

Abstract
There is evidence that the rate at which oxygen uptake (V̇O2) rises at the transition to higher metabolic rates within the moderate exercise intensity domain is modulated by oxidative enzyme inertia, and also that nitric oxide regulates mitochondrial function through competitive inhibition of cytochrome c oxidase in the electron transport chain. We therefore hypothesised that inhibition of nitric oxide synthase (NOS) by nitro-L-arginine methyl ester (l-NAME) would alleviate the inhibition of mitochondrial V̇O2 by nitric oxide and result in a speeding of V̇O2 kinetics at the onset of moderate-intensity exercise. Seven males performed square-wave transitions from unloaded cycling to a work rate requiring 90 % of predetermined gas exchange threshold with and without prior intravenous infusion of l-NAME (4 mg kg−1 in 50 ml saline over 60 min). Pulmonary gas exchange was measured breath-by-breath and V̇O2 kinetics were determined from the averaged response to four exercise bouts performed in each condition using a mono-exponential function following elimination of the phase I response. There were no significant differences between the control and l-NAME conditions for baseline V̇O2 (means ± s.e.m. 797 ± 32 vs. 794 ± 29), the duration of phase I (15.4 ± 0.8 vs. 17.2 ± 0.6), or the steady-state increment in V̇O2 above baseline (1000 ± 83 vs. 990 ± 85 ml min−1), respectively. However, the phase II time constant of the V̇O2 response was significantly smaller following l-NAME infusion (22.1 ± 2.4 vs. 17.9 ± 2.3; P < 0.05). These data indicate that inhibition of NOS by l-NAME results in a significant (19 %) speeding of pulmonary V̇O2 kinetics in the transition to moderate-intensity cycle exercise in man. At least part of the intrinsic inertia to oxidative metabolism at the onset of moderate-intensity exercise may result from competitive inhibition of mitochondrial V̇O2 by nitric oxide at cytochrome c oxidase, although other mechanisms for the effect of l-NAME on V̇O2 kinetics remain to be explored.
In the transition to a higher metabolic rate, there is an inherent ‘lag’ in the rate at which oxygen uptake (V̇O2) rises to meet the increased energetic requirement. There is debate over whether this lag is caused by a limitation in the rate at which O2 is delivered to working muscle or by the rate at which mitochondria adjust oxidative ATP supply to meet demand (Whipp & Mahler, 1980; Grassi et al. 1998; Tschakovsky & Hughson, 1999; Grassi et al. 2000). While the V̇O2 kinetics are inevitably influenced by the interaction of both metabolic substrate and O2 availability (Tschakovsky & Hughson, 1999), there is evidence that the kinetic adjustment of V̇O2 in the transition to a higher work rate within the moderate exercise intensity domain (i.e. exercise that does not cause a significant accumulation of muscle or blood lactate), is predominantly limited by an oxidative enzyme inertia rather than by muscle O2 supply (Whipp & Mahler, 1980; Yoshida & Whipp, 1994; Grassi et al. 1996, 1998). However, the precise locus of this oxidative enzyme inertia remains to be firmly established.
Nitric oxide has been implicated in a wide range of physiological functions including neurotransmission, platelet aggregation and the regulation of vasodilatation (Joyner & Dietz, 1997). It has also been demonstrated that nitric oxide may reversibly inhibit mitochondrial respiration, by binding to the O2-binding site at cytochrome c oxidase in the electron transport chain (Cleeter et al. 1994; Shen et al. 1994; Brown, 2000, 2001). Recently, Kindig et al. (2002) reported that inhibition of nitric oxide synthase (NOS) by the L-arginine analogue nitro-L-arginine methyl ester (L-NAME) caused a 32 % reduction in the time constant of the primary V̇O2 response to moderate-intensity treadmill running in the thoroughbred horse, with no change in the steady-state V̇O2. In humans, Frandsen et al. (2001) established that muscle V̇O2 after 10 and 20 min of sub-maximal exercise was not affected by infusion of l-NAME but they did not investigate the V̇O2 kinetic response at exercise onset. It therefore remains to be established whether l-NAME infusion causes a speeding of V̇O2 kinetics in man. We therefore hypothesised that if nitric oxide is partly responsible for determining the rate of the V̇O2 adjustment at the onset of moderate-intensity exercise in man, then NOS inhibition by l-NAME should lead to a significant speeding of the V̇O2 kinetics.
METHODS
Participants
Seven healthy males (mean ± S.D. age 25 ± 3 years, body mass 77.7 ± 8.3 kg) volunteered to participate in this study. All participants were informed of the experimental procedures, the potential risks and discomfort, and that they could withdraw from the study at any time. All participants gave their written informed consent. The experiments were approved by the Manchester Metropolitan University and South Cheshire Local Research Ethics Committees and conformed to the declaration of Helsinki.
Procedures
The participants were required to visit the laboratory on five occasions. On the first visit to the laboratory, the participants completed an incremental exercise test in order to determine the gas exchange threshold (GET) and V̇O2peak (see below). The remaining four visits to the laboratory were used to complete the experimentation. On each of these visits, the participants completed two bouts of moderate-intensity cycle exercise. On two occasions, the exercise bouts were preceded by infusion of l-NAME (see below). The participants therefore performed a total of four bouts of moderate-intensity exercise in each condition (i.e. two exercise bouts on each of two days). The conditions were presented in random order, and were separated by at least 48 h. All exercise tests were performed on an electronically braked cycle ergometer (Jaeger Ergoline E800, Germany).
