

Abstract
Acute inhibition of NO synthesis decreases left ventricular (LV) work and external efficiency, but it is unknown whether compensatory mechanisms can limit the alterations in LV mechanoenergetics after prolonged NO deficiency. Eight chronically instrumented male mongrel dogs received 35 mg kg−1 day−1 of Nω-nitro-L-arginine methyl ester orally for 10 days to inhibit NO synthesis. At spontaneous beating frequency, heart rate, coronary blood flow, peak LV pressure, end-diastolic LV pressure and the maximum derivative of LV pressure (dP/dtmax) were not significantly different vs. baseline, whereas LV end-diastolic diameter (32.5 ± 1.0 vs. 37.6 ± 1.4 mm) and LV stroke work (515 ± 38 vs. 650 ± 44 mmHg mm), were reduced (all P < 0.05). The slope of the LV end-systolic pressure-diameter relationship was increased at 10 days vs. baseline (13.9 ± 1.0 vs. 9.6 ± 0.9 mmHg mm−1, P < 0.05), while the end-diastolic LV diameter was smaller at matched LV end-diastolic pressures. At fixed heart rate (130 beats min−1), cardiac oxygen consumption was increased (12.2 ± 1.5 vs. 9.9 ± 1.0 ml min−1), and the ratio between stroke work and oxygen consumption was decreased by 33 ±7 % (all P < 0.05) after NO inhibition. We conclude that sustained inhibition of NO synthesis in dogs causes a decrease in LV work despite an increased contractility, which is most probably due to reduced diastolic compliance and a decrease in external efficiency. Thus, prolonged NO deficiency is not compensated for on the level of LV mechanoenergetics in vivo.
In normal hearts, the tonic release of NO directly influences several components of cardiac function. Acute inhibition of NO synthesis decreases contractile force in isolated rat hearts (Kojda et al. 1997), regional wall thickening in pigs (Heusch et al. 2000), left ventricular (LV) diameter shortening and stroke work in dogs (Puybasset et al. 1996; Recchia et al. 2002), and dP/dtmax in normal human hearts (Cotton et al. 2001). Strong evidence suggests that endogenous NO also supports diastolic LV function. In isolated hearts, NO synthase (NOS) inhibition elevates LV end-diastolic pressure, decreases the preload-recruitable rise in cardiac output and shifts the LV pressure-volume relationship leftwards and upwards (Prendergast et al. 1997). Finally, it has been shown in vitro that endogenous NO regulates cellular respiration (Clementi et al. 1996; Loke et al. 1999) and energy turnover (Gross et al. 1996; Shen et al. 2001). When this regulatory mechanism is lost due to acute inhibition of NO synthesis, cardiac mechanical efficiency decreases in isolated hearts (Shen et al. 2001), in anaesthetized pigs (Heusch et al. 2000; Post et al. 2001) and in conscious dogs (Bernstein et al. 1996; Recchia et al. 2002; Saavedra et al. 2002).
Whereas the changes in cardiac mechanics and oxygen consumption caused by acute NOS inhibition are well characterized, the effects of a sustained reduction in myocardial NO synthesis on LV mechanoenergetics have only partially been described. Hypothetically, a prolonged loss of NO-mediated modulation of ventricular efficiency might induce a gradual process of partial or total compensation. Puybasset et al. (1996) found that 7 days of NOS inhibition in chronically instrumented dogs causes a stable elevation in peripheral resistance counterbalanced by a decrease in cardiac output, but the authors did not measure cardiac contractility and oxygen consumption. Barouch et al. (2002) showed that the deletion of genes encoding endothelial and neuronal NOS alters cardiac mechanics in mice; however, the ratio between oxygen consumption and work was not determined. The aim of the present study was therefore to test whether alterations in cardiac function and mechanical efficiency are maintained or whether they are counterbalanced by compensatory mechanisms when the inhibition of NO synthesis is sustained for days. The experiments were performed in chronically instrumented dogs, which allowed simultaneous measurement of cardiac function and oxygen consumption, in vivo.
