

Abstract
There is evidence that reactive oxygen species (ROS) are produced and released during neuromuscular activity, but their role in synaptic transmission is not known. Using a two-electrode voltage-clamp technique, at frog neuromuscular junctions, the action H2O2 on end-plate currents (EPC) was studied to determine the targets for this membrane-permeable ROS. In curarized or cut muscles, micromolar concentrations of H2O2 increased the amplitude of EPCs. Higher (> 30 μm) doses inhibited EPCs and prolonged current decay. These effects were presynaptic since H2O2 did not change the amplitude or duration of miniature EPCs (although it reduced the rate of spontaneous release at high concentrations). Quantal analysis and deconvolution methods showed that facilitation of EPCs was due to increased quantal release, while depression was accompanied by temporal dispersion of evoked release. Extracellular recordings revealed prolonged presynaptic Ca2+ entry in the presence of high H2O2. Both low and high H2O2 increased presynaptic potentiation during high-frequency stimulation. Pro-oxidant Fe2+ did not affect facilitation by low doses of H2O2 but augmented the inhibition of EPCs by high H2O2, indicating involvement of hydroxyl radicals. High Mg2+ and the ROS scavenger N-acetylcysteine eliminated both the facilitatory and depressant effects of H2O2. The facilitatory effect of H2O2 was prevented by protein kinase C (PKC) inhibitors and 4β-phorbol 12-myristate, 13-acetate (PMA), an activator of PKC. PKC inhibitors but not PMA also abolished the depressant effect of H2O2. Our data suggest complex presynaptic actions of H2O2, which could serve as a fast feedback modulator of intense neuromuscular transmission.
Reactive oxygen species (ROS) such as superoxide anion (O2−), hydroxyl radical (OH•), and hydrogen peroxide (H2O2) are continuously produced by mitochondrial respiration, the membrane-associated NADPH oxidase complex (Vignais, 2002), or by the action of hormones, neurotransmitters and neurotrophins (Finkel, 1998; Oh et al. 2000).
H2O2 is of particular interest among other ROS because: (a) H2O2 is the most stable and long-lived ROS (Kress et al. 1995; Li & Jackson, 2002); (b) being a mild oxidant, H2O2 application for < 30 min does not produce non-specific peroxidation of membrane lipids (Tretter & Adam-Vizi, 1996); (c) H2O2 passes through cell membranes (Weiss, 1986; Waldron & Rozengurt, 2000); (d) H2O2 is more stable in extracellular than in intracellular space (Murrant & Reid, 2001) and it might serve, therefore, not only as an intracellular but also as an intercellular messenger (Oh et al. 2000; Servitja et al. 2000).
The mechanism of H2O2 action on target cells is not known. Since H2O2 is able to permeate membranes and can modify cysteine, methionine (Knapp & Klann, 2000) and tyrosine groups (Konishi et al. 1997) of proteins, it could modify intracellular enzymes in host or neighbouring cells. For example, PKC, a key enzyme participating in synaptic plasticity and transmitter release, is an important intracellular target for H2O2 (Konishi et al. 1997; Suzuki et al. 1997; Servitja et al. 2000).
The question arises of what role, if any, ROS play in synaptic transmission, since skeletal muscle fibres produce and release superoxide and H2O2 (O'Neill et al. 1996; Hasegawa et al. 1997; Supinski et al. 1999; Reid & Durham, 2002). Recent experiments suggest that ROS influence muscle fatigue (Hasegawa et al. 1997; Kolbeck et al. 1997; Reid & Durham, 2002). When applied to skeletal muscle in millimolar concentrations, H2O2 can lead to an increase or a decrease in muscle tension (Oba et al. 1996; Plant et al. 2002; Lamb & Posterino, 2003). Interestingly, Crosland (1995) has shown that exogenous ROS selectively impair nerve-mediated muscle contraction (but not that induced by direct muscle stimulation), suggesting vulnerability of synaptic transmission to ROS. In the hippocampus, ROS causes a PKC-dependent increase (Knapp & Klann, 2002) or depression of evoked excitatory postsynaptic potentials (Auerbach & Segal, 1997). In striatum, H2O2 induced an inhibitory action on dopamine release (Chen et al. 2001). However, no direct evidence has been reported on the action of ROS on signal transmission from motoneurons to skeletal muscle.
In the current study we analysed the effect of a wide range of H2O2 concentrations on pre- and postsynaptic mechanisms at the frog neuromuscular junction. We found that low (3 μM) concentrations of H2O2 produced facilitation while high (>30 μM) concentrations led to depression of cholinergic transmission. Our study provides direct evidence for the action of H2O2 on presynaptic terminals at the neuromuscular junction and suggests that ROS could serve as endogenous modulators of neurotransmission.
METHODS
Preparation and solutions
Experiments were carried out on frog sartorius muscle preparations in vitro at room temperature (for details see Giniatullin et al. 1997). Briefly, muscles were dissected from tricaine-anaesthetized frogs in accordance with the European Communities Council Directive (24th November 1986; 86/609/EEC). To prevent muscle contractions we used the following preparations: (a) transverse cutting (‘cut preparation’) of muscle fibres (Barstad & Lilleheil, 1968; Glavinovic, 1979); (b) postsynaptic block of ACh receptors with 3-5 μM (+)-tubocurarine (curarized preparation); (c) addition of 6 mM Mg2+ with reduction of Ca2+ to 0.9 mM (magnesium preparation). For recording the presynaptic Ca2+ currents associated with evoked quantal ACh release, the extracellular solution contained 1.8 mM Ca2+, 30 μM (+)-tubocurarine, 100 μM 4-aminopyridine and 1 mM TEA (McArdle et al. 1981; Silinsky & Solsona, 1992).
Prior to recording, the cut muscle was rinsed for at least 40 min with physiological solution. The cutting procedure does not produce significant changes in cable properties and, in combination with the voltage clamp technique, enables long-lasting stable recording of multiquantal end-plate currents (Glavinovic, 1979). Ringer solution contained (mM): NaCl 113, KCl 2.5, CaCl2 1.8, NaHCO3 2.4, pH adjusted to 7.3 with NaOH.
Chelerythrine was purchased from Tocris. All other drugs were from Sigma.