Incremental exercise test
Following 3 min of ‘unloaded’ cycling, the work rate was increased by 5 W every 10 s (i.e. 30 W min−1) until the participant was unable to continue. The participants cycled at a self-selected pedal rate (60-90 rev min−1) and this pedal rate and the saddle and handlebar height and configuration were recorded and reproduced in subsequent tests. Pulmonary gas exchange was measured on a breath-by-breath basis (see below). The V̇O2peak was determined as the highest value recorded in any 30 s period before the participant's volitional termination of the test. The GET was determined as the first disproportionate increase in V̇CO2 from visual inspection of individual plots of V̇CO2 versus V̇O2 by an experienced reviewer, and the work rate that would require 90 % of the V̇O2 at GET was calculated. In the present study, it was important that the work rate was below the GET in order that the results were not complicated by possible changes in O2 delivery resulting from NOS inhibition, since V̇O2 kinetics above the GET may be determined, in part, by O2 delivery (Tschakovsky & Hughson, 1999).
Square-wave exercise tests
The moderate-intensity exercise bouts were performed with and without prior infusion of L-NAME. Our protocol for the l-NAME infusion was based on that described by Frandsen et al. (2001) who demonstrated that this infusion protocol resulted in a 67 % reduction in NOS activity in skeletal muscle. Participants first rested for a 20 min period, before a cannula was placed in a hand vein and l-NAME (4 mg (kg body mass)−1 in 50 ml saline) was infused over 60 min. Throughout the infusion, blood pressure and heart rate were monitored. Following an additional 30 min rest, the participants mounted the cycle ergometer and the exercise protocol commenced. The exercise protocol began with 3 min of baseline pedalling at 20 W (the lowest available work rate on the cycle ergometer), followed by an abrupt transition to the 90 % GET work rate for 6 min. Following 3 min rest, this procedure (3 min of baseline pedalling and 6 min exercise at 90 % GET) was repeated. This ‘double square-wave’ protocol was completed on four separate days, twice with and twice without prior infusion of L-NAME.
Pulmonary gas exchange was measured breath-by-breath and heart rate was monitored by short-range telemetry (Polar Electro Oy, Kempele, Finland) throughout all exercise tests. Subjects wore a nose-clip and breathed through a low dead space, low resistance mouthpiece and volume sensor assembly. Pulmonary gas exchange was measured with a mass spectrometer and volume turbine system (Morgan EX670, Morgan Medical Limited, Gillingham, Kent). The system was calibrated prior to each test with gases of known concentration. A fingertip blood sample was collected into a capillary tube immediately before and after one of the exercise bouts in each condition and subsequently analysed for blood [lactate] (YSI 1500 Sport lactate analyser, Yellow Springs Instruments, Ohio, USA).
Analysis of V̇O2 kinetics
The breath-by-breath V̇O2 data for each transition were interpolated to give second-by-second values and time-aligned to the start of exercise. For each participant and each condition, the four repeat transitions were then averaged in order to enhance the underlying response characteristics. The baseline V̇O2 was defined as the average V̇O2 measured during unloaded cycling between 160 and 20 s before the start of exercise. The cardio-dynamic component (phase I) was ignored by eliminating the first 20 s of data after the onset of exercise. Subsequently, non-linear regression techniques were used to fit the remaining V̇O2 data with an exponential function:
This model includes an amplitude (A), a time constant (τ) and a delay time (TD). An iterative process was used in order to minimise the sum of squared error between the fitted function and the observed values.
To provide an indication of the overall rate of V̇O2 adaptation, the ‘mean response time’ (MRT; the sum of the time constant and time delay values) and the O2 deficit (product of the MRT and the steady-state amplitude of the V̇O2 response above baseline) were calculated.
Statistics
Paired-samples t tests were used to test for significant differences in the V̇O2 kinetic parameters between the control and l-NAME conditions with significance declared when P < 0.05. Results are reported as means ± s.e.m. unless otherwise stated.
RESULTS
The participants' mean ± S.D. V̇O2peak was 49.4 ± 5.6 ml kg−1 min−1 with GET occurring at 51 ± 8 % V̇O2peak. The mean ± S.D. increase in work rate above baseline cycling (20 W) to 90 % GET was 104 ± 26 W.
As would be expected for exercise in the moderate-intensity domain, blood [lactate] did not rise significantly above pre-exercise values either in the control or l-NAME conditions (Δ[lactate] 0.2 ± 0.1 mM and 0.1 ± 0.1 mM respectively; Table 1). Furthermore, the V̇O2 data were well fitted by a mono-exponential function with delay, i.e. the exercise did not elicit the V̇O2 ‘slow component’ phenomenon that is evident during exercise above the GET.
Table 1.
