

Abstract
We have studied the modulatory effect of dehydroepiandrosterone (DHEA), the most abundant neurosteroid produced by glial cells and neurones, on membrane currents induced by the activation of ionotropic ATP (P2X) receptors in neonatal rat dorsal root ganglion neurones. ATP (1 μm) induced three types of currents/responses termed F (fast and transient), S (slowly desensitizing) and M (mixed, sum of F- and S-type responses). DHEA (10 nm to 100 μm) concentration-dependently increased the amplitude of plateau-like currents of S- and M-type responses evoked by submaximal (1 μm) but not saturating (100 μm or 1 mM) concentrations of ATP. αβ-Methylene ATP (αβme-ATP, 5 μm) also evoked F-, S- and M-type responses, the plateau phases of which were potentiated by lowering external pH (6.3) and by ivermectin (IVM, 3 μm), indicating the presence heteromeric P2X2-containing receptors and possibly of functional native P2X4/6 receptors. There was a strict correlation between the potentiating effects of low pH and DHEA on αβme-ATP responses but not between that of IVM and DHEA, suggesting that DHEA selectively modulated P2X2-containing receptors. DHEA also potentiated putative homomeric P2X2 receptor responses recorded in the continuous presence of 1 μm 2′-(or 3′)-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP). Our results constitute the first demonstration of a fast potentiation of P2X receptors by a neurosteroid and suggest that DHEA could be an endogenous modulator of P2X2-containing receptors thereby contributing to the facilitation of the detection and/or the transmission of nociceptive messages, particularly under conditions of inflammatory pain where the P2X receptor signalling pathway appears to be upregulated.
Adenosine 5′-triphosphate (ATP) plays an important role as an extracellular signalling molecule, in particular in relation with nociception (Burnstock, 2000; Chizh & Illes, 2001; Dunn et al. 2001). Indeed, the transduction of painful stimuli in the periphery and the modulation of their transmission in the superficial dorsal horn of the spinal cord involves ionotropic ATP receptors (P2X receptors) (Bardoni et al. 1997; Gu & MacDermott, 1997; Li et al. 1998; Jo & Schlichter, 1999; Hugel & Schlichter, 2000) which are cation-permeable ion channels formed by the homomeric or heteromeric assembly of P2X subunits (North & Surprenant, 2000; Dunn et al. 2001; Khakh et al. 2001). Therefore, a selective modulation of the P2X receptors present on the peripheral and/or central endings of nociceptors could influence the detection and/or the transmission of pain messages by the somatosensory system.
To date seven different P2X subunits (P2X1-P2X7) have been cloned and six of them (P2X1-P2X6) appear to be present in sensory neurones of the dorsal root ganglia (DRG) (Collo et al. 1996) although, on a functional basis, DRG neurones conveying nociceptive information seem to preferentially express homomeric P2X3 and heteromeric P2X2/3 receptors (Burgard et al. 1999; Chizh & Illes, 2001; Dunn et al. 2001). In fact, relatively little is known about acute and fast functional modulation of P2X receptors under physiological conditions. Studies on recombinant and native P2X receptors have demonstrated more or less subtype-specific modulations by divalent cations (Nakazawa & Hess, 1993; Li et al. 1996b, 1997; Cook & McCleskey, 1997; North & Surprenant, 2000) some of which might be relevant to physiological/pathological situations, although this point remains to be established. In addition, a selective potentiation of P2X2-containing receptors by extracellular acidification has been described (King et al. 1996; Li et al. 1996a; Nakazawa et al. 1997; Stoop et al. 1997) and might play a fundamental role during the development of pain states associated with peripheral inflammation (Burnstock, 2000; Chizh & Illes, 2001). Moreover it has been shown recently that, among different subtypes of P2X receptors, ivermectin (IVM) exerts a selective allosteric potentiation of homomeric P2X4 and possibly of heteromeric P2X4/6 receptors (Khakh et al. 1999). Finally, there is evidence that the functional properties of P2X receptors can be modulated by phosphorylation/dephosphorylation (Boue-Grabot et al. 2000), a phenomenon which might possibly occur following the activation of neurokinin 1 and bradykinin receptors (Paukert et al. 2001) involved in peripheral sensitization of nociceptors (Millan, 1999) or activation of other metabotropic receptors such as GABAB receptors (Gomez-Villafuertes et al. 2003).
Neurosteroids are a family of cholesterol-derived molecules synthesized by glial cells and neurones which, in addition to their long-term genomic effects, can also rapidly modulate the activity of ionotropic neurotransmitter-gated channels such as NMDA-type glutamate receptors or GABAA receptors underlying fast synaptic transmission (Baulieu, 1997; Rupprecht & Holsboer, 1999; Compagnone & Mellon, 2000). Thus, we asked whether neurosteroids might also modulate the function of P2X receptors and found that dehydroepiandrosterone (DHEA), the most abundant neurosteroid in the nervous system (Baulieu, 1997), selectively potentiated P2X2-containing ATP receptors.
METHODS
Tissue culture
The culture procedure used was essentially the same as that previously described (Bowie et al. 1994). Animal treatment was in compliance with European Union directives (86/609/EEC). Neonatal (2- to 4-day-old) Wistar rat pups were killed under deep diethylether anaesthesia and dorsal root ganglia (DRG) from thoracic and lumbar levels were dissected out and collected in phosphate-buffered solution (PBS). After removal of the attached dorsal and ventral roots under a stereomicroscope, the ganglia were washed with divalent-free PBS and incubated with trypsin (0.5 g l−1, Seromed, Germany) for 25 min at 37 °C. Enzymatic dissociation was terminated by removing the dissociation medium and adding an excess volume of culture medium, the composition of which was the following: minimum essential medium (MEM) alpha (Gibco, France), heat-inactivated horse serum (10 % v/v, Gibco, France), penicillin and streptomycin (50 i.u. ml−1 each, Gibco). Mechanical dissociation of the DRGs into single cells was performed in culture medium by mild trituration with fire-polished Pasteur pipettes of decreasing tip diameters. The dissociated primary sensory neurones were then plated in the central compartment of 35 mm plastic culture dishes (Corning, France). This compartment was obtained by attaching a sterile glass ring (diameter: 16 mm) to the bottom of the dish with molten paraffin wax. The bottom of this compartment was coated with poly-L-lysine (10 μg ml−1, Sigma) before seeding the neurones. Cultures were maintained in a water-saturated atmosphere (95 % air, 5 % CO2) at 37 °C for 1-48 h before use. Under these culture conditions, DRG neurones appeared essentially as round cells with minimal neurite outgrowth. For each neurone from which we recorded, we determined the diameter of the soma by means of a calibrated graticule in the eye-piece of the microscope of the experimental set-up.
Electrophysiological recordings
Experiments were performed at room temperature (20-22 °C). The glass ring delimiting the central compartment of the culture dishes was removed at the beginning of the electrophysiological recording session. Perforated-patch clamp recordings were made with an Axopatch 200A amplifier (Axon Instruments, USA) and low resistance (3-4 MΩ) electrodes using amphotericin B as the pore-forming agent (Rae et al. 1991). The final series resistance during electrophysiological recordings was between 5 and 15 MΩ. Membrane currents were recorded under voltage clamp at a steady holding potential of −70 mV which also corresponded to the equilibrium potential for Cl− ions under our experimental conditions. The external solution contained (mM): NaCl 135, KCl 5, CaCl2 2.5, MgCl2 1, Hepes 5, glucose 10, pH 7.3 (with NaOH) and tetrodotoxin (TTX, 0.5 μM). The culture dish (total bath volume: 1.5 ml) was continuously superfused with extracellular solution at a rate of 3 ml min−1. The pipette solution contained (mM): CsCl 9, Cs2SO4 87, Hepes 10, EGTA 10, pH 7.3 (with CsOH) and amphotericin B (150 μg ml−1, Sigma, France). The amphotericin B stock solution (30 mg ml−1) was prepared in dimethylsulfoxide (DMSO) just before the recording session (Jo & Schlichter, 1999; Hugel & Schlichter, 2000). Voltage and current traces were stored digitally on a video tape recorder (sampling rate 20 kHz) and/or on a personal computer after filtering at 5 kHz by the Axopatch 200A. Acquisition and analysis were performed with the program pCLAMP (Axon Instruments, USA).