METHODS
Surgical instrumentation
Eight adult male mongrel dogs (body weight, 23-27 kg) were sedated with acepromazine maleate (1 mg kg−1 I.M.), anaesthetized with sodium pentobarbitone (25 mg kg−1 I.V.), and ventilated with room air. An adequate level of anaesthesia was maintained by intravenous injection of supplemental doses of pentobarbitone (3 mg kg−1 I.V.) every hour. The entire surgical procedure was carried out under aseptic conditions. A thoracotomy was performed in the left fifth intercostal space. One catheter was placed in the descending thoracic aorta, and a second catheter was placed in the coronary sinus with the tip leading away from the right atrium. A solid-state pressure gauge (P6.5; Konigsberg Instruments) was inserted into the left ventricle through the apex. A Doppler flow transducer (Craig Hartley) was placed around the left circumflex coronary artery, and a pair of 3 MHz piezoelectric crystals was fixed on opposing endocardial surfaces at the base of the left ventricle. A human, screw-type, unipolar myocardial pacing lead was fixed in the LV wall. A hydraulic occluder was placed around the inferior vena cava. Wires and catheters were run subcutaneously to the intrascapular region, the chest was closed in layers, and the pneumothorax was reduced. After surgery, dogs underwent analgesic therapy for 3 days (buprenorphine, 0.1 mg kg−1 I.M.) and antibiotic therapy for 10 days (amoxicillin, 16 mg kg−1 day−1 I.M.). Dogs responded very well to this therapeutic regimen, therefore post-surgical fever and weight loss were only transient. Ten to fourteen days were sufficient for a full recovery, which was indicated by normal ambulation, appetite, and faecal and urine output. Once this phase was completed, dogs were trained to lie quietly on the laboratory table. Rectal temperature was monitored daily for the entire duration of the protocol and, if found to be higher than 39 °C, amoxicillin (16 mg kg−1) or cefazolin (40 mg kg−1) was given I.M. We have used these methods in previous studies (Recchia et al. 1998, 2002; Osorio et al. 2002). Once the experimental protocol was completed, the dogs were killed with an overdose of sodium pentobarbitone (100 mg kg−1 I.V.). The protocols were approved by the Institutional Animal Care and Use Committee of the New York Medical College and conform to the guiding principles for the care and use of laboratory animals published by the National Institutes of Health.
Haemodynamic recordings and blood gas measurements
The aortic catheter was attached to a P23ID strain-gauge transducer for measurement of aortic pressure. LV pressure was measured using the solid-state pressure gauge. The first derivative of LV pressure (LV dP/dt) was obtained using an operational amplifier (National Semiconductor LM 324). Blood flow in the left circumflex coronary artery was measured with a pulsed Doppler flowmeter (model 100; Triton Technology). LV diameter was measured by connecting the implanted piezoelectric crystals to an ultrasonic dimension gauge. All signals were recorded on an eight-channel direct-writing oscillograph (model RS 3800; Gould). The analog signals were also stored digitally on an AT-type computer via an analog-digital interface (National Instruments) at a sampling rate of 250 Hz. Blood gases were measured using a blood gas analyser (Instruments Laboratory) and oxygen concentration was measured using an haemoglobin analyser (CO-Oximeter, Instrumentation Laboratory).
Protocol
The experiments were conducted in conscious dogs trained to lie quietly on the laboratory table. After recording of systemic haemodynamics, coronary blood flow and LV diameter, two to three occlusions of the inferior vena cava, separated by 5 min intervals, were performed to obtain end-diastolic and end-systolic LV pressure-diameter data (Senzaki et al. 2000). Once the baseline haemodynamics were re-established, the heart was paced at 130 beats min−1, haemodynamics were recorded and paired blood samples were taken from the aortic and the coronary sinus catheter. Cardiac pacing was used to eliminate the confounding effect of changes in heart rate on myocardial oxygen consumption that could have affected the comparison between measurements obtained before and after NOS inhibition. NOS activity was inhibited by administering 35 mg kg−1 day−1 of Nω-nitro-L-arginine methyl ester (L-NAME, Sigma Aldrich) for 10 days. L-NAME was packed in capsules and administered orally once a day in the morning. After 10 days of L-NAME administration, measurements were repeated as described above.
The dose of L-NAME was chosen based on the well-known efficacy of 35 mg kg−1 in dogs (Puybasset et al. 1996; Recchia et al. 2002). In pilot experiments (n = 3), we performed an intra-atrial injection of veratrine (5 μg kg−1), which increases coronary blood flow by stimulating NO release (Zhao et al. 1995). After 10 days of L-NAME treatment, the absolute increase of left circumflex coronary blood flow fell from 24 ± 7 to 4 ± 3 ml min−1, confirming a persistent NO synthase inhibition. Moreover, we injected 35 mg kg−1 of L-NAME intravenously after 10 days of L-NAME administration and we did not observe any haemodynamic change, confirming that systemic NO synthase was already inhibited by saturating levels of L-NAME.