Heart rate (HR) and blood (lactate) responses following infusion of l-NAME compared to control
| Baseline HR | End-exercise HR | ΔbLOOD (lactate) | ||||
|---|---|---|---|---|---|---|
| Subject number | Control | l-NAME | Control | l-NAME | Control | l-NAME |
| (beats min−1) | (beats min−1) | (mm) | ||||
| 1 | 94 | 73 | 115 | 108 | 0.7 | 0.6 |
| 2 | 66 | 65 | 107 | 113 | 0.1 | 0.1 |
| 3 | 76 | 65 | 113 | 107 | 0.5 | 0.3 |
| 4 | 100 | 72 | 127 | 109 | 0.0 | −0.2 |
| 5 | 88 | 75 | 125 | 115 | 0.1 | −0.1 |
| 6 | 87 | 67 | 109 | 105 | 0.3 | 0.4 |
| 7 | 84 | 77 | 109 | 107 | −0.2 | −0.5 |
| Mean | 85 | 71* | 115 | 109 | 0.2 | 0.1 |
| s.e.m. | 4 | 2 | 3 | 1 | 0.1 | 0.1 |
Significantly different from control condition (P < 0.01).
At rest, during the l-NAME infusion, blood pressure was significantly higher, and heart rate significantly lower compared to the control condition (P < 0.05). Compared to control, heart rate was lower following l-NAME infusion during unloaded cycling (84 ± 4 vs. 71 ± 2 beats min−1; P < 0.01) while the end-exercise heart rate tended to be lower (115 ± 3 vs. 109 ± 1 beats min−1), (Table 1).
The V̇O2 kinetic response data are presented in Table 2, and the response of a typical subject is shown in Fig. 1. Infusion of l-NAME did not significantly affect baseline V̇O2, the steady-state V̇O2 amplitude, or the time delay. However, l-NAME infusion resulted in a significant speeding of the V̇O2 kinetic response (τ reduced from 22.1 ± 2.4 to 17.9 ± 2.3 s; P < 0.05). The MRT of the response (from 37.5 ± 2.4 s to 35.1 ± 2.6 s; P < 0.05) and the O2 deficit (617 ± 60 vs. 588 ± 69 ml; P < 0.05) were significantly reduced in the l-NAME condition.
Table 2.
V̇o2 kinetics in the transition to moderate intensity exercise following infusion of l-NAME compared to control
| Baseline V̇o2 | V̇o2 amplitude | End-exercise V̇o2 | Time delay | V̇o2 time constant | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Subject number | Control | l-NAME | Control | l-NAME | Control | l-NAME | Control | l-NAME | Control | l-NAME |
| (ml min−1) | (ml min−1) | (ml min−1) | (s) | (s) | ||||||
| 1 | 773 | 804 | 1098 | 1016 | 1891 | 1820 | 17.0 | 19.6 | 28.5 | 26.1 |
| 2 | 737 | 720 | 840 | 796 | 1577 | 1516 | 13.3 | 16.1 | 13.4 | 8.1 |
| 3 | 789 | 795 | 1212 | 1195 | 2001 | 1990 | 12.7 | 17.0 | 25.0 | 14.7 |
| 4 | 940 | 892 | 1128 | 1184 | 2068 | 2076 | 15.9 | 16.0 | 22.0 | 21.5 |
| 5 | 844 | 854 | 1232 | 1245 | 2076 | 2099 | 17.8 | 15.0 | 16.9 | 17.3 |
| 6 | 673 | 668 | 685 | 714 | 1358 | 1382 | 13.6 | 17.6 | 30.8 | 22.8 |
| 7 | 825 | 828 | 808 | 782 | 1633 | 1610 | 17.8 | 18.8 | 18.0 | 14.9 |
| Mean | 797 | 794 | 1000 | 990 | 1801 | 1785 | 15.4 | 17.2 | 22.1 | 17.9* |
| s.e.m. | 32 | 29 | 83 | 85 | 106 | 108 | 0.8 | 0.6 | 2.4 | 2.3 |
Significantly different from control condition (P < 0.05).
Figure 1. V̇O2 kinetic response in the transition to moderate intensity exercise following infusion of l-NAME (○) and in control condition (•).
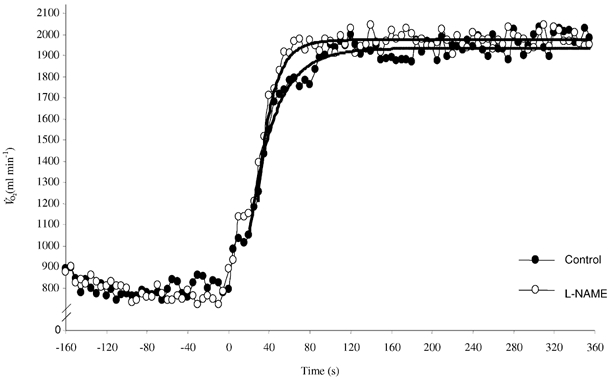
Notice the faster adjustment in the l-NAME condition. Continuous lines represent mono-exponential curve fits to the data.
