with VIP(10−28) made it impossible for investigators to determine the thresholds at those sites.
Linear regression analysis was applied to the CVC responses for each site and subject. Those regressions were performed from the onset of vasodilatation at the control sites until the responses began to level off. Slopes of the linear regression of the response after the onset of AVD at the four treatment sites were 1.76 ± 0.28, 1.44 ± 0.28, 0.82 ± 0.21, and 0.84 ± 0.16 % maximum CVC min−1 at the control, atropine-treated, VIP(10−28)-treated, and combination VIP(10−28) + atropine-treated sites, respectively. The sensitivity of the vasodilatation was significantly reduced by VIP(10−28) treatment (P = 0.036). An effect of atropine was not detected (P = 0.21). We also did not find a significant interaction between the two main effects of VIP blockade and ACh blockade (P = 0.097). A Tukey post hoc test on a repeated measures ANOVA showed the slopes at the control sites were significantly higher than at either the VIP(10−28)-treated sites or the combination VIP(10−28) + atropine-treated sites (P < 0.05).
Analysis of the individual linear regression data from the whole subject pool is presented in Fig. 6. An attenuation in slope relative to that at the control sites was evident at all sites treated with the VIP antagonist, whether atropine was present or not, across all subjects (Fig. 6B and C). Statistical analysis did not reveal a difference in slope between VIP(10−28)-sites and the combination VIP(10−28) + atropine-treated sites (Fig. 6E). This figure also shows the similarity of the responses at these two treatment sites across the subject pool. Figure 6A shows that the slopes at atropine-treated sites were reduced in all subjects compared to those at the control sites. Although statistical significance was approached, it was not achieved (P = 0.097).
Figure 6. Results from linear regression analyses of the CVC: time relationships for protocol 2.
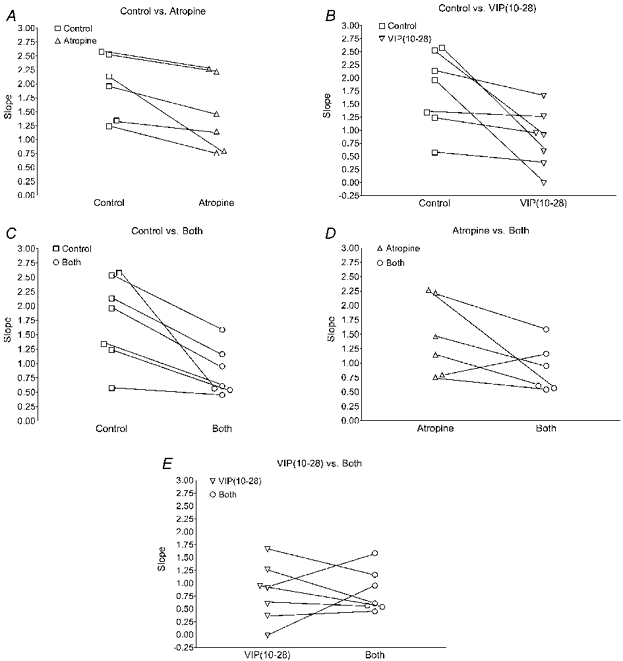
Individual slopes are shown for all subjects for pairwise comparisons of the responses between treatment sites. Antagonist-treated sites showed a reduced slope compared to control sites (A-C). VIP(10−28) (B) and combination atropine and VIP(10−28) (C) caused a statistically significant reduction in slope compared to control (P < 0.036). Although statistical analysis of the group data did not show an effect of atropine treatment, the slopes of the responses for each individual were reduced relative to control (A). B, atropine vs. combination atropine + VIP(10−28)-treated sites, shows a consistent reduction in slope with VIP(10−28) treatment. E shows similar responses at the VIP(10−28) and the combination atropine + VIP(10−28)-treated sites, suggesting no significant effect of atropine in the presence of VIP antagonism. Note that in one subject the atropine-treated site was not useful for technical reasons and data from that site are not included here or in Fig. 5.