Electrophysiology
The holding potential for cut preparations was kept at −40 mV while for all other preparations it was −70 mV. On both cut and curarized preparations the voltage dependence of the peak amplitudes of the evoked end-plate currents (EPCs) was linear in the range from 0 to −100 mV, indicating the effectiveness of the voltage clamp. Bi-exponential EPC decay was observed at some synapses (≈20 %) of cut preparations. Since the nature of this phenomenon was unclear, only junctions with mono-exponential decay phase of currents were considered. EPCs were elicited every 20 s (cut or curarized preparations) or every 2-5 s (magnesium preparation) by a single supramaximal nerve stimulation. Recording of postsynaptic end-plate currents was performed using the standard two-electrode voltage clamp technique with intracellular electrodes (resistance 3-5 MΩ). Presynaptic Ca2+ currents following the Na+ nerve action potential were recorded using focal Ringer-filled micropipettes (5-7 MΩ resistance) inserted into the perineurium (detailed description of the method and origin of the complex action potential is provided by McArdle et al. 1981 and Silinsky & Solsona, 1992). The recorded EPCs were digitized at 50 kHz, stored in a PC and analysed off-line to calculate mean values of EPC amplitudes, rise times (from 10 to 90 % of the amplitude), and decay time constants (τ).
Analysis
Usually 120-150 spontaneous miniature end-plate currents (MEPCs), 10-15 multiquantal EPCs (cut or curarized preparations), 20-50 low-quantal EPCs (magnesium preparation) or 24-48 presynaptic action potentials (Ca2+ current experiments) were averaged to obtain mean values. In experiments with spontaneous miniature end-plate currents (MEPCs), using the method of sequential current averaging, these uniquantal currents (digitized at 50 kHz) were repeatedly averaged (120-150 events) for at least 15 min before infusion of H2O2 to ensure stable baseline conditions. Spontaneous events were detected using amplitude thresholds set as a multiple (4-5 times) of the S.D. of the noise. Each event was also visually inspected to prevent noise disturbance of the analysis.
Spontaneous and elicited currents were analysed off-line using Origin software (OriginLab Corp, version 6.0). Quantal content was determined either as the ratio of averaged EPCs and averaged MEPCs or as ratio of their current areas. The time course of transmitter release was evaluated by the deconvolution method (Van der Kloot, 1988) using the average EPC and the average MEPC as detailed by Rich et al. (2002). In brief, each averaged MEPC was first fitted with the eqn (1) of Rich et al. (2002), using the least-squares method. This fitted MEPC was first adjusted to have zero latency and then deconvoluted using an approximation of the Wiener filter (eqn (3) of Rich et al. 2002). The quality of transmitter time course determination was evaluated by comparing the experimental EPC with one obtained by convolution (Van der Kloot, 1988). Measured features of the biexponential transmitter release function included the decay time constant of fast and slow components (Bukharaeva et al. 1999) and the ratio of amplitudes of two components.
The data are presented as the mean ± s.e.m. (n = number of synapses), with statistical significance assessed by Student's paired t test (for parametric data) or Wilcoxon's test (for non-parametric data). Statistical significance of data was assessed using SigmaStat (Jandel Scientific; version 2.0). A P value of < 0.05 was accepted as indicative of a statistically significant difference. All drugs were dissolved to a final concentration in the bathing solution and applied to a muscle maintained in the chamber (2.5 ml) via a superfusion system (2 ml min−1). To prepare stock solutions of PMA and staurosporine, these drugs were initially dissolved in DMSO. All other compounds were water-soluble. The final concentration of DMSO in the bathing solution was 0.01 %, which did not change the amplitude or the duration of the synaptic currents and did not modify the action of H2O2. We therefore used these data as the additional control. Measurements were started after 20 min in the presence (or washout) of various drugs, unless otherwise stated.
RESULTS
Bimodal effects of H2O2 on EPCs
On cut preparations of sartorius muscle, we tested the action of H2O2 on multiquantal end-plate currents (EPCs) elicited at a frequency of 0.05 Hz and miniature EPCs (MEPCs) occurring spontaneously in the same fibre. H2O2 at a low (3 μM) concentration increased the amplitude of EPCs from 123 ± 7 nA in control to 153 ± 11 nA (n = 6; P < 0.05 by paired t test, Fig. 1Aa and b). This reversible facilitation was stabilized after 15-20 min in the presence of H2O2. Enhancement of evoked currents was not accompanied by changes in the amplitude of MEPCs (1.13 ± 0.07 nA in control vs 1.12 ± 0.07 nA in H2O2; n = 6; P > 0.05, Fig. 1Ab). Furthermore, 3 μM H2O2 did not change the decay time constant of EPCs (τEPC = 1.21 ± 0.09 ms in control vs. 1.27 ± 0.10 ms in H2O2; n = 6; P > 0.05), or of MEPCs (τMEPC = 1.08 ± 0.13 ms in control vs. 1.02 ± 0.13 ms in H2O2; n = 6; P > 0.05). Estimation of the quantal content of EPCs based either on a simple ratio of peak current amplitudes, or the ratio of current areas (Fig. 1Ab; for details see Methods) revealed an enhancement of quantal release. With the first estimate (ratio of amplitudes), the quantal release was 110 ± 12 quanta in control and 139 ± 10 quanta in the presence of 3 μM H2O2 (n = 6; P < 0.05). The ratio of current integrals showed release of 160 ± 21 quanta in control and 224 ± 24 quanta in the presence of 3 μM H2O2 (n = 6; P < 0.05).
Figure 1. Opposite effects of low and high concentrations of H2O2 on multiquantal end-plate currents (EPCs).
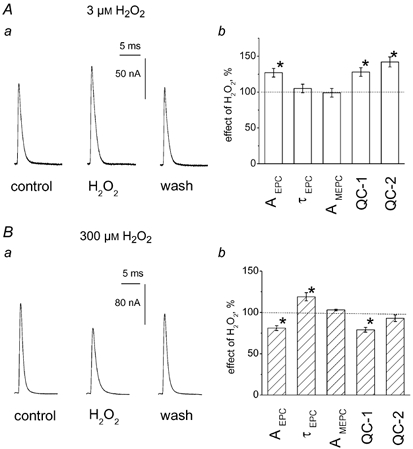
A, facilitating effect of 3 μM H2O2 on multiquantal EPCs. Aa, representative EPCs in control, in the presence of 3 μM H2O2 and after washout. Ab, histograms showing the action of 3 μM H2O2 on peak amplitude of EPCs (AEPC), decay time constant of EPCs (τEPC), amplitude of MEPCs (AMEPC) and quantal content estimated either as the ratio of peak current amplitudes of averaged EPCs and MEPCs (QC-1) or as the ratio of current integrals (QC-2) on cut preparation (* P < 0.05; n = 6). Data in this and subsequent histograms are presented as the mean ± s.e.m. (n = number of synapses). B, depressant action of 300 μM H2O2 on multiquantal EPCs. Ba, EPCs in control, in the presence of 300 μM H2O2 and after washout. Bb, histograms showing the action of 300 μM H2O2 on the same parameters of EPCs and MEPCs as in Ab (* P < 0.05; n = 6). Note that depression of QC-1 was stronger (P < 0.05) than that of QC-2 (P > 0.05).