DISCUSSION
We have demonstrated that infusion of l-NAME leads to a 19 % speeding of the phase II pulmonary V̇O2 kinetic response in the transition to a higher metabolic rate within the moderate exercise intensity domain in man. To our knowledge, this is the only intervention that has ever been shown to cause a speeding of pulmonary V̇O2 kinetics during moderate intensity upright cycle exercise in young, healthy participants. The mean response time was also significantly reduced by l-NAME despite the fact that the duration of phase I (reflected in the time delay term) was increased. These results extend the earlier findings of Kindig et al. (2001) who demonstrated that infusion of l-NAME resulted in a 32 % speeding of V̇O2 kinetics during moderate intensity treadmill running in the thoroughbred horse. Collectively, these studies indicate that reducing nitric oxide inhibition of cytochrome c oxidase may remove some of the inertia to mitochondrial respiration and allow an acceleration of V̇O2 kinetics in the transition to a higher metabolic rate within the moderate exercise intensity domain.
It has been reported that nitric oxide activates soluble guanylate cyclase in vascular smooth muscle resulting in vascular relaxation and increased tissue blood flow (Moncada et al. 1991). However, it is presently unclear whether NOS inhibition results in a reduction in skeletal muscle blood flow during exercise. In a number of species, there is evidence that cardiac output and/or muscle blood flow are reduced during exercise, and that O2 extraction at the muscle is correspondingly increased following NOS inhibition (Hirai et al. 1994; O'Leary et al. 1994; Kindig et al. 2000; Shen et al. 2000). In contrast, several studies in humans have reported no difference in skeletal muscle blood flow during exercise, at least in the steady state, between NOS-inhibited and control conditions (Bradley et al. 1999; Radegran & Saltin, 1999; Frandsen et al. 2001). Although one study with human volunteers indicated that NOS inhibition reduced skeletal muscle blood flow (Dyke et al. 1995), it should be noted that blood flow was estimated using plethysmography during pauses in contraction in that study. This is consistent with evidence that NOS inhibition might indeed reduce skeletal muscle blood flow during recovery from exercise (Frandsen et al. 2001). We did not measure muscle blood flow in the present study. However, the ≈13 beats min−1 lower heart rate at the onset of exercise in the l-NAME condition suggests that there might have been a slightly lower cardiac output and muscle blood flow early in the transition from rest to exercise. The longer duration of phase I, while not statistically significant, is also consistent with there having been a more sluggish cardiac output response at exercise onset. It is important to emphasise here that despite the increase in blood pressure that is noted following l-NAME infusion, our results cannot be explained by an increased bulk muscle blood flow or O2 delivery (Frandsen et al. 2001). Therefore, NOS inhibition speeded the pulmonary V̇O2 kinetics in phase II despite the probability that there was no change in (or even a slightly lower and/or slower) bulk O2 delivery to the working muscle in the transition from unloaded pedalling to moderate intensity exercise. It is possible, however, that the lower heart rate in the l-NAME condition, presumably resulting from baroreceptor-mediated sympathetic withdrawal, reflected a reduction in blood flow to areas with low metabolic activity due to vasoconstriction in the vascular beds of these tissues, and that blood flow to active muscle was preserved. Although very unlikely (see Frandsen et al. 2001), it is possible that blood flow to active muscle was greater and/or faster following l-NAME infusion. However, even if this were true, an increased muscle blood flow and/or O2 availability would not be expected to result in faster V̇O2 kinetics during exercise of this type (Gerbino et al. 1996; MacDonald et al. 1997; Grassi et al. 1998; Burnley et al. 2000).
It is pertinent here to consider the extent to which the measurement of V̇O2 kinetics at the lung reflects the kinetics of O2 consumption at the muscle. Where there are no perturbations to muscle blood flow, there is evidence that pulmonary V̇O2 kinetics in phase II provide a close estimate (to within ≈10 %) of muscle V̇O2 kinetics (Barstow et al. 1990; Grassi et al. 1996; Rossiter et al. 1999). However, this relationship becomes more complicated where there are changes in the cardiovascular response to exercise (Barstow et al. 1990; Essfeld et al. 1991), as may be the case following NOS inhibition. Using a mathematical modelling approach, Barstow et al. (1990) predicted that for the same muscle V̇O2 kinetics, slower cardiac output kinetics would result in a lengthening of phase I and apparently faster phase II pulmonary V̇O2 kinetics (it is important to note, here, however, that these effects were quantitatively rather small and cannot fully explain the difference we observed between the l-NAME and control conditions in the present study). Interestingly, when the relationship between muscle and pulmonary V̇O2 kinetics was distorted by changes in cardiac output kinetics, the calculated O2 deficit was unaffected (Barstow et al. 1990) suggesting that in these conditions the O2 deficit might still be used to infer changes in muscle V̇O2 kinetics. In the present study, the O2 deficit was significantly reduced following l-NAME infusion which, given that there were no differences in either baseline or steady-state V̇O2, may be taken as evidence that muscle V̇O2 kinetics were speeded if it is assumed that: (1) steady-state blood flow, and (2) changes in venous O2 stores, were similar between conditions (Barstow et al. 1990). The first of these assumptions seems reasonable (Frandsen et al. 2001), but the likelihood of there being a reduction in venous volume owing to vasoconstriction in non-working vascular beds following l-NAME infusion makes it difficult to estimate the latter. However, a large reduction in venous volume would be expected to result in a reduction in the duration of phase I (which we did not observe) and to have relatively little impact on the phase II time constant (where we observed a significant speeding), (Barstow et al. 1990). In the present study, it can be estimated (Δ[O2 stores] = ΔV̇O2 ss (MRT –τ); Whipp & Ward, 1982) that there was relatively little difference between the change in O2 stores in the control condition (257 ml O2) and the l-NAME condition (284 ml O2). Therefore, we believe that the changes in pulmonary V̇O2 kinetics that we observed following l-NAME infusion –which included reductions in the phase II time constant, mean response time, and O2 deficit –reflected a speeding of muscle V̇O2 kinetics. However, further studies involving direct measures of muscle blood flow and O2 extraction across a working muscle are required to confirm this.