As shown in Fig. 5, there was a distinct difference in the extent of the increase in CVC during heating, depending upon the treatment. From the onset of active vasodilatation to the end of heating, the increase in CVC at control sites averaged 35.0 ± 15.3 % of maximum CVC. The corresponding increases in CVC observed at the atropine-treated, VIP(10−28)-treated, and combination VIP(10−28) + atropine-treated sites averaged 38.9 ± 12.1 %, 27.9 ± 21.8 %, and 20.9 ± 11.9 % of maximum CVC, respectively. A repeated measures analysis of the magnitude of change in CVC during the last 2 min of heat stress revealed a trend that did not quite achieve statistical significance among treatment sites (P = 0.08, Fig. 5A). However, one subject displayed a dramatic, unexplained vasodilatation during the last 7 min of heat stress at the VIP(10−28)-treatment only site. With data from that site excluded, significant differences in the final level of CVC among sites were detected (P < 0.0001) (Fig. 5B). Post hoc analysis found control and atropine only-treated sites to have significantly higher final values than sites treated with VIP(10−28) (P < 0.001) or a combination of VIP(10−28) and atropine (P < 0.05).
DISCUSSION
A majority of the reflex cutaneous vasodilator response by non-glabrous skin to body heating is due to activation of an active vasodilator system (Roddie et al. 1957b; Johnson & Proppe, 1996). That this system is powerful is unquestionable; however, discovery of its mechanism of action has been elusive. It is dependent on NO synthesis for its full expression (Kellogg et al. 1998; Shastry et al. 1998). The results of the current study point to a role for VIP in AVD of the human skin. The antagonist, VIP(10−28), significantly attenuated the rate of increase in CVC normally achieved during heat stress. This was true whether VIP(10−28) was administered alone or in combination with atropine, a cholinergic muscarinic receptor antagonist. Figure 6B and C depicts the consistency of the VIP antagonism across the subject pool.
Although the inhibitory effects of the antagonist on AVD strongly suggest a role for VIP, a similar role for ACh was not as clear. Previous findings from atropine-treated skin suggested a role for ACh in the onset of AVD (Roddie et al. 1957a; Kellogg, et al. 1995). However, established AVD does not appear to be atropine sensitive (Roddie et al. 1957a; Kellogg et al. 1995; Shastry, et al. 2000). Although some evidence for an effect of atropine on AVD was noted in the current study, none of the data achieved statistical significance. The reasons for these discrepancies are not clear, but taken together, the suggestion is that any role for ACh is more subtle or complex than would be expected from our original hypothesis. For example, our failure to demonstrate a clear role for ACh in AVD in the human cutaneous vasculature may be due to the prolonged heat stress protocol. Lundberg et al. (1981a) demonstrated VIP release was frequency dependent, with the greatest release at the highest stimulation frequency. ACh output has been demonstrated to reach maximal levels at lower frequencies and shorter durations of stimulation (Lundberg et al. 1981a). As suggested by Shastry et al. (2000), it may be that ACh plays a role early in the heat stress, but as body temperature rises, the high levels of VIP release may mask the effects due to ACh.
Parallels have been drawn between the secretory and vasomotor functions of exocrine tissue, such as salivary glands, and human skin (Fox & Hilton, 1958). Upon nerve stimulation there is a copious secretion from both types of glandular tissue accompanied by dramatic dilatation of blood vessels. In studies of the cat submandibular salivary glands, Lundberg et al. (1981a) showed concomitant with this response a 30- to 50-fold increase in VIP concentration in the venous effluent. Additionally, similar to the findings that sweating in human skin was blocked by atropine but AVD was not (Roddie et al. 1957a; Kellogg et al. 1995), Lundberg (1981) showed that salivary secretion was atropine sensitive but dilatation of the surrounding blood vessels was atropine-resistant. Extrapolating such parallels to vasomotor and secretory regulation, Lundberg's extensive work indicates a complex relationship mediated by ACh and VIP that may be applicable to human skin as well. During high frequency nerve stimulation, atropine treatment increased the duration of the vasodilatation as well as the amount of VIP released. This may suggest a feedback mechanism whereby the presence of ACh inhibits the release of VIP via cholinergic autoreceptors. Additionally, local infusion of VIP and ACh into the cat submandibular artery (Lundberg et al. 1980, 1982a) showed that the vasomotor and secretory responses may not be simply additive. Infusion of ACh alone resulted in vasodilatation and salivary secretion. VIP infusion resulted in an atropine-resistant vasodilatation but no salivary secretion. Interestingly, infusion of ACh and VIP together resulted in at least an additive vasodilatation (Lundberg et al. 1982a) and, in some studies, potentiation (Lundberg et al. 1980). This potentiation was blocked by atropine treatment.