Drugs and application of substances
All substances were prepared as 1000 times concentrated stock solutions relative to the final concentrations to be used in electrophysiological experiments. Tetrodotoxin (TTX, Latoxan, France), adenosine 5′-triphosphate (ATP, Sigma, France), αβ-methylene ATP (αβme-ATP, Sigma, France), suramin (Sigma, France), pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS, Tocris Cookson, distributed by Bioblock, France) and reactive blue 2 (Sigma, France) were prepared in distilled water and stored at −20 °C. TNP-ATP (2′-(or 3′)-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate; Molecular Probes, USA) was purchased as a 5 mg ml−1 stock solution in 0.1 M Tris buffer and was also stored at −20 °C. Ivermectin (IVM, Sigma, France) was prepared in DMSO, and dehydroepiandrosterone (DHEA, Sigma, France) and capsaicin (Sigma, France) were prepared in absolute ethanol and stored at 4 °C. The stock solution of DHEA was prepared freshly every 2 weeks. Moreover, we noticed a relative instability of DHEA in powder form once the vial had been opened. Therefore, we renewed the powder stock every 6 months.
All substances to be tested were diluted to their final concentrations in extracellular solution just before the recording session, and pH was carefully checked and readjusted if necessary. The final concentration of solvent in the physiological solution was ≤ 0.1 % (1/1000). At such a concentration, neither DMSO nor ethanol had any effect on P2X receptor-mediated currents. The different substances were applied or coapplied locally by means of a U-tube (Fenwick et al. 1982) allowing a relatively fast solution exchange around the cell (< 50 ms). In experiments in which we tested the effects of IVM, suramin, PPADS, as well as the effect of low pH (6.3) on the ATP or αβme-ATP responses, these substances were applied by general bath perfusion in addition to the coapplication via the U-tube. PPADS was applied by bath for at least 5 min before evaluating its effect on P2X responses. In the case of DHEA, we noticed that the effect of DHEA on the P2X response was similar when coapplied with the P2X agonist in the absence or in the continuous presence of DHEA in the bath. Therefore, DHEA was always simply coapplied with the agonist.
In the figures only single representative current traces of agonist-induced currents are illustrated for each experimental condition. However three to four applications were always performed under each condition and only reproducible and stable responses were taken into consideration.
All statistical results are given as means ± s.e.m. (standard error of the mean). Statistical differences between results were assessed with a confidence level of 0.95 using the following tests (when appropriate, see results): Kruskall-Wallis one way ANOVA on ranks followed by Dunn's test, Kolmogorov-Smirnov test, χ2 test.
RESULTS
The results presented here were obtained from a total of 203 neurones recorded under voltage clamp with the perforated-patch clamp technique, which allows minimal disturbance of the intracellular compartment of the neurones with respect to endogenous second messenger systems and phosphorylation factors.
ATP elicits different types of fast membrane currents in DRG neurones
ATP was applied locally for a duration of 3 s at a concentration of 1 μM. This concentration corresponds approximately to the EC50 of recombinant and native P2X3 and P2X2/3 receptors, which were initially the main targets of our study (Khakh et al. 2001). It must, however, be emphasized that these EC50 values might vary with the density of receptor expression as clearly demonstrated in the case of P2X2 receptors (Clyne et al. 2003). Moreover, since homomeric P2X3 receptors undergo pronounced and rapid desensitization (Khakh et al. 2001; North, 2002) successive applications of ATP were separated by a delay of 4 min.
Among the 203 neurones recorded, 189 (93 %) responded with an inward current at a holding potential (Vh) of −70 mV during the application of ATP (1 μM). The three types of ATP responses observed are illustrated in Fig. 1A. In 39 % of the neurones responding to ATP (n = 189), we observed a rapidly activating and fast desensitizing inward current (termed F for ‘fast’). The amplitude of this current decayed rapidly to baseline levels during the 3 s lasting ATP application and was strongly reduced when the delay between two subsequent applications of ATP was below 4 min. In another category of neurones (23 %), ATP induced a slowly decaying inward current (termed S for ‘slow’). Finally, in 38 % of the neurones we observed a biphasic current displaying an initial fast and transient phase and a late slowly decaying phase. These responses appeared to be a combination of the F- and S-type responses described above and were termed M (for ‘mixed’). This conclusion was supported by the fact that when two ATP applications were performed at a short time interval (< 30 s) the transient phase was completely suppressed whereas the slowly decaying component was relatively unaffected (data not shown). This observation was consistent with those made on cultured embryonic, neonatal and adult DRG neurones in other studies (Burgard et al. 1999; Grubb & Evans, 1999; Labrakakis et al. 2000; Dunn et al. 2001; Pankratov et al. 2001) and indicated that (i) F-type responses were due to the activation of homomeric P2X3 receptors, (ii) S-type responses involved mainly heteromeric P2X2/3 receptors (but see below) and (iii) M-type responses represented the algebraic sum of independent F- and S-type responses. Among all neurones tested (n = 203), 14 did not respond to ATP at a concentration of 1 μM and were classified as NR (for ‘non-responsive’). It is important to emphasize that this conclusion was only valid under our control experimental conditions, since most NR neurones could develop a response to ATP (1 μM) in the presence of DHEA (see below).
Figure 1. Diversity of P2X receptor-mediated membrane currents and their relationship to somatic cell diameter and capsaicin sensitivity in neonatal DRG neurones.
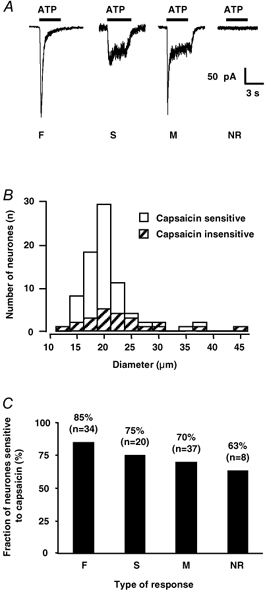
A, local application of ATP (1 μM) over 3 s (horizontal black bars), induced three types of membrane currents. F-type currents were characterized by a fast activation and complete desensitization whereas S-type currents were only slowly desensitizing. M-type currents reflected a mixed F and S phenotype. Finally, some neurones did not respond to ATP at the concentration tested and were termed NR for ‘non-responding’. All recordings were made at a holding potential (Vh) of −70 mV. B, histogram showing the distribution of capsaicin-sensitive (open bars, n = 76) and capsaicin-insensitive neurones (hatched bars, n = 24) as a function of their somatic diameter. Bin width: 2.5 μm. C, bar graph showing the percentage of neurones sensitive to capsaicin as a function of the type of ATP response recorded. Note that neurones displaying F-type responses had a greater tendency to include a larger proportion of capsaicin-sensitive cells than neurones which did not respond to ATP. However, statistical analysis (χ2 test) showed that there was no correlation between the type of ATP response and the presence of a response to capsaicin.
In 12 out 12 neurones tested, ATP responses (regardless of their type) were reversibly inhibited by more than 90 % by P2X receptor antagonists. We tested three different antagonists and their mean inhibitory effects were as follows: suramin (30 μM) −95 ± 2 % (n = 6), PPADS (50 μM) −92 ± 3 % (n = 3) and reactive blue (30 μM) −92 ± 3 % (n = 3).
To summarize, our results indicate that a majority (93 %) of neonatal DRG neurones freshly dissociated or maintained in short term culture (< 2 days) expressed functional P2X receptors. Among the responsive neurones, 77 % displayed a fast desensitizing phase (F neurones + M neurones) and 61 % displayed a slow plateau-like phase (S neurones + M neurones) during a 3 s lasting application of ATP (1 μM).
Distribution of F-, S- and M-type ATP responses among the DRG neuronal population in relation to somatic diameter and capsaicin sensitivity
Somatic diameter
The mean somatic diameters of each type of ATP responsive neurones or for non-responding neurones were as follows: NR 18.6 ± 1.0 μm (n = 14); S-type 20.4 ± 0.8 μm (n = 44); F-type 21.1 ± 0.6 μm (n = 74); M-type 23.1 ± 0.7 μm (n = 71). There was no statistically significant difference (Kruskall-Wallis one-way ANOVA on ranks followed by Dunn's test) in the mean somatic diameters of neurones displaying F-type responses or NR neurones (P > 0.05). However, the somatic diameter of neurones displaying M-type ATP responses was significantly larger than that of NR neurones and neurones with S-type responses. It is also interesting to note that the non-responding (NR) neurones were among the neurones with the smallest somatic diameters.