There was considerable inter-individual variability in the effect of l-NAME on V̇O2 kinetics (0-41 % speeding; Table 2). The reason for the lack of effect in two of our seven participants is unclear. One possibility is that l-NAME did not cause a substantial inhibition of NOS in these participants. It is interesting to note, however, that the two participants whose V̇O2 kinetics were not speeded by l-NAME had the greatest reductions in exercise heart rate (numbers 4 and 5; see Tables 1 and 2). It is therefore possible that the effect of NOS inhibition on relieving mitochondrial inertia was masked by a substantial reduction in O2 delivery to skeletal muscle in these participants. On the other hand, as discussed above, the lower heart rate following l-NAME infusion might also be related to a reduction in the requirement for blood flow because of an increased O2 extraction at the muscle or as a consequence of peripheral vasoconstriction in tissue with relatively low metabolic activity.
It is well known that muscle PO2 can modulate tissue respiratory control so that an appropriate V̇O2 is attained during submaximal exercise (Wilson et al. 1977). However, at least during moderate intensity exercise, it appears that muscle PO2 does not become sufficiently low that muscle V̇O2 is compromised. Therefore, O2 availability cannot be considered limiting in this situation. Indeed, there is evidence that, under normal control conditions, cardiac output and/or muscle blood flow kinetics are faster than muscle V̇O2 kinetics, and that O2 delivery is maintained such that it is likely in excess of muscle O2 requirement at any point in the transition to moderate intensity exercise (Yoshida & Whipp, 1994; Grassi et al. 1996; MacDonald et al. 1998; see also Bangsbo et al. 2000 for higher intensity exercise). Consistent with this, it has been shown that eliminating any delay in O2 delivery by pump-perfusing canine gastrocnemius muscle at the required steady-state blood flow across the rest-to-exercise transition did not significantly influence V̇O2 kinetics (Grassi et al. 1998). Furthermore, increasing O2 delivery and/or blood flow to muscle through the performance of prior heavy exercise (Gerbino et al. 1996; Burnley et al. 2000) or the inspiration of hyperoxic gas mixtures (MacDonald et al. 1997) do not result in a speeding of phase II V̇O2 kinetics during moderate intensity cycle exercise in man. It therefore appears that, under the conditions of the present study, the speed of the V̇O2 adjustment in the transition to a higher metabolic rate is primarily limited by an inertia within the mitochondrial oxidative machinery.
The exact location of the intra-muscular limitation to V̇O2 kinetics during moderate intensity exercise remains to be firmly established. Significant research attention has been directed to the possible role of pyruvate dehydrogenase complex (PDC) activation and/or the availability of acetyl groups in limiting mitochondrial ATP production at exercise onset (Timmons et al. 1998; Howlett et al. 1999). These studies demonstrated that prior activation of the PDC by infusion of dichloroacetate (DCA) resulted in a marked attenuation of substrate-level phosphorylation, presumably due to an increased mitochondrial ATP production. However, a number of recent studies have reported that DCA infusion did not affect muscle V̇O2 kinetics or substrate-level phosphorylation during exercise. In dogs, Grassi et al. (2002) reported that V̇O2 kinetics were unchanged despite significant accumulation of acetyl groups before muscle contraction at ≈65 % V̇O2peak. In humans, DCA infusion did not reduce substrate-level phosphorylation (Bangsbo et al. 2002; Savasi et al. 2002) nor alter muscle V̇O2 kinetics (Bangsbo et al. 2002) during maximal intensity exercise. It has recently been suggested that the PDC and/or acetyl group availability may only limit mitochondrial respiration at high intensities of submaximal exercise (≈65-90 % V̇O2peak) where there is a mismatch between the demands of the TCA cycle for substrate and the degree of PDC activation (Roberts et al. 2002). While the influence of PDC activation on V̇O2 kinetics during moderate intensity exercise in humans requires further attention, it is likely that acetyl group availability is not limiting during low to moderate intensity exercise (< 50 % V̇O2peak) such as that used in the present study (Evans et al. 2001; Watt et al. 2002).