Our failure to completely block AVD during heat stress using VIP(10−28) alone or in combination with atropine may be due to our inability to obtain a complete blockade of the actions of VIP, which could be attributable to the significantly lower affinity of VIP(10−28) for the receptor when compared to the agonist. Approximately a 100-fold molar excess of VIP(10−28) is required to inhibit a given concentration of VIP (Turner et al. 1986; Hill et al. 1995). We were able to obtain approximately a 54 % blockade of a 7.5 μm VIP-induced vasodilatation using 214 μm VIP(10−28). Antagonist-induced vasodilatation precluded the use of higher concentrations of VIP(10−28). The initial vasodilatation with 214 μm VIP(10−28) was small, as indicated by the ordinate value on Fig. 3.
Another explanation for the lack of complete blockade of AVD is the possible involvement of another neurotransmitter. For example, calcitonin gene-related peptide (CGRP) has been localized to cholinergic sympathetic fibres innervating sweat glands of the rat (Landis & Fredieu, 1986). The same fibres also contained VIP, although not all VIP-containing fibres also contained CGRP. Additionally, Lindh, et al. (1988) found substance P localized to sympathetic nerves of the cat footpads that also contained VIP, ACh and CGRP.
Also, it should be noted that VIP(10−28), while inhibiting the effects of VIP, may also inhibit the effects of other neurotransmitters. VIP(10−28) antagonizes the effects of peptide histidine isoleucine/methionine (PHI/PHM) (Grider & Rivier, 1990). PHI in rodents and PHM in humans are structurally related to VIP and are post-translational proteolytic products of the same prepropeptide (Itoh et al. 1983). Lundberg et al. (1984) found PHI- and VIP-immunoreactivity in the same autonomic neurons across many tissues and species, including humans. While there have been some reports that PHM is a weak agonist for VIP receptors in some human tissues, e.g. colonic epithelial cells, we do not have any data indicating its binding affinity in VIP receptors in human skin (Laburthe et al. 1985), where it is known that PHM and VIP have similar distribution patterns. While PHM-immunoreactive fibres are believed to be found predominantly around the eccrine sweat glands in axillary skin, reports indicate their presence in and around the vasculature of the skin as well (Johansson, 1986; Eedy et al. 1990). Therefore, although PHI/PHM is a less potent vasodilator than VIP, it is not possible with the current antagonist to discount a role in AVD.
Further evidence for a role for VIP in relaxation of vascular smooth muscle is found in pathophysiology. Inadequate VIP innervation has been implied in erectile dysfunction and impaired oesphophageal relaxation (Aggestrup et al. 1983). Excessive VIP production is associated with certain pancreatic tumours (VIPomas) (Frase et al. 1987). This latter disease is characterized by profuse watery diarrhoea, hypovolaemia, acidosis, and hypokalaemia. Although numerous other clinical symptoms often present with this condition, some of those most relevant to our hypothesis are excessive flushing of the skin, hypersecretion from the lacrimal and sweat glands, and hypotension.
In summary, we found strong evidence for a role for VIP in AVD in human skin. In healthy subjects, treatment with a VIP antagonist resulted in the attenuation of cutaneous vasodilatation associated with heat stress. VIP(10−28) caused a statistically significant attenuation of the change in CVC whether given alone or in combination with atropine, a muscarinic receptor antagonist. This conclusion is dependent upon the assumption that VIP(10−28) is selective for VIP receptors and that VIP receptors are sensitive only to VIP in human skin. The failure to achieve a complete antagonism of AVD with the VIP antagonist and atropine might be due to the involvement of a transmitter other than VIP or ACh. However, limitations associated with the efficacy of VIP(10−28) relative to VIP preclude the possibility of distinguishing that conclusion from one of incomplete blockade.
Acknowledgments
We gratefully acknowledge Wojciech A. Kosiba and Joan Zhao for their technical expertise and assistance. We also thank all the subjects for their participation and cooperation. This work was supported by National Institutes of Health Grants HL65599 (Dean L. Kellogg Jr) and HL59166 (John M. Johnson).