Haemodynamic and mechanoenergetic measurements
Digitized data were analysed off-line by custom-made software. The parameters were determined during one respiratory cycle and comprised heart rate, mean aortic pressure, LV end-diastolic, peak systolic and end-systolic pressure, mean blood flow in the left circumflex coronary artery, the maximum and minimum of the first derivative of LV pressure and end-diastolic and end-systolic LV diameter. The percentage shortening of the short axis diameter was calculated as the ratio of the difference between end-diastolic and end-systolic diameter (stroke dimension) to end-diastolic diameter (Suga, 1990; Saavedra et al. 2002). The slope of the LV end-systolic pressure-diameter relationship (Ees) was calculated using the first 7-10 beats during inferior vena cava occlusion before heart rate would increase (Senzaki et al. 2000; Saavedra et al. 2002). Effective arterial elastance (Ea), an index of LV afterload (Sunagawa et al. 1983; Senzaki et al. 2000), was calculated as the ratio of LV end-systolic pressure to stroke dimensions. To study diastolic function, end-diastolic pressure-diameter data points of one respiratory cycle and of the first 7-10 beats during inferior vena cava occlusion were grouped and averaged in intervals of 2 mmHg. Heart rate did not change during this initial period of vena cava occlusion. From each experiment we obtained data at four selected levels of end-diastolic pressure, necessary for this comparison: 0, 2, 4 and 6 mmHg (average values). At each level of end-diastolic pressure 5 ± 1 beats were averaged in each single experiment. A statistical comparison was then made between mean values of end-diastolic diameter, before and after NOS inhibition, corresponding to the same levels of end-diastolic pressure. An index of stroke work (SW) was determined by calculating the area of the LV pressure-diameter loop during a cardiac cycle (Recchia et al. 1998; Senzaki et al. 2000; Saavedra et al. 2002). The pressure-diameter area (PDA) was calculated and used as a substitute of the pressure-volume area, one of the best predictors of cardiac oxygen consumption (Suga, 1990). To calculate PDA, the difference between end-systolic diameter and the x-intercept of the end-systolic pressure-diameter relationship (D0) was multiplied by end-systolic pressure and the product divided by two. This value, corresponding to internal work, was then added to the pressure-diameter loop (external or stroke work) to determine PDA. Finally, myocardial oxygen consumption (MV̇O2) was calculated by multiplying the arterial-coronary sinus difference in oxygen content by total mean coronary blood flow, assumed to be double the mean blood flow in the left circumflex coronary artery (Recchia et al. 1998, 2002; Osorio et al. 2002). Left ventricular external and total mechanical efficiency were then calculated as SW/MV̇O2 and PDA/MV̇O2 ratio (Suga, 1990; Saavedra et al. 2002).
Statistical analysis
All data are mean values ± s.e.m. Data before and after 10 days of L-NAME treatment were compared using Student's paired t test (Sigma Stat 2.03).
RESULTS
Haemodynamics and LV function
After 10 days of NO synthase inhibition, heart rate, mean arterial pressure, LV pressure, dP/dtmax, the minimum derivative of LV pressure (dP/dtmin) and mean coronary blood flow were not significantly different from baseline (Table 1), in spite of the twofold increase in Ea, an index of LV afterload (Fig. 1). In contrast, LV stroke dimension, short-axis diameter shortening and stroke work were significantly decreased (Table 1). While these indices of LV function were reduced, Ees was increased, indicating an enhanced contractility after prolonged NOS inhibition (Fig. 1 and Fig. 2). The corresponding value of D0 was not significantly changed after L-NAME administration (Table 1). At matched levels of LV end-diastolic pressure, LV end-diastolic diameter was smaller after L-NAME vs. control (Fig. 3), indicating a reduced diastolic compliance. The end-diastolic pressure corresponding to the smallest diameter point after L-NAME is slightly below zero (−0.22 ± 0.14 mmHg) since slightly more data points within the respective interval of end-diastolic pressure, from −1 to +1 mmHg, had a negative rather than a positive value.
Table 1.