There is a growing body of evidence that nitric oxide regulates mitochondrial function by inhibiting cytochrome c oxidase, the terminal enzyme in the electron transport chain which is responsible for virtually all O2 consumption in mammals (Shen et al. 1994, 2000; Brown, 2000). Nitric oxide has a high affinity for the O2-binding site of cytochrome c oxidase when this site is reduced and therefore competes with O2 for the binding site. This competitive inhibition of V̇O2 by nitric oxide might serve to reduce the reliance on muscle O2 extraction in meeting the O2 requirement during exercise, therefore allowing maintenance of a higher intra-muscular PO2 (Shen et al. 2000). Indeed, it has been suggested that the competition between nitric oxide and O2 at cytochrome c oxidase might increase the sensitivity of mitochondrial respiration to O2 concentration because of an increase in the Michaelis-Menten constant (Km) of cytochrome c oxidase for O2 (Brown, 1995). It has also been suggested that nitric oxide might extend the zone of effective tissue respiration by increasing the O2 gradient away from the blood vessel (Thomas et al. 2001). Nitric oxide synthase (NOS), the enzyme responsible for synthesis of nitric oxide, is located within the skeletal muscle vascular endothelium as well as within myocytes (Frandsen et al. 1996). It is known that nitric oxide synthesis is greatly increased at the onset of exercise (Balon & Nadler, 1994) and that this may be attenuated with the use of L-arginine analogues such as L-NAME. The procedure we used in the present study for l-NAME infusion (4 mg (kg body mass)−1 infused over 60 min) has been shown to result in a 67 ± 8 % reduction in muscle NOS activity (Frandsen et al. 2001), and we therefore postulate that the speeding of V̇O2 kinetics that we observed resulted from a reduction of nitric oxide inhibition of mitochondrial V̇O2 at cytochrome c oxidase.
Interestingly, although the speed of the V̇O2 adaptation to exercise was greater in the l-NAME condition, the amplitude of the V̇O2 response above baseline was not different to the control condition. Our finding that the steady-state V̇O2 (established within ≈2 min) was not affected is consistent with other studies that have infused NOS inhibitors before exercise in humans (Radegran & Saltin, 1999; Frandsen et al. 2001) and other species (O'Leary et al. 1994; Mills et al. 1999; Kindig et al. 2002). For example, in the Frandsen et al. (2001) study, leg oxygen uptake was not significantly different between the l-NAME and control conditions after either 10 or 20 min of submaximal exercise. In horses, the steady-state V̇O2 amplitude was unaltered by l-NAME despite significantly faster V̇O2 kinetics (Kindig et al. 2002). It therefore appears that the inhibitory effect of nitric oxide on muscle V̇O2 might only be evident in the transition to a higher metabolic rate, and that regulation of muscle blood flow and O2 extraction enable the same steady-state V̇O2 to be attained. One report that the steady-state muscle V̇O2 was significantly increased during submaximal exercise in conscious dogs following NOS inhibition (Shen et al. 2000) is difficult to accept unless muscle efficiency is somehow influenced by nitric oxide in this species.
Although we favour the relief of nitric oxide inhibition of cytochrome c oxidase as an explanation for our results since the pernicious influence of nitric oxide at this site has been well documented (Shen et al. 1994; Brown, 2000), other possible effects of l-NAME should also be considered. Nitric oxide and its derivatives (reactive nitrogen species) inhibit a number of other enzymes that are involved in energy transduction including creatine kinase, glyceraldehyde-3-phosphate dehydrogenase and aconitase (Zhang & Snyder, 1995; Kaasik et al. 1999), as well as respiratory complexes I, II, III, and IV (Cassina & Radi, 1996; Brown, 1999). The inhibition of NOS by l-NAME therefore has the potential to influence exercise V̇O2 by a number of mechanisms. It should also be emphasised that although l-NAME infusion caused a substantial speeding of V̇O2 kinetics in the present study, the majority of the inertia in mitochondrial respiration in the transition to higher metabolic rates remains to be explained.
In conclusion, we have shown that inhibition of nitric oxide synthase by l-NAME resulted in a significant speeding of pulmonary V̇O2 kinetics in the transition to moderate intensity cycle exercise. This is the first study to demonstrate a speeding of pulmonary V̇O2 kinetics in young healthy subjects performing moderate-intensity upright cycle exercise. These data indicate that inhibition of mitochondrial V̇O2 by nitric oxide (possibly at cytochrome c oxidase) might contribute, in part, to the inertia in oxidative metabolism at the onset of moderate-intensity exercise.