A similar enhancement of EPCs (17 ± 5 %; n = 5; P < 0.05) without any change in the time course of the EPC was obtained in curarized preparations, suggesting that the facilitatory effect of H2O2 was not related to the mode of muscle contraction block.
These data indicate that low, physiological doses of H2O2 facilitated synaptic transmission by enhancement of transmitter release from motor nerve endings.
As shown on Fig. 1B, a higher dose of H2O2 (300 μM) produced the opposite effect, namely reversible depression of EPC amplitude. In six fibres, the effect of 300 μM H2O2 on multiquantal EPCs was compared with the action on spontaneous MEPCs recorded from the same fibre. H2O2 significantly depressed the amplitude of EPCs from 158 ± 12 to 130 ± 11 nA (n = 6; P < 0.01 by paired t test). As with the low dose, 300 μM H2O2 did not change the amplitude of MEPCs (1.39 ± 0.12 nA in control vs. 1.41 ± 0.13 nA in H2O2; n = 6; P > 0.05; Fig. 3A). Similarly, no changes were observed in time course of MEPCs (τMEPC = 0.90 ± 0.03 ms in control vs. 0.93 ± 0.04 ms in H2O2; n = 6; P > 0.05). Whereas 3 μM H2O2 did not change the MEPC frequency, 300 μM H2O2 strongly and reversibly depressed the frequency of MEPCs by 69 ± 5 % (n = 6; P < 0.05). Unlike the action of the low dose, 300 μM H2O2 prolonged the decay of multiquantal EPCs (1.11 ± 0.04 ms in control vs. 1.33 ± 0.06 ms in H2O2; n = 6; P > 0.05). The quantal content calculated as the ratio of the peak amplitudes of EPCs and MEPCs was significantly (n = 6; P < 0.05) reduced in the presence of 300 μM H2O2 (Fig. 1Bb). However, the ratio of current areas did not reveal a depressant action of 300 μM H2O2 on evoked quantal release (n = 6; P > 0.05; Fig. 1Bb).
Figure 3. The action of H2O2 on short-term plasticity.
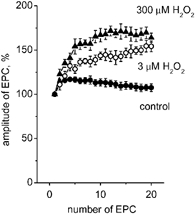
A, peak amplitudes of 20 successive EPCs during 60 Hz motor nerve stimulation of a cut preparation in control (•), and in the presence of 3 μM (○) or 300 μM (▴; n = 5-7) H2O2. Note the significantly enhanced potentiation of successive EPCs in the presence of both concentrations of H2O2.
The depression of EPCs by 300 μM H2O2 was reproduced on uncut curarized muscle (18 ± 6 %; n = 5; P < 0.05). The postsynaptic muscle block by curare did not allow recording of spontaneous synaptic events. In order to confirm the action of H2O2 on MEPCs of a different preparation, we performed experiments on uncut muscle without stimulation of the motor nerve. These experiments (including a test of the voltage dependence of τMEPC) confirmed the absence of any postsynaptic action of H2O2 obtained in the cut preparation (see Supplementary material available online at DOI: 10.1113/jphysiol. 2003. 050690).
Dose-response curve and high frequency stimulation
A systematic exploration of H2O2 action (0.3 μM-3 mM) on synaptic transmission of cut preparations is shown in Fig. 2. At a concentration of 0.3-1 μM, H2O2 had no effect, whereas at 3 μM, it elicited clear facilitatory effects on EPCs. At 30 μM, the effect of H2O2 was highly variable while concentrations exceeding 30 μM produced a depressant action on EPCs. The slowing of EPC decay occurred at concentrations of H2O2 greater than 30 μM (Fig. 2A). Unlike the dual action on the amplitude of EPCs, the effect of H2O2 on the time course of synaptic currents monotonically increased in a dose-dependent manner (Fig. 2B). This fact suggested that H2O2 changed not only the amplitude but also the time course of transmitter release (Van der Kloot 1988; Giniatullin et al. 1995). Indeed, the concentration dependence of the action of H2O2 on the area of EPCs (Fig. 2C) showed a pattern distinct from that of the amplitude changes (Fig. 2A). It should be noted that 3 mM H2O2 reduced both the amplitude (Fig. 2A) and the current area (Fig. 2C).
Figure 2. Concentration dependence of H2O2 action on EPCs.
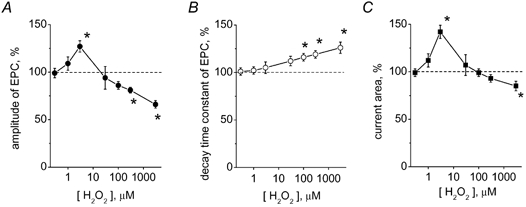
A, effect of various H2O2 concentrations on peak EPC amplitude in a cut preparation. B, effect of H2O2 on the decay time constant of EPCs. C, the action of H2O2 on the current integrals. (* P < 0.05; n = 4-8 synapses). Note the biphasic action of H2O2 on the amplitude and area of EPCs and monophasic action of H2O2 on the time course of EPCs.
The above tests were based on the evaluation of low-frequency (0.05 Hz), evoked synaptic events. In order to explore the action of H2O2 on short-term presynaptic plasticity (Zucker & Regehr, 2002), we used a high-frequency (60 Hz) stimulation of the motor nerve.
As shown in Fig. 3, a 60 Hz train of 20 stimuli in control conditions elicited initial presynaptic potentiation followed by a slight depression (•). At a depressant (300 μM) concentration, H2O2 increased presynaptic potentiation (Fig. 3, ▴). Surprisingly, 3 μM H2O2 also significantly promoted potentiation during high-frequency trains (Fig. 3, ○).
Effect of H2O2 on the time course of transmitter release
Since high doses of H2O2 selectively prolonged the decay phase of multiquantal EPCs, we next tested if this action was based on changes in the time course of transmitter release. To this end we evaluated the transmitter release functions by the deconvolution method, using averaged EPCs and averaged MEPCs (Van der Kloot, 1998; Rich et al. 2002; for detail see Methods). As exemplified in Fig. 4A for signals used for deconvolution, 300 μM H2O2 prolonged the decay of EPCs without changing the time course of MEPCs (note that signals were scaled for comparison). Our analysis revealed that the quantal release function for multiquantal control EPCs consists of two exponential components (see also Bukharaeva et al. 1999) with decay time constants of 0.18 ± 0.04 and 1.02 ± 0.11 ms, respectively (n = 6; Fig. 4B). The ratio of release component amplitudes (A1/A2) was 1.27 ± 0.25 in control (n = 6). The application of 300 μM H2O2 did not change the fast component of release (0.18 ± 0.05 ms; n = 6; P > 0.05; Fig. 4C). The A1/A2 ratio tended to decrease in the presence of H2O2 (Fig. 4C) but this difference did not reach statistical significance (n = 6; P > 0.05). Nevertheless, the slow phase of release was significantly prolonged (1.34 ± 0.13 ms; n = 6; P < 0.05; Fig. 4), suggesting a mechanism responsible for the slowing of the time course of multiquantal EPCs by high H2O2.