Haemodynamics before and after 10 days of l-NAME administration, with spontaneous heart rate
| Baseline | 10 days l-NAME | |
|---|---|---|
| HR (beats min−1) | 89 ± 6 | 96 ± 7 |
| MAP (mmHg) | 106 ± 3 | 111 ± 5 |
| LVPmax (mmHg) | 132 ± 4 | 142 ± 5 |
| LVPes (mmHg) | 117 ± 6 | 126 ± 3 |
| LVPed (mmHg) | 6 ± 1 | 4 ± 1 |
| LcxCBF (ml min−1) | 32.3 ± 5.2 | 25.5 ± 5.1 |
| dP/dtmax (mmHg s−1) | 3151 ± 176 | 3302 ± 122 |
| dP/dtmin (mmHg s−1) | 2815 ± 141 | 3305 ± 154 |
| DIAed (mm) | 37.6 ± 1.4 | 32.5 ± 1.0* |
| DIAes (mm) | 31.6 ± 1.4 | 29.3 ± 1.1* |
| ΔDIA (mm) | 5.9 ± 0.5 | 3.3 ± 0.4* |
| %ΔDIA | 15.8 ± 1.4 | 10.1 ± 1.3* |
| SW (mm mmHg) | 650 ± 44 | 515 ± 38* |
| D0 (mm) | 18.0 ± 1.6 | 20.2 ± 1.1 |
Data are presented as means ± s.e.m. HR, heart rate; MAP, mean arterial pressure; LVPmax, peak LV pressure; LVPes end-systolic LV pressure; LVPed, end-diastolic LV pressure; LcxCBF, mean blood flow in the left circumflex coronary artery; dP/dtmax, maximum first derivative of LV pressure; dP/dtmin, minimum first derivative of LV pressure; DIAed, end-diastolic LV diameter; DIAes, end-systolic LV diameter; ΔDIA, LV diameter shortening; %ΔDIA, percentage LV diameter shortening; SW, LV stroke work; D0-, x-intercept fo the end-systolic pressure-diameter relationship. n = 8
P < 0.05 vs. baseline.
Figure 1. Representative pressure-diameter loops from a dog before and after 10 days of l-NAME treatment.
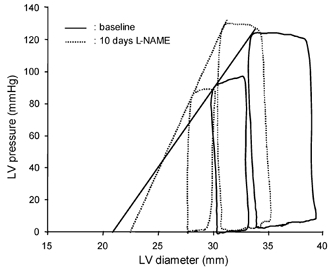
After l-NAME, the slope of the end-systolic pressure-diameter (Ees) is increased, but the pressure-diameter loop is shifted to the left and diameter shortening is decreased.
Figure 2.
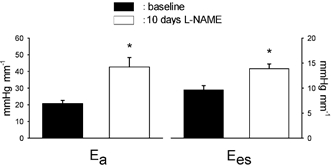
Effective arterial elastance (Ea) and slope of the end-systolic pressure-diameter relationship (Ees) are increased after 10 days of NOS inhibition. n = 8, * P < 0.05 vs. baseline.
Figure 3. End-diastolic pressure-diameter data obtained during inferior vena cava occlusion before and after 10 days of NOS inhibition at spontaneous heart rate.
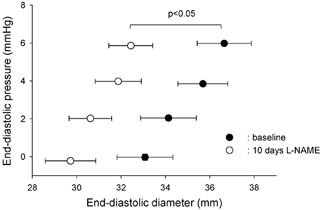
At matched end-diastolic pressures, LV diameter is significantly smaller after l-NAME, indicating reduced diastolic compliance. n = 8, * P < 0.05 vs. baseline.
Cardiac oxygen consumption and mechanical efficiency
When heart rate was fixed by cardiac pacing at 130 beats min−1, LV pressure, aortic pressure and dP/dtmax were not significantly different after 10 days of L-NAME administration compared to baseline, whereas SW was still reduced: 379 ± 25 vs. 473 ± 17 mmHg mm (P < 0.05). PDA was not significantly different (1108 ± 109 vs. 1328 ± 73 mmHg mm) vs. baseline. Values of coronary blood flow, blood oxygen content and MV̇O2 at fixed heart rate are presented in Table 2. After L-NAME treatment, coronary blood flow and arterial oxygen content did not change significantly vs. baseline. On the other hand, the arterial-coronary sinus difference in blood oxygen content was significantly higher in L-NAME-treated dogs. This caused a significant increase in MV̇O2 after L-NAME. Total mechanical efficiency, given by the ratio PDA/MV̇O2, decreased from 141 ± 19.0 to 96 ± 11.5 mmHg mm min ml−1 after NOS inhibition (P < 0.05), and external efficiency, represented by the ratio SW/MV̇O2, fell from 49.1 ± 5.0 at baseline to 32.7 ± 4.8 mmHg mm min ml−1 after NOS inhibition (P < 0.05). Figure 4 summarizes the percentage changes vs. baseline of SW, PDA, MV̇O2, SW/MV̇O2 and PDA/MV̇O2.