Capsaicin responsiveness
We checked for the presence of an inward current during the local application of capsaicin (1 μM) in the different categories of DRG neurones presented above. Among a total population of 99 neurones tested, 76 responded to capsaicin. Thus 77 % of the neurones possessed functional vanilloid type 1 (VR1/TRPV1) receptors. Figure 1B illustrates the general distribution of somatic diameters of capsaicin-sensitive and capsaicin-insensitive neurones. There was no significant difference between the two populations (Kolmogorov-Smirnov test, P = 0.172). The mean diameter of capsaicin-sensitive neurones was 20.2 ± 0.5 μm (n = 76) and that of capsaicin-insensitive neurones was 22.9 ± 1.5 μm (n = 23). There was no relationship between capsaicin sensitivity and type of ATP response since, within a given ATP response category, we systematically found both capsaicin-sensitive and capsaicin-insensitive neurones (Fig. 1C). Although the NR neurones tended to include less capsaicin-sensitive neurones than that displaying F-type responses, statistical analysis (χ2 test) showed no relationship between capsaicin sensitivity and a given ATP response type or between capsaicin sensitivity and absence of ATP response.
Effect of DHEA on P2X receptor-mediated ATP responses
Dehydroepiandrosterone (DHEA) applied alone at concentrations up to 100 μM never elicited any detectable membrane current at a Vh of −70 mV (n = 49). In contrast, DHEA modulated P2X receptor-mediated responses induced by local application of ATP. The effect of DHEA was fast in onset and completely reversible. The observed modulation of P2X currents was similar during acute coapplication of ATP and DHEA and steady state superfusion (bath application) of DHEA (data not shown). All results presented below have been obtained by locally coapplying both substances at the concentrations indicated.
At a concentration of 10 μM, DHEA modulated the response to ATP in 72 % (45 out of 64) of the neurones tested but there was a difference in the sign of the effect on the transient versus the sustained components of the inward current induced by ATP. Figure 2 illustrates the effect of 10 μM DHEA on F-, S- and M-type responses induced by 1 μM ATP. DHEA reduced the peak amplitude of F-type responses by 35 ± 8 % in 56 % (10 out of 18) of the neurones tested (Fig. 2A) and did not affect the current in the remaining 44 % of neurones. Moreover, in 50 % (n = 10) of the neurones in which DHEA inhibited the amplitude of the F-type response, it also induced the appearance of a plateau-like phase, thereby transforming the F-type response into an M-type response. Among the neurones in which the F-response was inhibited by DHEA, 80 % (n = 10) responded to capsaicin with an inward current. By contrast, under the same experimental conditions, DHEA potentiated S-type responses by 83 ± 19 % in 67 % (8 out of 12) of the neurones tested (Fig. 2B) and did not modify the amplitude of the response in the remaining 33 % of neurones. We tested the effect of capsaicin in four neurones in which DHEA potentiated the S-type response. Three out of the four neurones tested (75 %) possessed functional capsaicin receptors. In the case of M-type responses, DHEA inhibited the transient peak response while it potentiated the plateau phase. The potentiating effect of DHEA on the plateau phase was observed in 77 % (20 out of 26) of the neurones tested. In the remaining 23 % of neurones, the plateau phase was not modified by DHEA. The mean increase in the plateau amplitude was of 70 ± 18 % (n = 20) and among the neurones showing a potentiation of the plateau phase by DHEA, 77 % displayed also an inward current in response to capsaicin application. The effect of DHEA on the initial transient phase of M-type responses was difficult to quantify because of the large variability of the peak amplitude even under control conditions. Nevertheless, in six neurones displaying M-type responses with a reproducible and stable transient phase, we observed that DHEA inhibited the amplitude of the transient current component in three neurones (50 %) by 46 ± 18 % and had no effect on the transient component in the remaining neurones. It must also be emphasized that, concerning the modulation of M-type responses by DHEA, all combinations of effects were observed among the neurones from which we recorded, i.e. inhibition of the peak alone, potentiation of the plateau alone or inhibition of the peak and potentiation of the plateau phase. This observation suggested that the effects of DHEA on the two phases were independent.
Figure 2. Modulatory effect of dehydroepiandrosterone (DHEA) on F-type, S-type and M-type ATP responses.
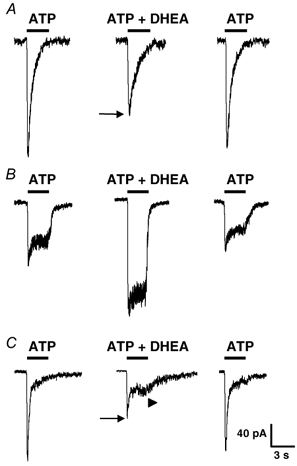
P2X receptor-mediated responses were elicited by local applications of ATP (1 μM) and when DHEA (10 μM) was coapplied with ATP. A, application of ATP elicited an F-type response which was reversibly inhibited (black arrow) by DHEA. B, in a neurone displaying a slowly desensitizing (S-type) current during application of ATP, DHEA potentiated markedly and reversibly the amplitude of the P2X receptor-mediated response. C, in this neurone ATP induced a mixed (M-type) response consisting of an initial transient and fast component followed by a slower plateau phase. When DHEA was coapplied with ATP, the amplitude of the transient initial peak current was inhibited (black arrow) while the amplitude of the plateau phase was increased (arrowhead). This effect of DHEA was fully reversible. Vh = −70 mV. Horizontal black bars indicate duration of application of substances.
Taken together, our results indicate that DHEA (10 μM) apparently inhibited P2X receptors underlying F-type responses, whereas it potentiated the activity of P2X receptors responsible for the S-type responses. These modulatory effects of DHEA were similar on the transient and plateau phases of M-type responses, respectively. Since the potentiating effect of DHEA on slowly desensitizing currents was the most frequent and reproducible, we decided to investigate this effect of DHEA in more detail.
Effect of DHEA on submaximal versus saturating ATP responses
Figure 3A illustrates the effect of DHEA on P2X receptor-mediated responses induced by the application of a submaximally effective concentration of ATP (1 μM) and by a saturating ATP concentration (100 μM or 1 mM) in the same neurone. In five neurones, DHEA (50 μM) potentiated the response to 1 μM ATP by 112 ± 29 %, while it had no significant effect on the amplitude of the response induced by ATP at concentrations of 100 μM or 1 mM (2 ± 1 %, n = 5). This result indicated that DHEA probably increased the potency rather than the efficacy of ATP at the P2X receptors modulated. Complete concentration-response curves to ATP and their displacement by DHEA were not constructed due to the heterogeneity and the variable contribution of different P2X receptors subtypes to the sustained plateau-like current of ATP responses (see below).
Figure 3. DHEA potentiates submaximal but not saturating responses to ATP.
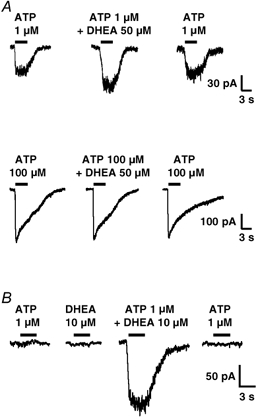
A, the effect of DHEA (50 μM) was tested on the responses induced by a low (submaximal) concentration of ATP (1 μM, top traces) and by a maximally effective concentration of ATP (100 μM, bottom traces) in the same neurone. DHEA (50 μM) reversibly potentiated the responses elicited by 1 μM ATP but had no effect on the amplitude of those triggered by 100 μM ATP, suggesting that DHEA probably increased the potency but not the efficacy of ATP on P2X receptors. B, in the neurone illustrated, neither ATP (1 μM) nor DHEA (10 μM) elicited a detectable membrane current when applied alone. However, when coapplied, they induced a clear inward current associated with an increase in membrane noise. This effect was perfectly reversible and indicated that DHEA probably increased the affinity of certain subtypes of P2X receptors which were not significantly activated by a low concentration of ATP (1 μM) under control conditions (i.e. in the absence of DHEA). Vh = −70 mV.