REFERENCES
- Aggestrup S, Uddman R, Sundler F, Fahrenkrug J, Hakanson R, Sorensen HR, Hambraeus G. Lack of vasoactive intestinal polypeptide nerves in esophageal achalasia. Gastroenterol. 1983;84:924–927. [PubMed] [Google Scholar]
- Anderson C, Andersson T, Wardell K. Changes in skin circulation after insertion of a microdialysis probe visualized by laser Doppler perfusion imaging. J Investig Dermatol. 1994;102:807–811. doi: 10.1111/1523-1747.ep12378630. [DOI] [PubMed] [Google Scholar]
- Chakder S, Rattan S. The entire vasoactive intestinal polypeptide molecule is required for the activation of the vasoactive intestinal polypeptide receptor: functional and binding studies on opossum internal anal sphincter smooth muscle. J Pharmacol Exp Ther. 1993;266:392–399. [PubMed] [Google Scholar]
- Eedy DJ, Shaw C, Armstrong EP, Johnston CF, Buchanan KD. Vasoactive intestinal peptide (VIP) and peptide histidine methionine (PHM) in human eccrine sweat glands: demonstration of innervation, specific binding sites and presence in secretions. Br J Dermatol. 1990;123:65–76. doi: 10.1111/j.1365-2133.1990.tb01825.x. [DOI] [PubMed] [Google Scholar]
- Fox RH, Hilton SM. Bradykinin formation in human skin as a factor in heat vasodilation. J Physiol. 1958;142:219–232. doi: 10.1113/jphysiol.1958.sp006011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frase LL, Gaffney FA, Lane LD, Buckey JC, Said SI, Blomqvist CG, Krejs GJ. Cardiovascular effects of vasoactive intestinal peptide in healthy subjects. Am J Cardiol. 1987;60:1356–1361. doi: 10.1016/0002-9149(87)90619-9. [DOI] [PubMed] [Google Scholar]
- Friedman DB, Jensen FB, Matzen S, Secher NH. Non-invasive blood pressure monitoring during head-up tilt using the Penaz principle. Acta Anaesth Scand. 1990;34:519–522. doi: 10.1111/j.1399-6576.1990.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Grant RT, Holling HE. Further observations on the vascular responses of the human limb to body warming; evidence for sympathetic vasodilator nerves in the normal subject. Clin Sci. 1938;3:273–285. [Google Scholar]
- Grider JR, Rivier JR. Vasoactive intestinal peptide (VIP) as transmitter of inhibitory motor neurons of the gut: evidence from the use of selective VIP antagonists and VIP antiserum. J Pharmacol Exp Ther. 1990;253:738–742. [PubMed] [Google Scholar]
- Hartschuh W, Reinecke M, Weihe E, Yanaihara N. VIP-immunoreactivity in the skin of various mammals: immunohistochemical, radioimmunological and experimental evidence for a dual localization in cutaneous nerves and merkel cells. Peptides. 1984;5:239–245. doi: 10.1016/0196-9781(84)90213-4. [DOI] [PubMed] [Google Scholar]
- Heinz-Erian P, Dey RD, Flux M, Said SI. Deficient vasoactive intestinal peptide innervation in the sweat glands of cystic fibrosis patients. Science. 1985;229:1407–1408. doi: 10.1126/science.4035357. [DOI] [PubMed] [Google Scholar]
- Henning RJ, Sawmiller DR. Vasoactive intestinal peptide: cardiovascular effects. Cardiovasc Res. 2001;49:27–37. doi: 10.1016/s0008-6363(00)00229-7. [DOI] [PubMed] [Google Scholar]
- Hill MR, Wallick DW, Mongeon LR, Martin PJ, Levy MN. Vasoactive intestinal polypeptide antagonists attenuate vagally induced tachycardia in the anesthetized dog. Am J Physiol. 1995;269:H1467–1472. doi: 10.1152/ajpheart.1995.269.4.H1467. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Johansson O, Ljungdahl A, Lundberg JM, Schultzberg M. Peptidergic neurones. Nature. 1980;284:515–521. doi: 10.1038/284515a0. [DOI] [PubMed] [Google Scholar]
- Huang Q, Legradi G, Arimura A. Perfusion of the paraventricular nucleus with pituitary adenylate cyclase-activating polypeptide and vasoactive intestinal polypeptide stimulates local release of norepinephrine and its metabolite: microdialysis study in freely moving rats. Ann N Y Acad Sci. 1996;805:737–742. doi: 10.1111/j.1749-6632.1996.tb17550.x. [DOI] [PubMed] [Google Scholar]