Table 2.
CBF, arterial O2 content, A−CS O2 difference and MV̇O2 before and after 10 days of l-NAME administration, with heart rate fixed at 130 baats min−1
| Baseline | 10 days l-NAME | |
|---|---|---|
| LcxCBF (ml min−1) | 39.0 ± 4.1 | 43.5 ± 4.0 |
| Arterial O2 content (vol%) | 16.3 ± 0.7 | 16.8 ± 1.0 |
| CS O2 content (vol%) | 4.1 ± 0.3 | 3.5 ± 0.4 |
| A-CS O2 difference (vol%) | 12.2 ± 0.5 | 14.0 ± 0.8* |
| MV̇O2 (ml min−1) | 9.9 ± 1.0 | 12.4 ± 1.5* |
Data are presented as means ± s.e.m. CBF, coronary blood flow; LcxCBF, mean blood flow in the left circumflex coronary artery; CS, coronary sinus; A−CS, arterial-coronary sinus. n = 8
P < 0.05 vs. baseline.
Figure 4. Relative changes in MV̇O2, stroke work (SW), pressure-diameter area (PDA), SW/MV̇O2 ratio and PDA/MV̇O2 ratio after 10 days of NOS inhibition.
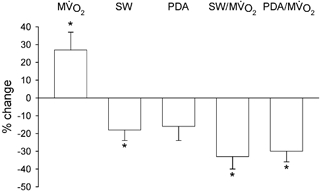
Heart rate was fixed at 130 beats min−1. SW/MV̇O2 and PDA/MV̇O2 were significantly decreased, indicating reduced LV efficiency. n = 8, * P < 0.05 vs. baseline.
DISCUSSION
The novel findings of our study are that, after 10 days of NOS inhibition: (1) LV systolic shortening and stroke work were reduced despite an increased contractility; (2) end-diastolic diameters were markedly reduced at matched filling pressures, indicating a decreased diastolic compliance, and (3) a concomitant decrease in LV stroke work and increase in cardiac oxygen consumption caused a fall in LV mechanical efficiency.
Mean arterial pressure was not significantly different from control after prolonged inhibition of NO synthesis, consistent with data reported by Puybasset et al. (1996). These authors have elegantly shown that mean arterial pressure returns to control levels within the first 24 h of NOS blockade, due to the reduction in cardiac output that compensates for the elevated peripheral resistance. However, they did not assess LV dimensions and contractility. Although we did not measure aortic flow, LV internal diameter sonomicrometry allowed us to determine changes in Ea, an established index that incorporates both steady and pulsatile components of LV afterload (Segers et al. 2002), and in Ees, a preload- and afterload-independent index of contractility (Suga, 1990). Ea was doubled after L-NAME treatment, confirming that a sustained inhibition of NO synthesis raises LV afterload. Moreover, the decrease in LV end-diastolic and end-systolic diameter found in the present study strongly suggests that a prolonged NOS inhibition caused a marked reduction in cardiac output. It is noteworthy that LV volume is a third-order function of LV diameter (Suga & Sagawa, 1974), therefore changes in short axis dimensions corresponded to much more pronounced changes in stroke volume. This finding, consistent with the reduced contractile function previously observed after acute NOS inhibition (Recchia et al. 2002; Saavedra et al. 2002), could have led to the erroneous conclusion that the chronic loss of NO synthesis causes a decrease in cardiac contractility. However, by calculating Ees we showed that cardiac inotropic state was not depressed; rather it increased by about 40 % after prolonged inhibition of NO synthesis. It seems, therefore, that the most prominent mechanism by which sustained NOS inhibition reduced cardiac output was not a diminished contractility but a combined effect of LV afterload and preload. In fact, the diminished LV diameter shortening indicated that LV ejection was limited, due to a higher afterload not sufficiently counterbalanced by an improved contractility, while the leftward shift of the end-diastolic pressure-diameter relationship limited stroke volume and cardiac output by restricting cardiac filling. On the other hand, dP/dtmax did not change significantly after 10 days of L-NAME administration, probably due to the net effect of a lower preload and increased contractility. The mechanisms that potentiated cardiac contractility could be numerous. Bartunek et al. (2000), for instance, have demonstrated an increased contractile responsiveness to calcium in hearts isolated from rats after 6 weeks of NOS inhibition. NOS I isoform knockout mice show baseline hypercontractility in vivo (Ashley et al. 2002; Barouch et al. 2002) along with an increased calcium current (ICa) and a higher calcium load of the sarcoplasmatic reticulum (Sears et al. 2003).