An additional interesting observation came from neurones which did not respond to ATP at a concentration of 1 μM and which were therefore classified as non-responsive (NR, see above). As illustrated in Fig. 3B, in NR neurones, neither ATP (1 μM) nor DHEA (10 μM) applied alone triggered a detectable membrane current. However, when both were coapplied they induced an inward current associated with an increase in membrane noise. This phenomenon was perfectly reversible. In a total of eight NR neurones tested, all of which were capsaicin sensitive, a current appeared during the coapplication of DHEA (10 μM) and ATP (1 μM) in seven cases (88 %). The mean amplitude of this current was of −36 ± 17 pA at a Vh of −70 mV. This observation suggested that these neurones expressed functional P2X receptors, but that, under control conditions, the concentration of ATP used (1 μM) was subthreshold for their activation. However, in the presence of DHEA the affinity of these receptors might have been increased leading to a detectable inward current during the coapplication of DHEA and ATP. This result was consistent with the observation that DHEA potentiated submaximal but not maximal P2X responses and suggested that DHEA was probably increasing the affinity of P2X receptors for their agonist (see above and Fig. 3A).
DHEA modulates P2X receptors in a concentration-dependent manner
We next determined the effect of various concentrations of DHEA (1 nM to 100 μM) on the amplitude of S-type responses or of the plateau phase of M-type responses induced by the application of ATP (1 μM). Concentrations of DHEA greater than 100 μM could not be tested due to poor solubility of DHEA at such concentrations. The results are presented in Fig. 4. The threshold for the DHEA-induced potentiation of ATP plateau currents was between 1 nM and 10 nM. There was a clear concentration-dependent effect of DHEA, but the apparently multiphasic aspect of the concentration-response curve (Fig. 4B) suggested that this concentration dependence was not simple. This phenomenon could reflect a complex mode of interaction of DHEA with P2X channels or the differential and/or selective modulation of multiple components of the current response constituting the plateau phase of the response. The latter might indeed implicate different combinations of homo- and/or heteromeric P2X receptors (see below). We therefore decided to characterize in more detail the subtypes of P2X receptors contributing to the slowly desensitizing (plateau) component of P2X responses.
Figure 4. The potentiating effect of DHEA on the slow component of ATP responses is concentration dependent.
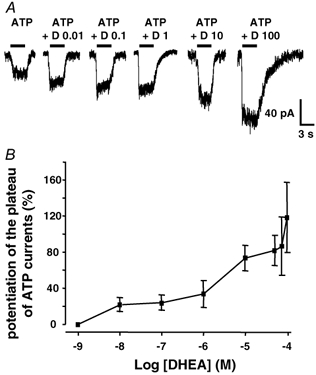
A, ATP was applied at a concentration of 1 μM and induced a non-desensitizing (S-type) response. When DHEA (D) was coapplied at increasing concentrations (expressed in μM) with ATP (1 μM), a concentration-dependent increase in the amplitude of the ATP-induced current was observed. B, summary of the concentration dependence of the effect of DHEA on P2X receptor-mediated inward currents. The effects of various concentrations of DHEA (10−9-10−4 M) on the amplitude of S-type or on the plateau phase of M-type responses triggered by 1 μM ATP were determined. Each point represents the mean ± s.e.m. and was obtained from at least 4 different neurones (range 4-38). The threshold of the DHEA effect was between 1 and 10 nM. Although, there was a clear concentration dependence of the DHEA effect, the complex shape of concentration-response relationship suggested a selective and/or differential modulation of P2X receptor subtypes underlying the macroscopic ATP-induced current. Vh = −70 mV.
Characterization of the subtypes of P2X receptors modulated by DHEA
Since DRG neurones can express six different P2X subunits (Collo et al. 1996), several homo- and/or heteromeric receptor combinations are likely to occur (Torres et al. 1999) and to coexist in a given neurone. Moreover, the relative proportion of each receptor type might vary from cell to cell. At present, there are no P2X subunit-specific antagonists or agonists available, a situation which seriously limits the identification of native homomeric and heteromeric P2X receptors (Ralevic & Burnstock, 1998; Burnstock, 2000; North & Surprenant, 2000; Dunn et al. 2001; Khakh et al. 2001; North, 2002). The only way to tentatively approach the nature of P2X receptors involved in a given response is to combine several arguments which include: (i) functional properties (degree of desensitization), (ii) sensitivity to the agonist αβme-ATP and (iii) effects of allosteric modulators such as protons which specifically potentiate P2X2-containing receptors (Li et al. 1996a; Nakazawa et al. 1997; Stoop et al. 1997; Burnstock, 2000), or IVM which potentiates P2X4 and P2X4/6 receptors (Khakh et al. 1999).
Neuronal responses to αβme-ATP
αβMe-ATP is an agonist which, at concentrations below 10 μM, significantly activates homo- or heteromeric receptors including P2X1 or P2X3 subunits, but only weakly activates P2X4/6 receptors (North & Surprenant, 2000; Khakh et al. 2001). Moreover, the low concentration of αβme-ATP used here was unlikely to activate rat P2X2 receptors at pH 7.3 (Spelta et al. 2002). Local application of αβme-ATP (1-5 μM) triggered membrane currents comparable to those induced by ATP. As with ATP, the responses to αβme-ATP could be classified as F-, S- or M-type, and non-responsive (NR) neurones were also found (Fig. 5A). In an initial phase of our experiments we used αβme-ATP at a concentration of 1 μM. We noticed, however, that under these conditions a relatively high proportion of neurones (18 %, n = 17) apparently did not respond to αβme-ATP. We therefore decided to increase the concentration of αβme-ATP to 5 μM. Under these conditions, 99 % (71 out of 72) of the neurones responded with an inward current. The relative percentage of the different types of current among the responsive neurones (n = 71) was the following: F 17 %; S 43.5 %; and M 39.5 %. As compared to the responses to 1 μM ATP, there was a significant decrease (P < 0.001, χ2 test) in the proportion of F-type currents and a corresponding increase in S-type responses. This can be at least partially ascribed to more pronounced desensitization of homomeric P2X3 receptors (responsible for F-type responses) versus that of heteromeric P2X2/3 receptors (mediating S-type and plateau phases of M-type responses) at the concentration of αβme-ATP used (5 μM). Among the responsive neurones, 56.5 % displayed a fast desensitizing phase (F neurones + M neurones) and 83 % displayed a slow plateau-like phase (S neurones + M neurones) during a 3 s application of αβme-ATP (5 μM). In 30 neurones responding to αβme-ATP, we also applied capsaicin (1 μM), and 87 % of them responded with an inward current.
Figure 5. Modulation of α,β-methylene ATP (αβme-ATP)-activated P2X receptor currents by DHEA, ivermectin and low pH solution.
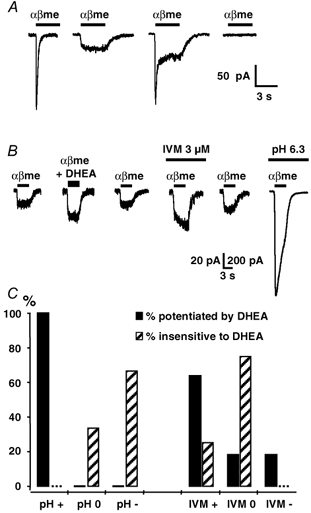
A, local application of 5 μM αβme-ATP (αβme) for 3 s induced membrane currents similar to the F-, S- and M-type currents induced by ATP (see Fig. 1A). B, illustration of the protocol used to identify the subtype of αβme-ATP-sensitive P2X receptor modulated by DHEA. We first checked for the presence (or the absence) of a potentiating effect of DHEA (50 μM) on the inward current induced by 5 μM αβme-ATP (αβme). Then in the same neurone, we determined sequentially the modulatory effect of ivermectin (IVM, 3 μM) and of an external solution at pH 6.3. The example illustrated shows a reversible potentiation of the αβme-ATP response by DHEA and IVM. Moreover, this neurone expressed αβme-ATP-sensitive P2X receptors which were strongly potentiated under low pH conditions (note the difference in calibration bar values: vertical calibration bar of 20 pA applies to the five traces on the left, whereas 200 pA applies to the last trace on the right, i.e. recorded under low pH conditions). C, correlation between sensitivity to DHEA and sensitivity to low pH (left panel) and ivermectin (IVM, right panel). Filled bars: αβme-ATP responses potentiated by DHEA; hatched bars: responses insensitive to DHEA. For each category (sensitive or insensitive to DHEA) we determined the percentage of αβme-ATP responses which were potentiated by (+), insensitive to (0) or inhibited by (−) the low pH solution or by IVM. Vh = −70 mV, n = 15.