Figure 4. Effect of H2O2 on the time course of transmitter release.
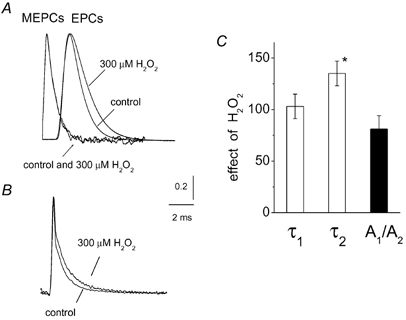
A, examples of averaged MEPCs (n = 120) or averaged multiquantal EPCs (n = 10) used for deconvolution in control and after 300 μM H2O2. All signals were normalized for comparison. Note that H2O2 selectively prolonged the decay of EPCs, while in the presence of 300 μM H2O2 the shape of MEPCs was the same as in control. B illustrates the results of deconvolution of the averaged EPC from A (transmitter release functions) with the fitted MEPCs (for detail see Methods). Deconvolution revealed two exponential components of transmitter release with decay time constants of 0.18 ± 0.04 and 1.02 ± 0.11 ms, respectively. Transmitter release functions were obtained by averaging data from 6 endplates. Note that 300 μM H2O2 increased the relative contribution of the slow phase of transmitter release function. C, histograms showing that 300 μM H2O2 significantly increased the decay time constant of the slow phase of the release function (τ2; * P < 0.05; n = 6), while it did not affect the fast phase (τ1; P > 0.05; n = 6) or the ratio of the amplitudes of the two components (A1/A2; P > 0.05; n = 6).
H2O2 at a low (3 μM) concentration did not change the time course of transmitter release (not shown), consistent with its lack of the action on the decay of EPCs.
Effect of H2O2 on the presynaptic Ca2+currents
The best approach for understanding the mechanism of the presynaptic action of H2O2 would be to record presynaptic Ca2+ currents coupled to transmitter release. The small size of motor nerve terminals does not allow intracellular recording of presynaptic depolarization or the presynaptic Ca2+ currents. Relatively high-resolution, extracellular recording of these currents was obtained using the method developed by McArdle et al. (1981) in which the Na+ action potential is detected as downward current flow preceding the upward Ca2+-mediated event (see example in Fig. 5A). The Ca2+-mediated nature of the late upward signal was confirmed by application of the Ca2+ channel blocker Cd2+ (100 μM), which almost completely removed this signal without changing the Na+-mediated signal (data not shown). This main Cd2+-sensitive component of Ca2+ current (Fig. 5A) decayed with a time constant of 1.33 ± 17 ms (n = 7), which was presumably prolonged due to the presence of K+ channel blockers. As shown in Fig. 5A and B, the application of 3 μM H2O2 changed neither the Na+ nor the Ca2+ phase of the complex action potential, implying that the increase in EPCs produced by H2O2 was located downstream from presynaptic depolarization and Ca2+ influx. The action of 300 μM H2O2 was more effective. In this case, as for 3 μM H2O2, no changes in the Na+ phase were detected (Fig. 5A and C). However, 300 μM H2O2 increased the decay time constant of the Ca2+ current (37 ± 7 % increase; n = 6; P < 0.05), while sparing the amplitude of this current (n = 6; P > 0.05; Fig. 5A and C).
Figure 5. Effect of H2O2 on the presynaptic Ca2+ current.
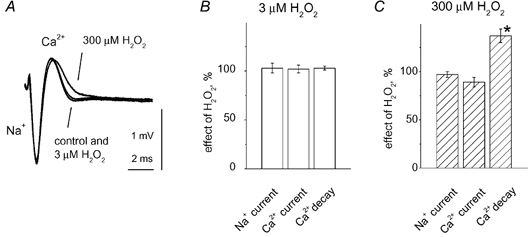
A, presynaptic action potential recorded by a microelectrode inserted into the perineurium adjacent to the nerve ending. The large initial downward part of the action potential represents the initial Na+ current while the late upward part represents the Ca2+ current. Each signal was obtained by averaging 24 records evoked at 0.05 Hz. The postsynaptic response was blocked by a high dose of (+)-tubocurarine (for details, see Methods). Low (3 μM) H2O2 did not change the Na+ or Ca2+ phase of the presynaptic signal, while high (100 μM) H2O2 slowed the decay of the Ca2+ current without producing significant changes in the Na+ phase. B and C, histograms showing the action of 3 or 300 μM H2O2 on the amplitude (% change vs. control) or the time constant of Ca2+ current decay. * P < 0.05, n = 6 for both H2O2 concentrations.
Thus, high H2O2 caused prolonged Ca2+ entry, consistent with the slowing down of the EPC time course.
Action of H2O2 on magnesium preparations
The actions of H2O2 are Ca2+ dependent in many experimental models (Lee et al. 2000; Oh et al. 2000). In order to understand the role of Ca2+ in the modulatory action of H2O2 on synaptic currents, we performed experiments using a ‘magnesium preparation’ (uncut muscle, −70 mV holding potential). Adding 6 mM Mg2+ to the external solution, while halving the Ca2+ concentration, reduced the amplitude of EPCs (12 ± 10 nA; n = 5) to levels roughly 10-fold smaller than in cut preparations. This effect was presumably due to strong depression of Ca2+ influx into the motor nerve terminals and a reduction in the quantal content of EPCs. As shown in Fig. 6A, application of 3 μM H2O2 to this magnesium preparation failed to enhance the peak amplitude (4 ± 3 %) or change the area (9 ± 5 %; n = 5; P > 0.05) of EPCs. Similarly, no depressant effect was obtained after 20 min with 300 μM H2O2 (1 ± 1 % change in amplitude or 10 ± 6 % change in area, respectively; n = 5; P > 0.05; Fig. 6B). Thus, the level of transmitter release appeared to play a crucial role in the modulatory action of H2O2.
Figure 6. Modification by Mg2+ of H2O2 action on EPCs.
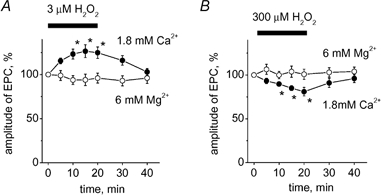
A, time profile for the facilitatory effect of 3 μM H2O2 on EPCs in control (•; [Ca2+]o = 1.8 mM) and in a magnesium preparation (○; [Mg2+]o = 6 mM; [Ca2+]o = 0.9 mM). * P < 0.05; n = 5. B, time profile for the depressant effect of 300 μM H2O2 on EPCs in control (•) and in a magnesium preparation (○; * P < 0.05; n = 5).