The reduced LV diastolic compliance was a second important finding of the present study. In contrast to the effects of acute NOS inhibition in dogs (Recchia et al. 2002; Saavedra et al. 2002), we observed a significant and pronounced decrease in end-diastolic diameter after sustained NOS inhibition. This decrease was independent of end-diastolic pressure and could therefore not be attributed to altered loading conditions, but strongly indicated a shift of the underlying end-diastolic pressure-diameter relationship towards smaller diameters. Similarly, rats undergoing 6 weeks of NOS inhibition display a decreased LV end-diastolic diameter at unchanged end-diastolic pressure in vivo (Bartunek et al. 2000). In addition, hearts isolated from rats after 8 weeks of NOS inhibition presented a leftward shift of the diastolic pressure-volume relationship (Matsubara et al. 1998). Barouch et al. (2002) have recently found that mice knockout for NOS III or NOS I/III genes had decreased ventricular volumes. Although none of these studies determined diastolic pressure-volume or pressure-diameter relationships in vivo, their results are in close concordance with our findings in conscious dogs subjected to prolonged blockade of all NOS isoforms. As recently pointed out by Burkhoff et al. (2003), a leftward shift of the end-diastolic pressure-volume or pressure-diameter relationship is the pathophysiological basis of decreased cardiac output induced by diastolic dysfunction. Interestingly, in dogs with pacing-induced heart failure, the critical increase in end-diastolic pressure during cardiac decompensation correlates closely with the loss of myocardial NO production (Recchia et al. 1998), and, in guinea-pigs with aortic banding, cardiac decompensation and lung congestion are paralleled by a decrease of eNOS expression and coronary vascular NO bioactivity (Grieve et al. 2001). Similarly, in patients with dilated cardiomyopathy, preload reserve is directly correlated to the expression of NO synthase isoforms (Heymes et al. 1999), while the administration of NO donors increases LV diastolic distensibility in normal (Paulus et al. 1994) and in hypertrophied human hearts (Matter et al. 1999).
Direct regulatory actions of NO on the mitochondrial respiratory chain, on enzyme activities and on calcium transients through the sarcoplasmic reticulum have been clearly defined in vitro (Gross et al. 1996; Han et al. 1998; Clementi et al. 1999; Loke et al. 1999; Shen et al. 2001), and a number of in vivo studies from our laboratory and others have showed an increased MV̇O2 after acute NOS inhibition (Recchia et al. 2002; Saavedra et al. 2002). However, MV̇O2 measurements have not been performed in previous studies of chronic NOS inhibition (Puybasset et al. 1996), or in NOS I/III knockout mice (Barouch et al. 2002), given the difficulty in sampling coronary sinus blood from mice. Very interestingly, the same increase in MV̇O2 observed after acute NOS inhibition (Recchia et al. 2002; Saavedra et al. 2002) was also found by us after days of reduced NO synthesis, when the sum of internal and external work (PDA) tended to be lower and LV external work was decreased. This indicates a fall in cardiac efficiency that could not be attenuated by compensatory mechanisms. Myocardial NO production therefore seems to be indispensable for the economy of myocardial oxygen usage. In this regard, we cannot exclude the possibility that the partial oxygen wastage, due to the lack of endogenous NO, limited oxygen availability in the sub-endocardial muscular layer, contributing to the reduced diastolic compliance. On the other hand, global MV̇O2 was sufficient to support a significant increase in LV contractility.
Although gene knockout mice constitute very powerful tools for investigating the physiological role of the various NOS isoforms, our dog model offers several advantages. First, the size of canine hearts allowed us to determine MV̇O2, in vivo, by employing established methods of direct O2 measurement in arterial and coronary sinus blood that are presently unfeasible in mice. Second, dogs were studied in the conscious state, therefore the experiments were devoid of possible confounding effects of anaesthesia, mechanical ventilation and surgical trauma that accompany invasive assessments of cardiac mechanics in mice. Third, chronic NO synthase blockade was generated in adult animals which did not develop any compensatory adaptation consequent to inborn genotypic alterations.
In conclusion, our results support an important role for endogenous NO in the maintenance of LV function in vivo. Sustained inhibition of NO synthesis causes a decrease in LV work despite an augmented contractility, primarily due to the combined effect of a higher afterload and impaired LV diastolic filling, which is furthermore accompanied by a fall in mechanical efficiency. Therefore, the prolonged loss of NO regulatory action on LV mechanoenergetics is not compensated for by long-term adaptive mechanisms.