Modulation of αβme-ATP responses by DHEA
DHEA (10 μM) modulated αβme-ATP responses in 50 % (11 out of 22) of the cases. Most importantly we did not observe any significant modulation for concentrations of DHEA below 10 μM, indicating that the threshold concentration for the modulation of αβme-ATP responses was between 1 and 10 μM. This observation was in contrast with the results obtained with ATP as an agonist, where the threshold concentration for the modulatory effect of DHEA was around 10 nM (see above and Fig. 4B). DHEA (10 μM) inhibited the peak amplitude of F-type responses by 24 ± 10 % in 83 % (5 out of 6) of the cases and potentiated S-type responses by 36 ± 4 % in 50 % (7 out of 14) of the neurones tested. In the remaining neurones, F- and S-type responses were not affected by DHEA. In the case of M-type responses, the peak current was inhibited by 20 ± 1 % and the plateau phase was potentiated by 36 ± 8 % in all neurones tested (n = 3). Moreover, DHEA (10 μM) induced the appearance of a plateau phase (mean amplitude: −27 ± 12 pA) in 50 % (4 out of 8) of the neurones displaying initially an F-type response.
When coapplied with αβme-ATP (5 μM), DHEA at concentrations of 10, 50 or 100 μM increased the amplitude of S-type responses or of the plateau phase of M-type responses by 36 ± 4 % (n = 7), 124 ± 35 % (n = 12) and 306 ± 164 % (n = 4), respectively, indicating that the effect of DHEA was apparently concentration dependent.
Identification of the P2X receptor subtypes activated by αβme-ATP and modulated by DHEA
As indicated above, at low concentrations αβme-ATP activates preferentially homo- and heteromeric P2X receptors which include P2X1 or P2X3 subunits, as well as heteromeric P2X4/6 receptors. In order to discriminate between the P2X receptor subtypes activated by αβme-ATP (5 μM), we tested, in the same neurones, the effect of two allosteric modulators on the αβme-ATP responses. The allosteric modulators used were (i) IVM, which potentiates P2X4 and P2X4/6 receptors (Khakh et al. 1999), and (ii) protons, which induce a strong potentiation of homomeric P2X2 and heteromeric P2X2-containing receptors (Burnstock, 2000; North & Surprenant, 2000; Khakh et al. 2001). The involvement of homomeric P2X4 receptors could already be excluded in our preparation since αβme-ATP does not activate these receptors at low concentration (Collo et al. 1996; Le et al. 1998). Moreover, the P2X responses recorded during our experiments were largely inhibited by PPADS and suramin, whereas homomeric P2X4 receptors are not inhibited by these antagonists (Collo et al. 1996; Le et al. 1998).
IVM (3 μM) potentiated αβme-ATP responses in 54 % of the cases, inhibited the response in 33 % of the cases and had no effect in 13 % of the cells (n = 15), suggesting that 54 % of the neurones might eventually express functional P2X4/6 receptors contributing at least to a fraction of the current induced by αβme-ATP (5 μM). Under low pH conditions (pH 6.3) we observed a potentiation of the αβme-ATP response in 77 % of the cases, an inhibition in 15 % of the cases and no effect in 8 % of the neurones (n = 13). Since αβme-ATP does not activate homomeric P2X2 receptors at the low concentration used here (Spelta et al. 2002), this result indicated that a majority of neurones expressed heteromeric P2X receptors containing the P2X2 subunit, which were most likely to be P2X2/3 receptors (Burnstock, 2000; North & Surprenant, 2000; Chizh & Illes, 2001; Khakh et al. 2001). Moreover, in 73 % of the neurones in which P2X responses were potentiated by low external pH (6.3) we also found a potentiation by IVM, indicating a high degree of colocalization of P2X2-containing receptors and of putative P2X4/6 receptors.
We next tried to correlate the sensitivity of the αβme-ATP response to IVM and low pH with the potentiating effect of DHEA (50 μM) in the same neurones. The protocol used and a typical result are illustrated in Fig. 5B. We first determined the presence or absence of a modulatory effect of DHEA on the αβme-ATP response (plateau phase). Then we determined the sensitivity of the αβme-ATP-induced current to IVM (3 μM) and low pH (6.3). With respect to the effect of DHEA, we considered two situations, i.e. ‘potentiated by DHEA’ or ‘insensitive to DHEA’. Concerning the sensitivity of αβme-ATP responses to IVM or low pH, we distinguished between ‘potentiation’, ‘no effect’ or ‘inhibition’. Figure 5C summarizes the results we obtained. All αβme-ATP responses potentiated by DHEA (100 %, n = 11) were also potentiated by low pH, whereas responses which were unaffected by DHEA were either insensitive (33 %, n = 3) or inhibited (67 %, n = 3) by low pH. Concerning IVM, 64 % (n = 11) of the αβme-ATP currents modulated by DHEA were potentiated by IVM (mean potentiation: 88 ± 14 %, n = 7), but interestingly 18 % (n = 2) were insensitive and 18 % were inhibited (n = 2, inhibition: 93 % and 100 %). Moreover, the αβme-ATP responses which were not modulated by DHEA were either potentiated by IVM (25 %) or insensitive to IVM (75 %). Thus, there was a strict correlation between the presence of a modulatory effect of DHEA and the potentiation by the low pH solution of the αβme-ATP responses. This indicated that DHEA most probably interacted with a receptor containing the P2X2 subunit.
Effect of DHEA on putative native homomeric P2X2 receptors
We next wondered whether it would be possible to detect the presence of putative homomeric P2X2 receptors in DRG neurones and to check for their eventual modulation by DHEA. In these experiments we used ATP as the agonist, and we recorded the responses to ATP in the presence of 2′-(or 3′)-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) which, at a concentration of 1 μM, completely blocks homomeric and heteromeric receptors containing P2X1 and P2X3 subunits, but only partly blocks (≈50 %) homomeric P2X2 receptors (Thomas et al. 1998; Virginio et al. 1998). TNP-ATP (1 μM) inhibited the amplitude of ATP (1 μM) responses by 86 ± 7 % (n = 8) (Fig. 6A) and that of αβme-ATP (5 μM) responses by 90 ± 3 % (n = 10). When ATP was applied at a concentration of 20 μM in the continuous presence of TNP-ATP (1 μM), we recorded a slow non-desensitizing inward current (Fig. 6A and B). At a concentration of 50 μM, DHEA potentiated this current by 165 ± 72 % in three out of five neurones tested. All neurones potentiated by DHEA (50 μM) were also potentiated by the low pH solution, whereas the ATP responses which were not modulated by DHEA were insensitive to low pH. These results suggested that DHEA probably also modulated homomeric P2X2 receptors. Moreover, the effect of DHEA was concentration dependent with a threshold between 10 nM and 100 nM (Fig. 6B and C).
Figure 6. Effect of DHEA on putative native homomeric P2X2 receptors.
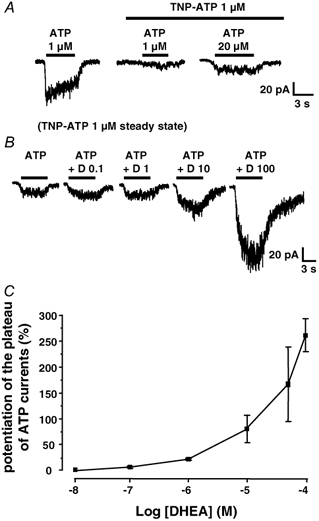
A, in a neurone displaying an S-type response to 1 μM ATP, TNP-ATP (1 μM) almost completely blocked the response to ATP (middle trace). However, in the continuous presence of TNP-ATP (1 μM) a small non-desensitizing current appeared when the concentration of ATP was increased to 20 μM, a phenomenon which probably reflected the activation of homomeric P2X2 receptors. B, the membrane current induced by ATP (20 μM) in the continuous presence of TNP-ATP (1 μM) was potentiated by DHEA (D) at concentrations between 0.1 and 100 μM. C, concentration-response relationship of the potentiating effect of DHEA on the current induced by ATP (20 μM) in the steady presence of TNP-ATP (1 μM). Each point represents the mean ± s.e.m. and was obtained from at least 3 different neurones. The threshold of the DHEA effect was between 10 and 100 nM.