Effects of Fe2+ and N-acetylcysteine
In many cases the biological action of H2O2 is related to the generation of hydroxyl radicals (OH•) (Winterbourn, 1995). To understand the mechanism of the action of H2O2, it is therefore important to know whether its effects were mediated by hydroxyl radicals. It is known that in the presence of Fe2+, H2O2 is readily converted into the hydroxyl radical and this provides a reliable tool for ascertaining the mechanism of action (Winterbourn, 1995; Kress et al. 2002).
Preconditioning of muscles with 100 μM FeSO4 depressed multiquantal EPCs by 25 ± 5 % (n = 10; P < 0.05), probably because of Fe2+ interference with the presynaptic voltage-activated Ca2+ channels. Fe2+ did not change the facilitatory effect of 3 μM H2O2, since in the presence of iron this ROS produced the same reversible enhancement of EPCs (26 ± 5 %; n = 5; P < 0.05; Fig. 7Aa) and increase in current area (27 ± 10 %; n = 5; P < 0.05; Fig. 7Ab) as in controls. However, application of 300 μM H2O2 in the presence of Fe2+ evoked a dramatic reduction in EPC amplitude of 71 ± 4 % (n = 6; P < 0.001; Fig. 7Ba) compared with a 19 % reduction in controls (P < 0.01). In the presence of pro-oxidant Fe2+, 300 μM H2O2 reduced the current area by 55 ± 5 % (n = 6; P < 0.05; Fig. 7Bb). Unlike the strong depressant effect of H2O2 on peak amplitude, the effect on current area was modest, presumably because the decay of EPCs was prolonged by 300 μM H2O2 (by 32 ± 10 %; n = 6; P < 0.05). The effect of the high concentration of H2O2 was slowly reversible, in contrast to the quickly reversible action of 3 μM H2O2. Thus, only a partial recovery to 52 ± 5 % of control EPC amplitude was achieved after 30 min.
Figure 7. Fe2+ selectively promoted the depressant effect of 300 μM H2O2.
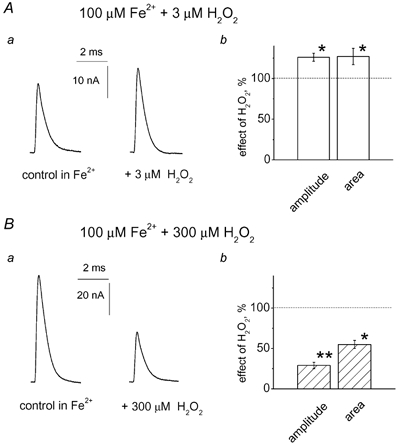
Aa, effect of 3 μM H2O2 in the presence of 100 μM Fe2+ on multiquantal EPCs in a cut preparation. Ab, histograms showing the facilitatory action of 3 μM H2O2 on peak EPC amplitude or current area. Note that the facilitatory action of a low dose of H2O2 was not changed by Fe2+ (26 ± 5 % enhancement in Fe2+; * P < 0.05; n = 5; compare with 28 ± 7 % in control). Ba, the depressant action of 300 μM H2O2 on EPCs was strongly enhanced by 100 μM Fe2+. Bb, histograms showing the action of 300 μM H2O2 on peak EPC amplitude or current area in the presence of 100 μM Fe2+ (** P < 0.01; significantly different from the action of 300 μM H2O2 in control; n = 5).
These data suggest that the enhancement of EPCs by low H2O2 was based on the action of H2O2 as such, while the strong enhancement by Fe2+ of high H2O2-induced synaptic inhibition indicated the involvement of hydroxyl radicals generated via the Fenton reaction (Winterbourn, 1995).
Next, we tested the action of H2O2 in the presence of N-acetylcysteine, a ROS scavenger, which removes muscle fatigue created by ROS accumulation (Reid et al. 1994). The application of 100 μM N-acetylcysteine increased the amplitude of EPCs by 40 ± 6 % (n = 10; P < 0.05). Subsequent application of 3 μM H2O2 did not change the amplitude of EPCs (7 ± 4 %) or current areas (4 ± 4 % change; n = 5; P > 0.05). Similarly, 100 μM N-acetylcysteine prevented the depressant action of 300 μM H2O2 on the amplitude (3 ± 3 %; n = 5; P > 0.05) and area of EPCs (3 ± 4 % change; n = 5; P > 0.05).
Thus, a typical scavenger of ROS, N-acetylcysteine, abolished the effects of H2O2, indicating that the enhancing and depressant effects were indeed produced by ROS.
The role of PKC in the action of H2O2
To further explore the mechanisms of the action of H2O2 on motor nerve terminals, we tested the role of PKC, since the intracellular action of H2O2 is mediated by PKC in many cells (Gopalakrishna & Anderson, 1989; Konishi et al. 1997; Suzuki et al. 1997; Servitja et al. 2000).
We first blocked the activity of PKC using the non-specific inhibitor staurosporine. Staurosporine (500 nM) per se produced a slight increase in EPC amplitude (22 ± 4 %; n = 10; P < 0.05). Subsequent application of 3 μM H2O2 did not affect the amplitude (3 ± 5 % change; n = 5; P > 0.05) or area of EPCs (Fig. 8A). Chelerythrine (10 μM), a more specific inhibitor of PKC, also abolished the facilitatory effect of 3 μM H2O2 (1 ± 4 % change of amplitude and 2 ± 4 % change of area; n = 5; P > 0.05; Fig. 8A). Interestingly, pre-treatment of the muscle with 500 nM PMA, a PKC activator, also prevented the facilitatory action of H2O2 (Fig. 8A). PMA itself enhanced the amplitude of EPCs by 59 ± 8 % (n = 6; P < 0.05).
Figure 8. Testing the role of PKC in the action of H2O2.
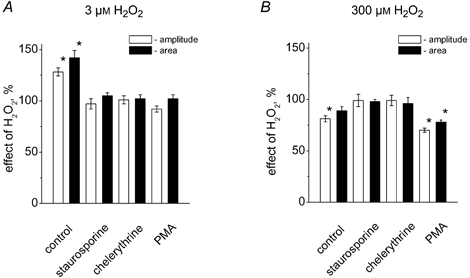
A, histograms showing the action of 3 μM H2O2 on the peak amplitude of EPCs (□) and the current area (▪) in control (* P < 0.05; n = 5), in the presence of 0.5 μM staurosporine (P > 0.05; n = 5), 10 μM chelerythrine (P > 0.05; n = 5), and 0.5 μM PMA (P > 0.05; n = 5). B, the same data for the action of 300 μM H2O2 (n = 4-7). Note, that unlike the facilitatory action of 3 μM H2O2, the depressant effect of 300 μM H2O2 was not only preserved in the presence of PMA but even enhanced compared with control (P < 0.05).