Acknowledgments
This study was supported by the National Heart, Lung and Blood Institute grant RO1 HL-62573 (F.A.R.) and in part by PO1 HL-43023 (T.H.H.). H.P. was the recipient of a grant of the Deutsche Forschungsgemeinschaft (PO 612 1/1).
REFERENCES
- Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105:3011–3016. doi: 10.1161/01.cir.0000019516.31040.2d. [DOI] [PubMed] [Google Scholar]
- Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke BA, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- Bartunek J, Weinberg EO, Tajima M, Rohrbach S, Katz SE, Douglas PS, Lorell BH. Chronic N(G)-nitro-L-arginine methyl ester-induced hypertension: novel molecular adaptation to systolic load in absence of hypertrophy. Circulation. 2000;101:423–429. doi: 10.1161/01.cir.101.4.423. [DOI] [PubMed] [Google Scholar]
- Bernstein RD, Ochoa FY, Xu X, Forfia P, Shen W, Thompson CI, Hintze TH. Function and production of nitric oxide in the coronary circulation of the conscious dog during exercise. Circ Res. 1996;79:840–848. doi: 10.1161/01.res.79.4.840. [DOI] [PubMed] [Google Scholar]
- Burkhoff D, Maurer MS, Packer M. Heart failure with normal ejection farction: is it really a disorder of diastolic function. Circulation. 2003;107:656–658. doi: 10.1161/01.cir.0000053947.82595.03. [DOI] [PubMed] [Google Scholar]
- Cotton JM, Kearney MT, MacCarthy PA, Grocott-Mason RM, McClean DR, Heymes C, Richardson PJ, Shah AM. Effects of nitric oxide synthase inhibition on basal function and the force-frequency relationship in the normal and failing human heart in vivo. Circulation. 2001;104:2318–2323. doi: 10.1161/hc4401.098515. [DOI] [PubMed] [Google Scholar]
- Grieve DJ, MacCarthy PA, Gall NP, Cave AC, Shah AM. Divergent biological actions of coronary endothelial nitric oxide during progression of cardiac hypertrophy. Hypertension. 2001;38:267–273. doi: 10.1161/01.hyp.38.2.267. [DOI] [PubMed] [Google Scholar]
- Gross WL, Bak MI, Ingwall JS, Arstall MA, Smith TW, Balligand JL, Kelly RA. Nitric oxide inhibits creatine kinase and regulates rat heart contractile reserve. Proc Natl Acad Sci U S A. 1996;93:5604–5609. doi: 10.1073/pnas.93.11.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, Huang P, Fishman MC, Michel T, Kelly RA. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:6510–6515. doi: 10.1073/pnas.95.11.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G, Post H, Michel MC, Kelm M, Schulz R. Endogenous nitric oxide and myocardial adaptation to ischemia. Circ Res. 2000;87:146–152. doi: 10.1161/01.res.87.2.146. [DOI] [PubMed] [Google Scholar]
- Heymes C, Vanderheyden M, Bronzwaer JG, Shah AM, Paulus WJ. Endomyocardial nitric oxide synthase and left ventricular preload reserve in dilated cardiomyopathy. Circulation. 1999;99:3009–3016. doi: 10.1161/01.cir.99.23.3009. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Noack E. Inhibition of nitric oxide synthase and soluble guanylate cyclase induces cardiodepressive effects in normal rat hearts. Eur J Pharmacol. 1997;334:181–190. doi: 10.1016/s0014-2999(97)01168-0. [DOI] [PubMed] [Google Scholar]
- Loke KE, McConnell PI, Tuzman JM, Shesely EG, Smith CJ, Stackpole CJ, Thompson CI, Kaley G, Wolin MS, Hintze TH. Endogenous endothelial nitric oxide synthase-derived nitric oxide is a physiological regulator of myocardial oxygen consumption. Circ Res. 1999;84:840–845. doi: 10.1161/01.res.84.7.840. [DOI] [PubMed] [Google Scholar]
- Matsubara BB, Matsubara LS, Zornoff LA, Franco M, Janicki JS. Left ventricular adaptation to chronic pressure overload induced by inhibition of nitric oxide synthase in rats. Basic Res Cardiol. 1998;93:173–181. doi: 10.1007/s003950050084. [DOI] [PubMed] [Google Scholar]