DISCUSSION
Our results indicate that the neurosteroid DHEA selectively potentiates slowly inactivating P2X receptor-mediated responses in neonatal DRG neurones. This modulatory effect seems to be specific for P2X2-containing homomeric and heteromeric receptors and most probably involves an increase in affinity for the agonist.
Diversity of P2X receptor-mediated responses in neonatal rat DRG neurones
Application of low concentrations of ATP (1 μM) or αβme-ATP (5 μM) induced rapid inward currents in the majority (≥ 93 %) of the neurones from which we recorded. Three types of responses were distinguished on the basis of their kinetic properties: fast desensitizing and transient (F-type), slowly desensitizing (S-type) and mixed (M-type) responses displaying a fast peak followed by a steady plateau phase. These responses were due to the activation of ionotropic P2X receptors since they were blocked by the P2X receptor antagonists suramin, PPADS and reactive blue 2 (Ralevic & Burnstock, 1998; North & Surprenant, 2000; Khakh et al. 2001). Previous studies on embryonic, neonatal and adult DRG neurones have provided convincing evidence that F-type responses were due to homomeric P2X3 receptors, that S-type currents involved mainly heteromeric P2X2/3 receptors and that M-type responses represented the sum of F- and S-type responses (Robertson et al. 1996; Rae et al. 1998; Burgard et al. 1999; Grubb & Evans, 1999; Li et al. 1999; Ueno et al. 1999; Cockayne et al. 2000; Labrakakis et al. 2000; Souslova et al. 2000; Pankratov et al. 2001; Zhong et al. 2001). Our results are consistent with these conclusions and in addition we show that a large fraction of DRG neurones (47 %) possess receptors which are activated by low concentrations of αβme-ATP and potentiated by IVM and could therefore represent native P2X4/6 receptors (Le et al. 1998; Khakh et al. 1999). This current component contributed to the slow plateau phase of S- and M-type P2X responses and was present in 73 % of the neurones expressing also P2X2-containing receptors.
Modulation of P2X receptors by DHEA
DHEA concentration-dependently increased the amplitude of the plateau phases of S- and M-type responses, suggesting that DHEA could possibly modulate P2X2, P2X2/3 and/or P2X4/6 receptors. Interestingly, DHEA (10 μM) potentiated only a subpopulation of S-type (67 %) and M-type (77 %) responses confirming the heterogeneity of these responses and indicating that DHEA, which is a lipophilic substance, did not exert its effect via a non-specific interaction with the membrane lipid bilayer. The effect of DHEA was rapid in onset and fully reversible during local coapplication with ATP or αβme-ATP, suggesting a direct and fast interaction of DHEA with P2X receptors. The simplest explanation of these observations would be that DHEA directly interacts with P2X receptors to modulate their functioning. DHEA also potentiated plateau responses to ATP when neurones were recorded in the whole-cell configuration and, when applied alone, DHEA never induced any membrane current or any increase in intracellular free calcium concentration (M. de Roo, J. L. Rodeau & R. Schlichter, unpublished observation), suggesting that the fast effect of DHEA on P2X responses was probably mainly independent of intracellular second messenger systems and that a specific membrane receptor for DHEA was probably not involved. However, it remains possible that the effect of DHEA might be influenced by the phosphorylation status of the modulated P2X receptor as it has been shown for the neurosteroid allopregnanolone on GABAA receptors (Koksma et al. 2003). We rather favour the hypothesis of an allosteric modulation of P2X receptors by DHEA since: (i) DHEA potentiated P2X responses induced by low but not by saturating concentrations of ATP and (ii) DHEA, when coapplied with ATP, led to the appearance of an inward current in a large fraction (88 %) of neurones which did not display any response to ATP or DHEA applied alone. The fact that the amplitude of P2X responses induced by a saturating concentration of ATP was not augmented suggested that DHEA increased the potency but not the efficacy of P2X receptor agonists. We also noticed that, at a concentration of 10 μM, DHEA inhibited F-type (P2X3) responses, an effect which was opposite to that on the plateau phases of S- and M-type responses. Unfortunately, due to the difficulty in obtaining stable and reproducible F-type responses upon repeated applications over a long time period, it was impossible to further study the mechanisms underlying this modulation.
DHEA modulates P2X receptors containing the P2X2 subunit
We observed a strict and absolute correlation between the potentiation of the P2X response by DHEA and by an acidification (pH 6.3) of the external medium. Such a facilitatory effect of acidic pH reveals the presence of a P2X2 subunit in the native receptor (Burnstock, 2000; North & Surprenant, 2000; Khakh et al. 2001). By contrast, there was no systematic coexistence of the potentiating effects of DHEA and IVM, suggesting that putative P2X4/6 receptors were not modulated by DHEA. DHEA potentiated responses to αβme-ATP most probably mediated by heteromeric P2X2/3 receptors, as well as responses to ATP in the presence of TNP-ATP (1 μM), a condition which blocks all P2X1- and P2X3-containing receptors (Thomas et al. 1998; Virginio et al. 1998; North & Surprenant, 2000; Khakh et al. 2001) and about half of homomeric P2X2 receptors (Virginio et al. 1998). The latter observations suggested that DHEA also potentiated homomeric P2X2 receptors and/or other combinations of P2X subunits containing P2X2 but excluding P2X3. It is interesting to note that, at a concentration of 10 μM, DHEA produced a similar degree of potentiation of the P2X responses induced by ATP (20 μM) in the presence of TNP-ATP (1 μM) or by ATP (1 μM) in the absence of TNP-ATP (compare Fig. 4 and Fig. 6). However, at higher DHEA concentrations (50 and 100 μM) the degree of potentiation was larger for the responses evoked by ATP (20 μM) in the presence of TNP-ATP (1 μM). This observation could indicate that, at high concentrations, DHEA might tend to relieve the TNP-ATP-induced block of certain P2X receptors. This point will have to be investigated more carefully, for example on homogeneous populations of recombinant P2X2 and/or P2X2/3 receptors.
The relationship between the concentration of DHEA and the potentiation of the ATP response was complicated and appeared to be multiphasic, although the multiphasic character could not be clearly established here. In particular, we noticed that the threshold for the effect of DHEA on the responses evoked by ATP in the absence or the presence of TNP-ATP was around 10 nM, whereas that on responses evoked by αβme-ATP was between 1 and 10 μM, suggesting that homomeric P2X2 or heteromeric P2X2-containing receptors which do not include the P2X3 subunit were more sensitive to the modulatory action of DHEA than P2X2/3 receptors. This observation may in part explain the complex aspect of the concentration-response curve to DHEA (Fig. 4B).
Our results are the first to describe a potentiation of P2X receptors by DHEA. However, it has been shown previously that the sulphated derivative of DHEA (DHEA-S) inhibited the increase in intracellular free calcium concentration induced by P2X receptor stimulation in PC12 cells (Liu et al. 2001). This effect was not reproduced by DHEA and therefore probably represents a different mode of modulation of P2X receptor signalling than that we describe in our study. It nevertheless indicates that P2X receptors and/or P2X receptor-dependent signalling pathways might represent important targets for the modulatory actions of neurosteroids.