The depressant effect of high H2O2 appeared to be PKC-mediated as well. Thus, staurosporine or chelerythrine completely eliminated the depressant action of 300 μM H2O2 on the amplitude of EPCs (2 ± 6 % and 1 ± 5 % change, respectively; for changes in current areas see Fig. 8B, ▪). In contrast to its blocking effect on the action of low doses of H2O2, PMA did not prevent the ability of 300 μM H2O2 to reduce EPCs amplitude. In fact, the depression was even stronger (30 ± 2 %; n = 5; Fig. 8B; □) than in control (19 ± 3 %; P < 0.05) and was accompanied by a significant reduction in current area (22 ± 2 %; n = 5; P < 0.05; Fig. 8B, ▪).
DISCUSSION
We have found that H2O2 produced opposing actions on transmission of motor signals to skeletal muscle fibres. At low physiological concentrations, H2O2 facilitated ACh secretion, while at high concentrations, the action of H2O2 resulted in synaptic depression. While the genome of the motoneuron is very susceptible to attack by ROS (Martin & Liu, 2002), current data represent another fast, presumably non-genomic, action of H2O2 on motoneuron terminals.
The mechanisms of H2O2 action on motor nerve terminals
Our data indicate that the motor nerve terminal is the preferential target for the fast action of exogenous (and potentially endogenous) H2O2. This was confirmed by the fact that the amplitude and decay of MEPCs were unchanged by H2O2. The high sensitivity of nerve terminals to H2O2 was consistent with earlier observations that the direct modulatory action of H2O2 on the muscle fibres needs much higher (mM) concentrations of this ROS (Oba et al. 1996; Lamb & Posterino, 2003).
Low H2O2 affected neither presynaptic Na+-mediated depolarization, nor subsequent Ca2+ current, indicating that the facilitatory action of H2O2 on ACh release was located downstream from Ca2+ influx (Hilfiker & Augustine, 1999). The potentiation of EPC amplitude by low doses of H2O2 during high-frequency stimulation could be explained by either enlargement of the readily releasable pool of quantal ACh (Stevens & Sullivan, 1998), or an increase in the level of residual Ca2+ (Zucker & Regehr, 2002).
Unlike the effect of low concentrations of H2O2, the actions of high doses of this ROS were more complex. First, high concentrations of H2O2 reduced the peak amplitude of signals but prolonged the duration of the falling phase of EPCs. Second, they prolonged Ca2+ influx into the motor nerve terminal. Third, high concentrations of H2O2, which only slightly depressed multiquantal EPCs, strongly reduced spontaneous release of ACh.
These distinct effects of high concentrations of H2O2 could be explained by assuming that the presynaptic action of this ROS consists of two components, one facilitatory and the other one inhibitory. Facilitation was probably related to the prolonged Ca2+ influx. Inhibition could be based on two mechanisms: first, on the temporal dispersion of evoked release (Van der Kloot, 1988; Giniatullin et al. 1995) consistent with the predominance of the slow component of transmitter release function found by deconvolution of EPCs and MEPCs, and second, on the impairment of releasing machinery (to explain the strong inhibition of spontaneous, Ca2+-independent release). Distinct factors such as Fe2+, PMA or an increase in H2O2 concentration could shift the equilibrium between facilitation and inhibition evoked by H2O2 and make depression the predominant phenomenon.
The effect of Fe2+suggests the involvement of distinct ROS
Pretreatment with Fe2+ dramatically increased the inhibitory potency of H2O2, suggesting that it was mediated by the hydroxyl radical (OH•). The OH• could be generated by the combined action of Fe2+ and H2O2 via the Fenton reaction: Fe2+ + H2O2 = Fe3+ + OH•+ OH− (Winterbourn, 1995; Kress et al. 2002). Hydroxyl radicals, while being short-lived, are the most reactive form of ROS (Winterbourn, 1995) and participate in the depressant effects of high H2O2. The facilitatory action of low concentrations of H2O2 was unchanged by Fe2+, suggesting that the action was mediated by untransformed H2O2, with no contribution from hydroxyl radicals.
The transformation of H2O2 via the Fenton reaction probably occurred in the intracellular rather than the extracellular space. OH•, unlike H2O2, is a short-lived and cell-impermeant form of ROS that acts only at the site of its generation (Kress et al. 1995). Despite the fact that only a limited amount of free iron within the cell is in the reduced state (Kress et al. 2002), several other mechanisms could account for the transformation of H2O2 into OH• under physiological conditions (Liochev, 1996). If the rate of H2O2 breakdown was relatively low, it could explain why Fe2+ did not reduce the facilitatory effects of exogenous H2O2 when the concentration of this agent in solution was at steady state. However, breakdown could be one important factor in the action of endogenous H2O2 produced, for instance, during muscle activity.
Thus, the current study has revealed a synergistic interaction between Fe2+ and H2O2 in depression of synaptic transmission, indicating participation of hydroxyl radicals in this effect.
Second messenger cascades
PKC, a key enzyme for the expression of synaptic plasticity and control of transmitter release, is an important intracellular target for H2O2 (Gopalakrishna & Anderson, 1989; Konishi et al. 1997; Waldron & Rozengurt, 2000; Shibukawa et al. 2003).
Consistent with this notion, the facilitatory action of low H2O2 on ACh release was abolished by the PKC inhibitors staurosporine and chelerythrine. Furthermore, the facilitatory effect of H2O2 was prevented when PKC activity was up-regulated by PMA.
Unexpectedly, PKC was also involved in the depressant action of H2O2, as two distinct PKC inhibitors abolished the H2O2-mediated inhibition. However, unlike the facilitatory effect, the inhibitory action of H2O2 was preserved after PMA treatment.
There are several explanations for the mechanisms by which PKC might produce opposite effects. First, we cannot exclude the possibility that distinct isoforms of PKC participate in facilitation or depression, because H2O2 can activate either conventional, novel or atypical PKCs (Konishi et al. 1997). Remarkably, phorbol esters activate only conventional and novel, but not atypical, PKCs (Ron & Kazanietz, 1999). Second, H2O2 might activate PKC at low concentrations and inactivate it at high concentrations (Gopalakrishna & Anderson, 1989). The ability of PMA to counteract the facilitatory but not the inhibitory action of H2O2 supports this suggestion. It has been shown that the active form of PKC is more susceptible to oxidative inactivation (Gopalakrishna & Anderson, 1989), consistent with our observation that PMA pre-treatment increased the depressant action of high H2O2. In the nerve terminals of Calyx of Held, Ca2+-independent εPKC (a novel isoform) determines PMA-induced synaptic facilitation (Saitoh et al. 2001). Another isoform, γPKC, widely expressed in synapses, does not appear to be involved in the action of phorbol esters on transmitter release (Goda et al. 1996). It remains to be established what isoforms of PKC are present at motor nerve endings and are activated (or inhibited) by low or high doses of H2O2.