- Matter CM, Mandinov L, Kaufmann PA, Vassalli G, Jiang Z, Hess OM. Effect of NO donors on LV diastolic function in patients with severe pressure-overload hypertrophy. Circulation. 1999;99:2396–2401. doi: 10.1161/01.cir.99.18.2396. [DOI] [PubMed] [Google Scholar]
- Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia FA. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106:606–612. doi: 10.1161/01.cir.0000023531.22727.c1. [DOI] [PubMed] [Google Scholar]
- Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89:2070–2078. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- Post H, Schulz R, Gres P, Heusch G. No involvement of nitric oxide in the limitation of beta-adrenergic inotropic responsiveness during ischemia. Am J Physiol Heart Circ Physiol. 2001;281:H2392–2397. doi: 10.1152/ajpheart.2001.281.6.H2392. [DOI] [PubMed] [Google Scholar]
- Prendergast BD, Sagach VF, Shah AM. Basal release of nitric oxide augments the Frank-Starling response in the isolated heart. Circulation. 1997;96:1320–1329. doi: 10.1161/01.cir.96.4.1320. [DOI] [PubMed] [Google Scholar]
- Puybasset L, Bea ML, Ghaleh B, Giudicelli JF, Berdeaux A. Coronary and systemic hemodynamic effects of sustained inhibition of nitric oxide synthesis in conscious dogs. Evidence for cross talk between nitric oxide and cyclooxygenase in coronary vessels. Circ Res. 1996;79:343–357. doi: 10.1161/01.res.79.2.343. [DOI] [PubMed] [Google Scholar]
- Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. 1998;83:969–979. doi: 10.1161/01.res.83.10.969. [DOI] [PubMed] [Google Scholar]
- Recchia FA, Osorio JC, Chandler MP, Xu X, Panchal AR, Lopaschuk GD, Hintze TH, Stanley WC. Reduced synthesis of NO causes marked alterations in myocardial substrate metabolism in conscious dogs. Am J Physiol. 2002;282:E197–206. doi: 10.1152/ajpendo.2002.282.1.E197. [DOI] [PubMed] [Google Scholar]
- Saavedra WF, Paolocci N, St John ME, Skaf MW, Stewart GC, Xie JS, Harrison RW, Zeichner J, Mudrick D, Marban E, Kass DA, Hare JM. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res. 2002;90:297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:52–59. doi: 10.1161/01.RES.0000064585.95749.6D. [DOI] [PubMed] [Google Scholar]
- Segers P, Stergiopulos N, Westerhof N. Relation of effective arterial elastance to arterial system properties. Am J Physiol Heart Circ Physiol. 2003;282:H1041–1046. doi: 10.1152/ajpheart.00764.2001. [DOI] [PubMed] [Google Scholar]
- Senzaki H, Isoda T, Paolocci N, Ekelund U, Hare JM, Kass DA. Improved mechanoenergetics and cardiac rest and reserve function of in vivo failing heart by calcium sensitizer EMD-57033. Circulation. 2000;101:1040–1048. doi: 10.1161/01.cir.101.9.1040. [DOI] [PubMed] [Google Scholar]
- Shen W, Tian R, Saupe KW, Spindler M, Ingwall JS. Endogenous nitric oxide enhances coupling between O2 consumption and ATP synthesis in guinea pig hearts. Am J Physiol Heart Circ Physiol. 2001;281:H838–846. doi: 10.1152/ajpheart.2001.281.2.H838. [DOI] [PubMed] [Google Scholar]
- Suga H. Ventricular energetics. Physiol Rev. 1990;70:247–277. doi: 10.1152/physrev.1990.70.2.247. [DOI] [PubMed] [Google Scholar]
- Suga H, Sagawa K. Assessment of absolute volume from diameter of the intact canine left ventricular cavity. J Appl Physiol. 1974;36:496–499. doi: 10.1152/jappl.1974.36.4.496. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Maughan WL, Burkhoff D, Sagawa K. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol Heart Circ Physiol. 1983;245:H773–780. doi: 10.1152/ajpheart.1983.245.5.H773. [DOI] [PubMed] [Google Scholar]
- Zhao G, Shen W, Xu X, Ochoa M, Bernstein R, Hintze TH. Selective impairment of vagally mediated, nitric oxide-dependent coronary vasodilation in conscious dogs after pacing-induced heart failure. Circulation. 1995;91:2655–2663. doi: 10.1161/01.cir.91.10.2655. [DOI] [PubMed] [Google Scholar]