Physiological significance
Dehydroepiandrosterone (DHEA) is the most abundant neurosteroid in the central nervous system (Baulieu, 1997; Compagnone & Mellon, 2000) and is also secreted as a hormone (or rather a precursor of different steroid hormones) in the bloodstream following adrenocorticotrophin stimulation of the adrenal cortex (Kroboth et al. 1999). For several years neurosteroids have been known to interact with certain ligand-gated neurotransmitter receptors to exert fast allosteric modulations of these receptors (Baulieu, 1997; Rupprecht & Holsboer, 1999; Compagnone & Mellon, 2000). For example, tetrahydroprogesterone (allopregnanolone) or tetrahydrodeoxycorticosterone (THDOC) are very potent positive allosteric modulators of GABAA receptors, whereas DHEA and its sulphated derivative (DHEA-S) potentiate the activity of N-methyl-D-aspartate (NMDA)-type glutamate receptors (Baulieu, 1997; Rupprecht & Holsboer, 1999; Compagnone & Mellon, 2000). Here we show that ionotropic ATP receptors containing the P2X2 subunit can also be rapidly and positively modulated by DHEA. This effect is observed for low concentrations of DHEA (≈10 nM) which are likely to be reached locally under physiological conditions (Baulieu, 1997; Rupprecht & Holsboer, 1999). ATP and P2X receptors are modulators of the excitability of unmyelinated sensory nerve fibres and are involved in the communication between these fibres and Schwann cells (Irnich et al. 2001). If these receptors included the P2X2 subunit, elevation of circulating levels or increase in local production of DHEA would be likely to modulate the excitability of the axons of nociceptors and the communication between axons and glial cells. P2X2 subunits are present both at peripheral and central terminals of primary sensory afferent neurones and in particular on nociceptors (Vulchanova et al. 1997, 1998; Tsuda et al. 2000). A potentiation of P2X2-containing receptors by DHEA could therefore lead to a sensitization and/or an increased activation of nociceptors by ATP and/or to a facilitation of glutamate release from sensory neurones in the dorsal horn (DH) of the spinal cord. Interestingly, P2X2 subunits have been localized to the terminals of primary afferent nociceptors in laminae I-II of the DH where they rarely coexist with P2X3 subunits (Vulchanova et al. 1997). The P2X receptors expressed by these primary nociceptor terminals would therefore be very sensitive to the modulatory effect of DHEA. Such P2X2-containing receptors are also present on a subpopulation of laminae I-III interneurones (Jo & Schlichter, 1999; Hugel & Schlichter, 2000) and their modulation by DHEA may change the integration of nociceptive messages in the DH. It is known that the production of neurosteroids or of neuroactive steroids can be regulated (Mensah-Nyagan et al. 1999) but the exact conditions under which such a regulation could occur in the nociceptive system remains to be determined. P2X receptors seem to be of minor importance in acute nociception, but play an important role in the processing of nociceptive information during peripheral inflammation (Hamilton et al. 1999; Stanfa et al. 2000; Chizh & Illes, 2001; Hamilton et al. 2001). It is therefore tempting to speculate that neurosteroids such as DHEA may contribute, at least in part, to the recruitment of P2X receptor signalling during inflammatory pain.
In conclusion, our results show, for the first time, that P2X receptors containing P2X2 subunits are selectively potentiated by DHEA, which is the most abundant neurosteroid in the nervous system. This indicates the possibility of an endogenous modulation by DHEA of P2X receptor-mediated signalling in both the peripheral and central nervous system where P2X2-containing receptors are prominent. In addition, DHEA might prove a useful tool to identify and study the properties of native P2X receptors expressed by central and peripheral neurones.
Acknowledgments
We thank Mrs Catherine Moreau and Mrs Francine Herzog for excellent technical assistance. This work was supported by grants from the Institut UPSA de la Douleur and the Institut Universitaire de France. We acknowledge additional support from Université Louis Pasteur (Strasbourg) and Centre National de la Recherche Scientifique (CNRS, France).
REFERENCES
- Bardoni R, Goldstein PA, Lee CJ, Gu JG, MacDermott AB. ATP P2X receptors mediate fast synaptic transmission in the dorsal horn of the rat spinal cord. J Neurosci. 1997;17:5297–5304. doi: 10.1523/JNEUROSCI.17-14-05297.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulieu EE. Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res. 1997;52:1–32. [PubMed] [Google Scholar]
- Boue-Grabot E, Archambault V, Seguela P. A protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2X(2) ATP-gated channels. J Biol Chem. 2000;275:10190–10195. doi: 10.1074/jbc.275.14.10190. [DOI] [PubMed] [Google Scholar]
- Bowie D, Feltz P, Schlichter R. Subpopulations of neonatal rat sensory neurons express functional neurotransmitter receptors which elevate intracellular calcium. Neuroscience. 1994;58:141–149. doi: 10.1016/0306-4522(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Burgard EC, Niforatos W, van Biesen T, Lynch KJ, Touma E, Metzger RE, Kowaluk EA, Jarvis MF. P2X receptor-mediated ionic currents in dorsal root ganglion neurons. J Neurophysiol. 1999;82:1590–1598. doi: 10.1152/jn.1999.82.3.1590. [DOI] [PubMed] [Google Scholar]
- Burnstock G. P2X receptors in sensory neurones. Br J Anaesth. 2000;84:476–488. doi: 10.1093/oxfordjournals.bja.a013473. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Illes P. P2X receptors and nociception. Pharmacol Rev. 2001;53:553–568. [PubMed] [Google Scholar]
- Clyne JD, Brown TC, Hume RI. Expression level dependent changes in the properties of P2X(2) receptors. Neuropharmacology. 2003;44:403–412. doi: 10.1016/s0028-3908(02)00406-9. [DOI] [PubMed] [Google Scholar]
- Cockayne DA, Hamilton SG, Zhu QM, Dunn PM, Zhong Y, Novakovic S, Malmberg AB, Cain G, Berson A, Kassotakis L, Hedley L, Lachnit WG, Burnstock G, McMahon SB, Ford AP. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. doi: 10.1038/35039519. [DOI] [PubMed] [Google Scholar]
- Collo G, North RA, Kawashima E, Merlo-Pich E, Neidhart S, Surprenant A, Buell G. Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnone NA, Mellon SH. Neurosteroids: biosynthesis and function of these novel neuromodulators. Front Neuroendocrinol. 2000;21:1–56. doi: 10.1006/frne.1999.0188. [DOI] [PubMed] [Google Scholar]
- Cook SP, McCleskey EW. Desensitization, recovery and Ca(2+)-dependent modulation of ATP-gated P2X receptors in nociceptors. Neuropharmacology. 1997;36:1303–1308. doi: 10.1016/s0028-3908(97)00132-9. [DOI] [PubMed] [Google Scholar]
- Dunn PM, Zhong Y, Burnstock G. P2X receptors in peripheral neurons. Prog Neurobiol. 2001;65:107–134. doi: 10.1016/s0301-0082(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E. A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Villafuertes R, Pintor J, Gualix J, Miras-Portugal MT. GABAB receptor-mediated presynaptic potentiation of ATP ionotropic receptors in rat midbrain synaptosomes. Neuropharmacology. 2003;44:311–323. doi: 10.1016/s0028-3908(02)00379-9. [DOI] [PubMed] [Google Scholar]
- Grubb BD, Evans RJ. Characterization of cultured dorsal root ganglion neuron P2X receptors. Eur J Neurosci. 1999;11:149–154. doi: 10.1046/j.1460-9568.1999.00426.x. [DOI] [PubMed] [Google Scholar]
- Gu JG, MacDermott AB. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- Hamilton SG, McMahon SB, Lewin GR. Selective activation of nociceptors by P2X receptor agonists in normal and inflamed rat skin. J Physiol. 2001;534:437–445. doi: 10.1111/j.1469-7793.2001.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SG, Wade A, McMahon SB. The effects of inflammation and inflammatory mediators on nociceptive behaviour induced by ATP analogues in the rat. Br J Pharmacol. 1999;126:326–332. doi: 10.1038/sj.bjp.0702258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugel S, Schlichter R. Presynaptic P2X receptors facilitate inhibitory GABAergic transmission between cultured rat spinal cord dorsal horn neurons. J Neurosci. 2000;20:2121–2130. doi: 10.1523/JNEUROSCI.20-06-02121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irnich D, Burgstahler R, Bostock H, Grafe P. ATP affects both axons and Schwann cells of unmyelinated C fibres. Pain. 2001;92:343–350. doi: 10.1016/S0304-3959(01)00277-9. [DOI] [PubMed] [Google Scholar]