In summary, our results indicate a principal role of PKC in the action of H2O2 on transmitter release from the motor nerve endings.
Functional implications
Since the changes in synaptic currents were observed with very low concentrations of H2O2, our data suggest that ROS-mediated modulation of synaptic transmission might take place in physiological conditions. The production (O'Neill et al. 1996; Hasegawa et al. 1997; Supinski et al. 1999; Reid & Durham, 2002) and extracellular release (Reid et al. 1992) of endogenous ROS during muscle contraction provide the conditions for the feedback action of these compounds on the terminal axonal branches in skeletal muscle. At low doses, H2O2 could enhance ACh release, thus leading to better performance of neuromuscular transmission. In contrast, during prolonged muscle activity, a high level of H2O2 could participate in fatigue mechanisms. The functional output presumably depends on the concentration of H2O2 in the synaptic cleft. Mitochondrial respiration, one of the major sources of ROS production in skeletal muscle (Murrant & Reid, 2001) produces superoxide (the parent of H2O2) at a rate of approximately 1.2 nmol min−1 mg−1 (Boveris et al. 1972). An at least several-fold increase in ROS production during exercise has been well documented by many research groups (reviewed by Murrant & Reid, 2001). However, the exact value of the H2O2 concentration in the vicinity of nerve terminals remains unknown. For central synapses, high (up to millimolar) local concentrations of H2O2 have been suggested (Chen et al. 2001). The direct determination of ROS during reperfusion of the brain after short periods of ischaemia revealed about 200 μM H2O2 (Hyslop et al. 1995). Currently, it is unknown whether the presynaptic action of H2O2 is direct or involves other partners. For instance, the possibility exists that the action of H2O2 is mediated by glial cells in analogy with the well-established action of reactive nitrogen species (Castonguay et al. 2001).
Another interesting but still poorly understood phenomenon is the hypoxia- and re-oxygenation-related overproduction of ROS such as the superoxide anion (O2−) and H2O2 (Li & Jackson, 2002). The modulatory effect of H2O2 on synaptic transmission should become even more important under these conditions and potentially damaging to motor nerve endings.
Conclusions
The production of cell-permeable H2O2 by skeletal muscle during intense contractions could generate contrasting actions on motor nerve endings. Low doses of H2O2 in the synaptic cleft would be expected to enhance sustained ACh release from motor nerve endings, while high doses of H2O2 inhibit synaptic currents. Both processes are sensitive to Ca2+ entry and PKC activity and might be significantly modified by ROS transformation. We hypothesize that the opposite actions of H2O2 represent a fast feedback mechanism for fine-tuning transmitter release during muscle activity.
Acknowledgments
This work was supported by RFBI and INTAS. Authors thank Professors J. Nicholls, A. Nistri and W. Van der Kloot and Dr J. Yakel for helpful discussions.
Supplementary material
The online version of this paper can be found at:
DOI: 10.1113/jphysiol.2003.050690
and contains material entitled:
Effect of H2O2 on miniature end-plate currents (MEPCs)
REFERENCES
- Auerbach JM, Segal M. Peroxide modulation of slow onset potentiation in rat hippocampus. J Neurosci. 1997;17:8695–8701. doi: 10.1523/JNEUROSCI.17-22-08695.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barstad JA, Lilleheil G. Transversaly cut diaphragm preparation from rat. An adjuvant tool in the study of the physiology and pharmacology of the myoneural junction. Arch Int Pharmacodyn Ther. 1968;175:373–390. [PubMed] [Google Scholar]
- Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukcharaeva EA, Kim KC, Moravec J, Nikolsky EE, Vyskocil F. Noradrenaline synchronizes evoked quantal release at frog neuromuscular junctions. J Physiol. 1999;517:879–888. doi: 10.1111/j.1469-7793.1999.0879s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay A, Lévesque S, Robitaille R. Glial cells as active partners in synaptic functions. Prog Brain Res. 2001;132:227–240. doi: 10.1016/S0079-6123(01)32079-4. [DOI] [PubMed] [Google Scholar]
- Chen BT, Avshalumov MV, Rice ME. H2O2 is a novel, endogenous modulator of synaptic dopamine release. J Neurophysiol. 2001;85:2468–2476. doi: 10.1152/jn.2001.85.6.2468. [DOI] [PubMed] [Google Scholar]
- Crosland RD. Action of reactive oxygen species and their antagonism on twitch tension of the rat phrenic nerve-diaphragm. Pharmacol Toxical. 1995;77:231–237. doi: 10.1111/j.1600-0773.1995.tb01018.x. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxygen radicals and signalling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- Giniatullin RA, Khiroug LS, Vyskocil F. Modeling end-plate currents-dependence on quantum secretion probability and decay of miniature current. Eur Biophys J. 1995;23:443–446. doi: 10.1007/BF00196832. [DOI] [PubMed] [Google Scholar]
- Giniatullin RA, Talantova M, Vyskocil F. Desensitization shortens the high-quantal-content endplate current time course in frog muscle with intact cholinesterase. J Physiol. 1997;502:641–648. doi: 10.1111/j.1469-7793.1997.641bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glavinovic MI. Voltage clamping of unparalysed cut rat diaphragm for study of transmitter release. J Physiol. 1979;290:467–480. doi: 10.1113/jphysiol.1979.sp012784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF, Tonegawa S. Phorbol ester effects at hippocampal synapses act independently of the gamma isoform of PKC. Learn Mem. 1996;3:182–187. doi: 10.1101/lm.3.2-3.182. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R, Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa A, Suzuki S, Matsumoto Y, Okubo T. In vivo fatiguing contraction of rat diaphragm produces hydroxyl radicals. Free Radic Biol Med. 1997;22:349–354. doi: 10.1016/s0891-5849(96)00343-7. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Augustine GJ. Regulation of synaptic vesicle fusion by protein kinase C. J Physiol. 1999;515:1. doi: 10.1111/j.1469-7793.1999.001ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyslop PA, Zhang Z, Pearson DV, Phebus LA. Measurment of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vivo. Brain Res. 1995;671:181–186. doi: 10.1016/0006-8993(94)01291-o. [DOI] [PubMed] [Google Scholar]
- Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J Neurosci. 2002;22:674–683. doi: 10.1523/JNEUROSCI.22-03-00674.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbeck RC, She ZW, Callahan LA, Nosek TM. Increased superoxide production during fatigue in the perfused rat diaphragm. Am J Respir Crit Care Med. 1997;156:140–145. doi: 10.1164/ajrccm.156.1.9610041. [DOI] [PubMed] [Google Scholar]
- Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci. 2002;22:5848–5855. doi: 10.1523/JNEUROSCI.22-14-05848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress M, Riedl B, Reeh PW. Effect of oxygen radicals on nociceptive afferents in the rat skin in vitro. Pain. 1995;62:87–94. doi: 10.1016/0304-3959(94)00254-C. [DOI] [PubMed] [Google Scholar]
- Lamb GD, Posterino GS. Effects of oxidation and reduction on contractile function in skeletal muscle fibres of the rat. J Physiol. 2003;546:149–163. doi: 10.1113/jphysiol.2002.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BD, Kim JH, Lee SD, Kim Y, Suh PG, Ryu SH. Hydrogen peroxide-induced phospholipase D2 activation in lymphocytic leukemic L1210 cells. J Leukoc Biol. 2000;67:630–636. doi: 10.1002/jlb.67.5.630. [DOI] [PubMed] [Google Scholar]
- Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:227–241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- Liochev SL. The role of iron-sulfur clusters in vivo hydroxyl radical production. Free Radic Res. 1996;25:369–384. doi: 10.3109/10715769609149059. [DOI] [PubMed] [Google Scholar]
- McArdle JJ, Angaut-Petit D, Mallart A, Bournaud R, Faille L, Brigant JL. Advantages of the triangularis sterni muscle of the mouse for investigations of synaptic phenomena. J Neurosci Methods. 1981;4:109–115. doi: 10.1016/0165-0270(81)90044-3. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z. DNA damage profiling in motor neurons: a single-cell analysis by comet assay. Neurochem Res. 2002;27:1093–1104. doi: 10.1023/a:1020961006216. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Reid M. Detection of reactive oxygen and reactive nitrogen species in skeletal muscle. Microsc Res Tech. 2001;55:236–248. doi: 10.1002/jemt.1173. [DOI] [PubMed] [Google Scholar]
- Oba T, Koshita M, Yamaguchi M. H2O2 modulates twitch tension and increases Po of Ca2+ release channel in frog skeletal muscle. Am J Physiol. 1996;271:810–818. doi: 10.1152/ajpcell.1996.271.3.C810. [DOI] [PubMed] [Google Scholar]
- Oh SO, Hong JH, Kim YR, Yoo HS, Lee SH, Lim K, Hwang BD, Exton JH, Park SK. Regulation of phospholipase D2 by H2O2 in PC12 cells. J Neurochem. 2000;75:2445–2454. doi: 10.1046/j.1471-4159.2000.0752445.x. [DOI] [PubMed] [Google Scholar]
- O'Neill CA, Stebbins CL, Bonigut S, Halliwell B, Longhurst JC. Production of hydroxyl radicals in contracting skeletal muscle of cats. J Appl Physiol. 1996;81:1197–2106. doi: 10.1152/jappl.1996.81.3.1197. [DOI] [PubMed] [Google Scholar]
- Plant DR, Lynch GS, Williams DA. Hydrogen peroxide increases depolarization-induced contraction of mechanically skinned slow twitch fibres from rat skeletal muscles. J Physiol. 2002;539:883–981. doi: 10.1113/jphysiol.2001.013369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MB, Durham WJ. Generation of reactive oxygen and nitrogen species in contracting skeletal muscle: potential impact on aging. Ann N Y Acad Sci. 2002;959:108–116. doi: 10.1111/j.1749-6632.2002.tb02087.x. [DOI] [PubMed] [Google Scholar]
- Reid MB, Shoji T, Moody MR, Entman ML. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J Appl Physiol. 1992;73:1805–1809. doi: 10.1152/jappl.1992.73.5.1805. [DOI] [PubMed] [Google Scholar]
- Reid MB, Stokic DS, Koch SM, Khawli FA, Leis AA. N-acetylcysteine inhibits muscle fatigue in humans. J Clin Invest. 1994;94:2468–2474. doi: 10.1172/JCI117615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Wang X, Cope TC, Pinter MJ. Reduced neuromuscular quantal content with normal synaptic release time course and depression in canine motor neuron disease. J Neurophysiol. 2002;88:3305–3314. doi: 10.1152/jn.00271.2002. [DOI] [PubMed] [Google Scholar]
- Ron D, Kazanietz MG. New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 1999;13:1658–1676. [PubMed] [Google Scholar]
- Saitoh N, Hori T, Takahashi T. Activation of the epsilon isoform of protein kinase C in mammalian nerve terminals. Proc Natl Acad Sci U S A. 2001;98:14017–14021. doi: 10.1073/pnas.241333598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servitja J-M, Masgrau R, Pardo R, Sarri E, Picatoste F. Effects of oxidative stress on phospholipid signaling in rat cultured astrocytes and brain slices. J Neurochem. 2000;75:788–794. doi: 10.1046/j.1471-4159.2000.0750788.x. [DOI] [PubMed] [Google Scholar]
- Shibukawa Y, Takahashi M, Laffont I, Honke K, Taniguchi N. Downregulation of hydrogen peroxide-induced PKC-activation in N-acetylglucosaminyltransferase III transfected HeLaS3 cells. J Biol Chem. 2003;278:3197–3203. doi: 10.1074/jbc.M207870200. [DOI] [PubMed] [Google Scholar]
- Silinsky EM, Solsona CS. Calcium currents at motor nerve endings: absence of effects of adenosine receptor agonist in the frog. J Physiol. 1992;457:316–328. doi: 10.1113/jphysiol.1992.sp019380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Supinski G, Nethery D, Stofan D, Dimarco A. Extracellular calcium modulates generation of reactive oxygen species by the contracting diaphragm. J Appl Physiol. 1999;87:2177–2185. doi: 10.1152/jappl.1999.87.6.2177. [DOI] [PubMed] [Google Scholar]
- Suzuki YJ, Forman HJ, Sevanin A. Oxidants a stimulators of signal transduction. Free Rad Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Early events in free radicals-mediated damage of isolated nerve terminals: effects of peroxides on membrane potential and intracellular Na+ and Ca2+ concentrations. J Neurochem. 1996;66:2057–2066. doi: 10.1046/j.1471-4159.1996.66052057.x. [DOI] [PubMed] [Google Scholar]
- Van Der Kloot W. Estimating the timing of quantal releases during end-plate currents at the frog neuromuscular junction. J Physiol. 1988;402:595–604. doi: 10.1113/jphysiol.1988.sp017224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J Biol Chem. 2000;275:17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- Weiss JS. Oxygen, ischemia and inflammation. Acta Physiol Scand. 1986;548:9–37. [PubMed] [Google Scholar]
- Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82:969–974. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.