REFERENCES
- Balon TW, Nadler JL. Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol. 1994;366:233–249. doi: 10.1152/jappl.1994.77.6.2519. [DOI] [PubMed] [Google Scholar]
- Bangsbo J, Gibala MJ, Krustrup P, Gonzalez-Alonso J, Saltin B. Enhanced pyruvate dehydrogenase activity does not affect muscle O2 uptake at onset of intense exercise in humans. Am J Physiol Renal Physiol. 2002;282:R273–280. doi: 10.1152/ajpregu.2002.282.1.R273. [DOI] [PubMed] [Google Scholar]
- Bangsbo J, Krustrup P, Gonzalez-Alonso J, Saltin B. Muscle oxygen kinetics at onset of intense dynamic exercise in humans. Am J Physiol Renal Physiol. 2000;279:R899–906. doi: 10.1152/ajpregu.2000.279.3.R899. [DOI] [PubMed] [Google Scholar]
- Barstow TJ, Lamarra N, Whipp BJ. Modulation of muscle and pulmonary O2 uptakes by circulatory dynamics during exercise. J Appl Physiol. 1990;68:979–989. doi: 10.1152/jappl.1990.68.3.979. [DOI] [PubMed] [Google Scholar]
- Bradley SJ, Kingwell BA, Mcconell GK. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes. 1999;48:1815–1821. doi: 10.2337/diabetes.48.9.1815. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide as a competitive inhibitor of oxygen consumption in the mitochondrial respiratory chain. Acta Physiol Scand. 2000;168:667–674. doi: 10.1046/j.1365-201x.2000.00718.x. [DOI] [PubMed] [Google Scholar]
- Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504:46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- Burnley M, Jones AM, Carter H, Doust JH. Effect of prior heavy exercise on phase II pulmonary oxygen uptake kinetics and the slow component during heavy exercise in humans. J Appl Physiol. 2000;89:1387–1396. doi: 10.1152/jappl.2000.89.4.1387. [DOI] [PubMed] [Google Scholar]
- Cassina A, Radi R. Different inhibitory actions of NO and peroxynitrite on mitochondrial electron transport. Arch Biophys Biochem. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- Cleeter MWJ, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AhV. Reversible inhibition of cytochrome oxidase, the terminal enzyme of the mitochondrial respiratory chain, by NO. FEBS Letters. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- Dyke CK, Proctor DN, Dietz NM, Joyner MJ. Role of nitric oxide in exercise hyperaemia during prolonged rhythmic handgripping in humans. J Physiol. 1995;488:259–265. doi: 10.1113/jphysiol.1995.sp020964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essfeld D, Hoffmann U, Stegemann J. A model for studying the distortion of muscle oxygen uptake patterns by circulation parameters. Eur J Appl Physiol. 1991;62:83–90. doi: 10.1007/BF00626761. [DOI] [PubMed] [Google Scholar]
- Evans MK, Savasi I, Heigenhauser GJ, Spriet LL. Effects of acetate infusion and hyperoxia on muscle substrate phosphorylation after onset of moderate exercise. Am J Physiol Endocrinol Metab. 2001;281:E1144–1150. doi: 10.1152/ajpendo.2001.281.6.E1144. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with NG-nitro-L-arginine methyl ester in humans. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen U, Lopez-Figueroa M, Hellsten Y. Localization of nitric oxide synthase in human skeletal muscle. Biochem Biophys Res Comm. 1996;227:88–93. doi: 10.1006/bbrc.1996.1472. [DOI] [PubMed] [Google Scholar]
- Gerbino A, Ward SA, Whipp BJ. Effects of prior exercise on pulmonary gas-exchange kinetics during high-intensity exercise in humans. J Appl Physiol. 1996;80:99–107. doi: 10.1152/jappl.1996.80.1.99. [DOI] [PubMed] [Google Scholar]
- Grassi B, Gladden LB, Samaja M, Stary CM, Hogan MC. Faster adjustment of O2 delivery does not affect V̇O2 on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998;85:1394–1403. doi: 10.1152/jappl.1998.85.4.1394. [DOI] [PubMed] [Google Scholar]
- Grassi B, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Gladden LB. Oxygen uptake on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. J Physiol. 2002;538:195–207. doi: 10.1113/jphysiol.2001.012984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi B, Hogan MC, Kelley KM, Aschenbach WG, Hamann JJ, Evans RK, Patillo RE, Gladden LB. Role of convective O2 delivery in determining V̇O2on-kinetics in canine muscle contracting at peak V̇O2. J Appl Physiol. 2000;89:1293–1301. doi: 10.1152/jappl.2000.89.4.1293. [DOI] [PubMed] [Google Scholar]
- Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD. Muscle O2 uptake kinetics in humans: implications for metabolic control. J Appl Physiol. 1996;80:988–998. doi: 10.1152/jappl.1996.80.3.988. [DOI] [PubMed] [Google Scholar]
- Hirai T, Visnski MD, Kearns KJ, Zelis R, Musch TI. Effects of NO synthase inhibition on the muscular blood flow response to treadmill exercise in rats. J Appl Physiol. 1994;77:1288–1293. doi: 10.1152/jappl.1994.77.3.1288. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Heigenhauser GjF, Hultman E, Hollidhe-Horvath MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am J Physiol. 1999;277:E18–25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Dietz NM. Nitric oxide and vasodilation in human limbs. J Appl Physiol. 1997;81:1619–1626. doi: 10.1152/jappl.1997.83.6.1785. [DOI] [PubMed] [Google Scholar]