- Jo YH, Schlichter R. Synaptic corelease of ATP and GABA in cultured spinal neurons. Nat Neurosci. 1999;2:241–245. doi: 10.1038/6344. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Seguela P, Voigt M, Humphrey PP. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev. 2001;53:107–118. [PubMed] [Google Scholar]
- Khakh BS, Proctor WR, Dunwiddie TV, Labarca C, Lester HA. Allosteric control of gating and kinetics at P2X(4) receptor channels. J Neurosci. 1999;19:7289–7299. doi: 10.1523/JNEUROSCI.19-17-07289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, Ziganshina LE, Pintor J, Burnstock G. Full sensitivity of P2X2 purinoceptor to ATP revealed by changing extracellular pH. Br J Pharmacol. 1996;117:1371–1373. doi: 10.1111/j.1476-5381.1996.tb15293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koksma JJ, van Kesteren RE, Rosahl TW, Zwart R, Smit AB, Luddens H, Brussaard AB. Oxytocin regulates neurosteroid modulation of GABA(A) receptors in supraoptic nucleus around parturition. J Neurosci. 2003;23:788–797. doi: 10.1523/JNEUROSCI.23-03-00788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroboth PD, Salek FS, Pittenger AL, Fabian TJ, Frye RF. DHEA and DHEA-S: a review. J Clin Pharmacol. 1999;39:327–348. doi: 10.1177/00912709922007903. [DOI] [PubMed] [Google Scholar]
- Labrakakis C, Gerstner E, MacDermott AB. Adenosine triphosphate-evoked currents in cultured dorsal root ganglion neurons obtained from rat embryos: desensitization kinetics and modulation of glutamate release. Neuroscience. 2000;101:1117–1126. doi: 10.1016/s0306-4522(00)00373-0. [DOI] [PubMed] [Google Scholar]
- Le KT, Babinski K, Seguela P. Central P2X4 and P2X6 channel subunits coassemble into a novel heteromeric ATP receptor. J Neurosci. 1998;18:7152–7159. doi: 10.1523/JNEUROSCI.18-18-07152.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Peoples RW, Lanthorn TH, Li ZW, Weight FF. Distinct ATP-activated currents in different types of neurons dissociated from rat dorsal root ganglion. Neurosci Lett. 1999;263:57–60. doi: 10.1016/s0304-3940(99)00114-7. [DOI] [PubMed] [Google Scholar]
- Li C, Peoples RW, Weight FF. Acid pH augments excitatory action of ATP on a dissociated mammalian sensory neuron. Neuroreport. 1996a;7:2151–2154. doi: 10.1097/00001756-199609020-00018. [DOI] [PubMed] [Google Scholar]
- Li C, Peoples RW, Weight FF. Cu2+ potently enhances ATP-activated current in rat nodose ganglion neurons. Neurosci Lett. 1996b;219:45–48. doi: 10.1016/s0304-3940(96)13186-4. [DOI] [PubMed] [Google Scholar]
- Li C, Peoples RW, Weight FF. Mg2+ inhibition of ATP-activated current in rat nodose ganglion neurons: evidence that Mg2+ decreases the agonist affinity of the receptor. J Neurophysiol. 1997;77:3391–3395. doi: 10.1152/jn.1997.77.6.3391. [DOI] [PubMed] [Google Scholar]
- Li P, Calejesan AA, Zhuo M. ATP P2x receptors and sensory synaptic transmission between primary afferent fibers and spinal dorsal horn neurons in rats. J Neurophysiol. 1998;80:3356–3360. doi: 10.1152/jn.1998.80.6.3356. [DOI] [PubMed] [Google Scholar]
- Liu PS, Hsieh HL, Lin CM. Dehydroepiandrosterone sulfate (DHEAS) suppresses P2X purinoceptor-coupled responses in PC12 cells. Neurochem Int. 2001;39:193–198. doi: 10.1016/s0197-0186(01)00027-4. [DOI] [PubMed] [Google Scholar]
- Mensah-Nyagan AG, Do-Rego JL, Beaujean D, Luu-The V, Pelletier G, Vaudry H. Neurosteroids: expression of steroidogenic enzymes and regulation of steroid biosynthesis in the central nervous system. Pharmacol Rev. 1999;51:63–81. [PubMed] [Google Scholar]
- Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Hess P. Block by calcium of ATP-activated channels in pheochromocytoma cells. J Gen Physiol. 1993;101:377–392. doi: 10.1085/jgp.101.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Liu M, Inoue K, Ohno Y. pH dependence of facilitation by neurotransmitters and divalent cations of P2x2 purinoceptor/channels. Eur J Pharmacol. 1997;337:309–314. doi: 10.1016/s0014-2999(97)01293-4. [DOI] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- Pankratov YV, Lalo UV, Dashkin AN, Krishtal A. Heterogeneity of the functional expression of P2X3 and P2X2/3 receptors in the primary nociceptive neurons of rat. Neurochem Res. 2001;26:993–1000. doi: 10.1023/a:1012344803672. [DOI] [PubMed] [Google Scholar]
- Paukert M, Osteroth R, Geisler HS, Brandle U, Glowatzki E, Ruppersberg JP, Grunder S. Inflammatory mediators potentiate ATP-gated channels through the P2X(3) subunit. J Biol Chem. 2001;276:21077–21082. doi: 10.1074/jbc.M101465200. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Rae MG, Rowan EG, Kennedy C. Pharmacological properties of P2X3-receptors present in neurones of the rat dorsal root ganglia. Br J Pharmacol. 1998;124:176–180. doi: 10.1038/sj.bjp.0701803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Robertson SJ, Rae MG, Rowan EG, Kennedy C. Characterization of a P2X-purinoceptor in cultured neurones of the rat dorsal root ganglia. Br J Pharmacol. 1996;118:951–956. doi: 10.1111/j.1476-5381.1996.tb15491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht R, Holsboer F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999;22:410–416. doi: 10.1016/s0166-2236(99)01399-5. [DOI] [PubMed] [Google Scholar]
- Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa L, Suzuki R, Carpenter K, Dickenson A, Boyce S, Hill R, Nebenuis-Oosthuizen D, Smith AJ, Kidd EJ, Wood JN. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017. doi: 10.1038/35039526. [DOI] [PubMed] [Google Scholar]
- Spelta V, Jiang LH, Surprenant A, North RA. Kinetics of antagonist actions at rat P2X2/3 heteromeric receptors. Br J Pharmacol. 2002;135:1524–1530. doi: 10.1038/sj.bjp.0704591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfa LC, Kontinen VK, Dickenson AH. Effects of spinally administered P2X receptor agonists and antagonists on the responses of dorsal horn neurones recorded in normal, carrageenan-inflamed and neuropathic rats. Br J Pharmacol. 2000;129:351–359. doi: 10.1038/sj.bjp.0703047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop R, Surprenant A, North RA. Different sensitivities to pH of ATP-induced currents at four cloned P2X receptors. J Neurophysiol. 1997;78:1837–1840. doi: 10.1152/jn.1997.78.4.1837. [DOI] [PubMed] [Google Scholar]
- Thomas S, Virginio C, North RA, Surprenant A. The antagonist trinitrophenyl-ATP reveals co-existence of distinct P2X receptor channels in rat nodose neurones. J Physiol. 1998;509:411–417. doi: 10.1111/j.1469-7793.1998.411bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres GE, Egan TM, Voigt MM. Hetero-oligomeric assembly of P2X receptor subunits. Specificities exist with regard to possible partners. J Biol Chem. 1999;274:6653–6659. doi: 10.1074/jbc.274.10.6653. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Koizumi S, Kita A, Shigemoto Y, Ueno S, Inoue K. Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J Neurosci. 2000;20:RC90. doi: 10.1523/JNEUROSCI.20-15-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virginio C, Robertson G, Surprenant A, North RA. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Buell G, Surprenant A, North RA, Elde R. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36:1229–1242. doi: 10.1016/s0028-3908(97)00126-3. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Stone LS, Hargreaves KM, Buell G, Surprenant A, North RA, Elde R. P2X3 is expressed by DRG neurons that terminate in inner lamina II. Eur J Neurosci. 1998;10:3470–3478. doi: 10.1046/j.1460-9568.1998.00355.x. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Dunn PM, Bardini M, Ford AP, Cockayne DA, Burnstock G. Changes in P2X receptor responses of sensory neurons from P2X3-deficient mice. Eur J Neurosci. 2001;14:1784–1792. doi: 10.1046/j.0953-816x.2001.01805.x. [DOI] [PubMed] [Google Scholar]