- Itoh N, Obata K, Yanaihara N, Okamoto H. Human preprovasoactive intestinal polypeptide contains a novel PHI-27-like peptide, PHM-27. Nature. 1983;304:547–549. doi: 10.1038/304547a0. [DOI] [PubMed] [Google Scholar]
- Johansson O. Evidence for PHI-immunoreactive nerve fibres in the human skin: coexistence with VIP. Med Biol. 1986;64:67–73. [PubMed] [Google Scholar]
- Johnson JM, Brengelmann GL, Hales JR, Vanhoutte PM, Wenger CB. Regulation of the cutaneous circulation. Fed Proc. 1986;45:2841–2850. [PubMed] [Google Scholar]
- Johnson JM, Proppe DW. Cardiovascular adjustments to heat stress. In: Fregly M, Blatteis C, editors. Handbook of Physiology. Environmental Physiology. New York: Oxford University Press; 1996. pp. 215–243. [Google Scholar]
- Kellogg DL, Jr, CrandalL CG, Liu Y, Charkoudian N, Johnson JM. Nitric oxide and cutaneous active vasodilation during heat stress. J Appl Physiol. 1998;85:824–829. doi: 10.1152/jappl.1998.85.3.824. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Johnson JM, Kenney WL, Pérgola PE, Kosiba WA. Mechanisms of control of skin blood flow during prolonged exercise in humans. Am J Physiol. 1993;265:H562–568. doi: 10.1152/ajpheart.1993.265.2.H562. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Liu Y, Kosiba IF, O'Donnell D. Role of nitric oxide in the vascular effects of local warming of the skin in humans. J Appl Physiol. 1999;86:1185–1190. doi: 10.1152/jappl.1999.86.4.1185. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Pérgola PE, Piest KL, Kosiba WA, Crandall CG, Grossmann M, Johnson JM. Cutaneous active vasodilation in humans is mediated by cholinergic nerve cotransmission. Circ Res. 1995;77:1222–1228. doi: 10.1161/01.res.77.6.1222. [DOI] [PubMed] [Google Scholar]
- Laburthe M, Couvineau A, Rouyer-Fessard C, Moroder L. Interaction of PHM, PHI and 24-glutamine PHI with human VIP receptors from colonic epithelium: comparison with rat intestinal receptors. Life Sci. 1985;36:991–995. doi: 10.1016/0024-3205(85)90396-0. [DOI] [PubMed] [Google Scholar]
- Landis SC, Fredieu JR. Coexistence of calcitonin gene-related peptide and vasoactive intestinal peptide in cholinergic sympathetic innervation of rat sweat glands. Brain Res. 1986;377:177–181. doi: 10.1016/0006-8993(86)91205-9. [DOI] [PubMed] [Google Scholar]
- Lewis T, Pickering GW. Vasodilation in the limbs in response to warming the body: with evidence for vasodilator nerves in man. Heart. 1931;16:33–51. [Google Scholar]
- Lindh B, Haegerstrand A, Lundberg JM, Hökfelt T, Fahrenkrug J, Cuello AC, Graffi J, Massoulie J. Substance P-, VIP- and CGRP-like immunoreactivities coexist in a population of cholinergic postganglionic sympathetic nerves innervating sweat glands in the cat. Acta Physiol Scand. 1988;134:569–570. doi: 10.1111/j.1748-1716.1998.tb08536.x. [DOI] [PubMed] [Google Scholar]
- Lundberg JM. Evidence for coexistence of vasoactive intestinal polypeptide (VIP) and acetylcholine in neurons of cat exocrine glands. Acta Physiol Scand Suppl. 1981;496:1–57. [PubMed] [Google Scholar]
- Lundberg JM, Änggård A, Fahrenkrug J. Complementary role of vasoactive intestinal polypeptide (VIP) and acetylcholine for cat submandibular gland blood flow and secretion. I. VIP release. Acta Physiol Scand. 1981a;113:317–327. doi: 10.1111/j.1748-1716.1981.tb06902.x. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Änggård A, Fahrenkrug J. Complementary role of vasoactive intestinal polypeptide (VIP) and acetylcholine for cat submandibular gland blood flow and secretion. II. Effects of cholinergic antagonists and VIP antiserum. Acta Physiol Scand. 1981b;113:329–336. doi: 10.1111/j.1748-1716.1981.tb06903.x. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Änggård A, Fahrenkrug J. Complementary role of vasoactive intestinal polypeptide (VIP) and acetylcholine for cat submandibular gland blood flow and secretion. III Effects of local infusions. Acta Physiol Scand. 1982a;114:329–337. doi: 10.1111/j.1748-1716.1982.tb06992.x. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Änggård A, Fahrenkrug J, Hökfelt T, Mutt V. Vasoactive intestinal polypeptide in cholinergic neurons of exocrine glands: functional significance of coexisting transmitters for vasodilation and secretion. Proc Natl Acad Sci U S A. 1980;77:1651–1655. doi: 10.1073/pnas.77.3.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JM, Änggård A, Fahrenkrug J, Lundgren O, Holmstedt B. Corelease of VIP and acetylcholine in relation to blood flow and salivary secretion in cat submandibular salivary gland. Acta Physiol Scand. 1982b;115:525–528. [Google Scholar]
- Lundberg JM, Fahrenkrug J, Hökfelt T, Martling CR, Larsson O, Tatemoto K, Änggård A. Co-existence of peptide HI (PHI) and VIP in nerves regulating blood flow and bronchial smooth muscle tone in various mammals including man. Peptides. 1984;5:593–606. doi: 10.1016/0196-9781(84)90090-1. [DOI] [PubMed] [Google Scholar]
- Mutt V, Said SI, Bodanszky M, Klausner YS, Lin CY. Structure of the porcine vasoactive intestinal octacosapeptide. The amino-acid sequence. Use of kallikrein in its determination. Synthesis of the vasoactive intestinal peptide (VIP) Eur J Biochem. 1974;42:581–589. doi: 10.1111/j.1432-1033.1974.tb03373.x. [DOI] [PubMed] [Google Scholar]
- Öberg PA. Laser-Doppler flowmetry. Crit Rev Biomed Eng. 1990;18:125–163. [PubMed] [Google Scholar]
- Roddie IC, Shepherd JT, Whelan RF. The contribution of constrictor and dilator nerves to the skin vasodilation during body heating. J Physiol. 1957a;136:489–497. doi: 10.1113/jphysiol.1957.sp005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddie IC, Shepherd JT, Whelan RF. The vasomotor nerve supply to the skin and muscle of the human forearm. Clin Sci. 1957b;16:67–74. [PubMed] [Google Scholar]
- Saumet JL, Kellogg Dl JR, Taylor WF, Johnson JM. Cutaneous laser-Doppler flowmetry: influence of underlying muscle blood flow. J Appl Physiol. 1988;65:478–481. doi: 10.1152/jappl.1988.65.1.478. [DOI] [PubMed] [Google Scholar]
- Savage MV, Brengelmann GL, Buchan AM, Freund PR. Cystic fibrosis, vasoactive intestinal polypeptide, and active cutaneous vasodilation. J Appl Physiol. 1990;69:2149–2154. doi: 10.1152/jappl.1990.69.6.2149. [DOI] [PubMed] [Google Scholar]
- Shastry S, Minson CT, Wilson SA, Dietz NM, Joyner M. Effects of atropine and L-NAME on cutaneous blood flow during body heating in humans. J Appl Physiol. 2000;88:467–472. doi: 10.1152/jappl.2000.88.2.467. [DOI] [PubMed] [Google Scholar]
- Shastry S, Reed AS, Halliwill JR, Dietz NM, Joyner MJ. Effects of nitric oxide synthase inhibition on cutaneous vasodilation during body heating in humans. J Appl Physiol. 1998;85:830–834. doi: 10.1152/jappl.1998.85.3.830. [DOI] [PubMed] [Google Scholar]
- Taylor WF, Johnson JM, Kosiba WA, Kwan CM. Cutaneous vascular responses to isometric handgrip exercise. J Appl Physiol. 1989;66:1586–1592. doi: 10.1152/jappl.1989.66.4.1586. [DOI] [PubMed] [Google Scholar]
- Turner JT, Jones SB, Bylund DB. A fragment of vasoactive intestinal peptide, VIP(10–28), is an antagonist of VIP in the colon carcinoma cell line, HT29. Peptides. 1986;7:849–854. doi: 10.1016/0196-9781(86)90105-1. [DOI] [PubMed] [Google Scholar]
- Vaalasti A, Tainio A, Rechardt L. Vasoactive intestinal polypeptide (VIP)-like immunoreactivity in the nerves of human axillary sweat glands. J Investig Dermatol. 1985;85:246–248. doi: 10.1111/1523-1747.ep12276717. [DOI] [PubMed] [Google Scholar]
- Valtorta F, Arslan G. The pharmacology of botulinum toxin. Pharmacol Res. 1993;27:33–44. doi: 10.1006/phrs.1993.1003. [DOI] [PubMed] [Google Scholar]