- Kaasik A, Minajeva A, Desousa E, Ventura-Clapier R, Veksler V. Nitric oxide inhibits cardiac energy production via inhibition of mitochondrial creatine kinase. FEBS Lett. 1999;444:75–77. doi: 10.1016/s0014-5793(99)00033-2. [DOI] [PubMed] [Google Scholar]
- Kindig CA, Gallatin LL, Erickson HH, Fedde MR, Poole DC. Cardiorespiratory impact of the nitric oxide synthase inhibitor l-NAME in the exercising horse. Respir Physiol Neurobiol. 2000;120:151–166. doi: 10.1016/s0034-5687(00)00096-7. [DOI] [PubMed] [Google Scholar]
- Kindig CA, McDonough P, Erickson HH, Poole DC. Effect of L-NAME on oxygen uptake kinetics during heavy-intensity exercise in the horse. J Appl Physiol. 2001;91:891–896. doi: 10.1152/jappl.2001.91.2.891. [DOI] [PubMed] [Google Scholar]
- Kindig CA, McDonough P, Erickson HH, Poole DC. Nitric oxide synthase inhibition speeds oxygen uptake kinetics in horses during moderate domain running. Respir Physiol Neurobiol. 2002;132:169–178. doi: 10.1016/s1569-9048(02)00068-x. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Pedersen PK, Hughson RL. Acceleration of V̇O2 kinetics in heavy submaximal exercise by hyperoxia and prior high-intensity exercise. J Appl Physiol. 1997;83:1318–1325. doi: 10.1152/jappl.1997.83.4.1318. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Shoemaker JK, Tschakovsky ME, Hughson RL. Alveolar oxygen uptake and femoral artery blood flow dynamics in upright and supine leg exercise in humans. J Appl Physiol. 1998;85:1622–1628. doi: 10.1152/jappl.1998.85.5.1622. [DOI] [PubMed] [Google Scholar]
- Mills PC, Marlin DJ, Scott CM, Smith NC. Metabolic effects of nitric oxide synthase inhibition during exercise in the horse. Res Vet Sci. 1999;66:135–138. doi: 10.1053/rvsc.1998.0258. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- O'Leary DS, Dunlap RC, Glover KW. Role of endothelium-derived relaxing factor in hindlimb reactive and active hyperemia in conscious dogs. Am J Physiol. 1994;266:R1213–1219. doi: 10.1152/ajpregu.1994.266.4.R1213. [DOI] [PubMed] [Google Scholar]
- Radegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol. 1999;276:H1951–1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- Roberts PA, Loxham SJ, Poucher SM, Constantin-Teodosiu D, Greenhaff PL. The acetyl group deficit at the onset of contraction in ischaemic canine skeletal muscle. J Physiol. 2002;544:591–602. doi: 10.1113/jphysiol.2002.021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter HB, Ward SA, Doyle VL, Howe FA, Griffiths JR, Whipp BJ. Inferences from pulmonary O2 uptake with respect o intramuscular [phosphocreatine] kinetics during moderate exercise in humans. J Physiol. 1999;518:921–932. doi: 10.1111/j.1469-7793.1999.0921p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savasi I, Evans MK, Heigenhauser GJ, Spriet LL. Skeletal muscle metabolism is unaffected by DCA infusion and hyperoxia after onset of intense aerobic exercise. Am J Physiol Endocrinol Metab. 2002;283:E108–115. doi: 10.1152/ajpendo.00337.2001. [DOI] [PubMed] [Google Scholar]
- Shen W, Xu X, Ochoa M, Zhao G, Bernstein RD, Forfia P, Hintze TH. Endogenous nitric oxide in the control of skeletal muscle oxygen extraction during exercise. Acta Physiol Scand. 2000;168:675–686. doi: 10.1046/j.1365-201x.2000.00719.x. [DOI] [PubMed] [Google Scholar]
- Shen W, Xu X, Ochoa M, Zhao G, Wolin MS, Hintze TH. Role of nitric oxide in the regulation of oxygen consumption in conscious dogs. Circ Res. 1994;75:1086–1095. doi: 10.1161/01.res.75.6.1086. [DOI] [PubMed] [Google Scholar]
- Thomas DD, Liu X, Kantrow SP, Lancaster JR. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc Natl Acad Sci U S A. 2001;98:355–360. doi: 10.1073/pnas.011379598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol. 1998;274:E377–380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Hughson RL. Interaction of factors determining oxygen uptake at the onset of exercise. J Appl Physiol. 1999;86:1101–1113. doi: 10.1152/jappl.1999.86.4.1101. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Heigenhauser GJ, Stellingwerff T, Hargreaves M, Spriet LL. Carbohydrate ingestion reduces skeletal muscle acetylcarnitine availability but has no effect on substrate phosphorylation at the onset of exercise in man. J Physiol. 2002;544:949–956. doi: 10.1113/jphysiol.2002.026757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipp BJ, Mahler M. Dynamics of pulmonary gas exchange during exercise. In: West JB, editor. Pulmonary Gas Exchange. New York: Academic Press; 1980. pp. 33–96. [Google Scholar]
- Whipp BJ, Ward SA. Cardiopulmonary coupling during exercise. J Exp Biol. 1982;100:175–193. doi: 10.1242/jeb.100.1.175. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Ericinska M, Drown C, Silver IA. Effect of oxygen tension on cellular energetics. Am J Physiol. 1977;233:C135–C140. doi: 10.1152/ajpcell.1977.233.5.C135. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Whipp BJ. Dynamic asymmetries of cardiac output transients in response to muscular exercise in man. J Physiol. 1994;480:355–359. doi: 10.1113/jphysiol.1994.sp020365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Snyder SH. Nitric oxide and the nervous system. Annu Rev Pharmacol Toxicol. 1995;35:213–233. doi: 10.1146/annurev.pa.35.040195.001241. [DOI] [PubMed] [Google Scholar]