

Abstract
The aim of this study was to determine the effects of a clinically relevant single course of prenatal betamethasone in the rat on growth parameters with particular reference to brain cell proliferation and apoptosis. We report that administration of 170 μg kg−1 betamethasone twice within 4 h to E20 pregnant rats conveys moderate somatic growth retardation. Further, using a measure of brain cell proliferation independent of blood-brain barrier (BBB) permeability, we demonstrate for the first time that betamethasone is chronically anti-proliferative to brain cells without inducing caspase-3-mediated apoptosis. More importantly we show that there is a significant and sexually divergent rebound of neural proliferation which occurs earlier in males than in females and continues until at least 21 days of postnatal life. BBB permeability to [3H]thymidine was significantly increased by steroid treatment re-iterating the fact that tracer studies not correcting for BBB permeability, such as bromodeoxyuridine (BrdU), may be questionable in this type of study. Further, prenatal steroid treatment did not alter postnatal corticosterone levels. In summary we show that prenatal betamethasone conveys significant and long-lasting side effects and that its human clinical application in preterm labour needs more careful consideration as compared to the relative ease with which it is prescribed today.
Ever since the pioneering work of Liggins and Howie in the early 1970s, (Liggins & Howie, 1972) betamethasone has been used in the prevention of respiratory distress syndrome (RDS) in premature babies (between 25 and 34 weeks gestation). This aids maturation of the fetal lungs (Ballard & Ballard, 1995), lowers the risk of cystic periventricular leucomalacia (cPVL) and the incidence of intracranial haemorrhage in neonates (Crowley, 1995). In humans little is known about the long- or short-term side effects of a single course of prenatal betamethasone other than that it retards birth weight and head circumference at birth (Thorp et al. 2002).
In animal models, prenatal glucocorticoids have been shown to cause a range of serious side effects depending on the dosage, the gestational age of the fetus and the species used. Steroid treatment during the third trimester has been shown to cause: growth retardation (LaBorde et al. 1992), reduction of cerebellar DNA content (Velazquez & Romano, 1987), reduced brain growth (Huang et al. 1999), hypomyelination (Dunlop et al. 1997), increased susceptibility of cerebellar neurons to oxidative cell death and enhancement of the maturational state of neural cells already present during treatment (Fuentes-Pardo et al. 1990). Treatment of non-human primates with pharmacological doses of prenatal glucocorticoids has been shown to induce acute neural degeneration within the hippocampus which is still evident at birth and at 9 months of age (Uno et al. 1994). In a more recent primate study, using a clinically relevant treatment protocol of a single course of prenatal betamethasone, decreased levels of expression of neuronal cytoskeletal proteins and of the presynaptic marker synaptophysin were found in the fetal brain (Antonow-Schlorke et al. 2003).
The primary concern with extrapolating many of the aforementioned animal studies into the human situation is that very few of the studies have used a clinically relevant dose. Often, doses have been increased until significant results are found. Our primary aim in this study was to determine the effects of a clinically relevant single course of prenatal betamethasone on somatic growth parameters and in particular, on neural measures of brain growth and the balance between brain cell proliferation and apoptosis within the rat.
METHODS
Ethics
The animal studies described here were all approved by the Animal Ethics Board of the University of Maastricht, The Netherlands. At the end of the experiment all adult animals were killed by carbon dioxide inhalation (unless stated otherwise). They were initially exposed to a mixture of O2 and CO2 for 3-4 min (in a sealed container) after which the O2 was discontinued for 1-2 min. Animals were left in pure CO2 for ≈1 h to ensure they were dead.
Animals
Normal 3-month-old time pregnant Fisher 344 dams (Charles River, The Netherlands; pregnancy confirmed by vaginal plug) were delivered to our animal facility on day 14 of pregnancy (E14) and randomly assigned to a treatment group of vehicle, low dose or high dose. The animals were kept under standard laboratory conditions with 12 h light/12 h dark and standard rat chow and water ad libitum. Fisher 344 rats were chosen because these are a pure breeding inbred strain with low heterogeneity and they are known to be reasonably stress responsive. For this study we took the pups at postnatal days (P)1, P2 and P21. For the time between P2 and P21 the pups were all cross-fostered to dams who had given birth on the same day and had received vehicle-only treatment (to prevent a possible betamethasone effect on maternal behaviour influencing the results (Brabham et al. 2000)). All the litters were kept at eight pups per dam and the pups were kept with the mother until P21 (i.e. they were not weaned). The investigators were ignorant of the treatment groups as much as possible; however, due to growth retardation it was often obvious to which group the pups belonged.
Betamethasone treatment
As mentioned previously, women threatening to deliver preterm are still often administered 12 mg betamethasone (Celestone Chronodose) twice within 24 h. With an average weight of around 80 kg, this gives 170 μg kg−1 betamethasone with a plasma half-life of 6 h in the human. The second injection therefore occurs at an interval of two half-lives. Within the rat, betamethasone has a plasma half-life of 2 h (Tamvakopoulos et al. 2002). An equivalent dose of betamethasone for a rat would then be two doses of 170 μg kg−1, 4 h apart. A major concern with regard to the interpretation of animal studies examining the effects of glucocorticoids on neural measures is the comparison of the stage of brain development. Estimates of the rat equivalent age of a term human in respect of neural development have ranged from 7 to 24 days of postnatal age (Romijn et al. 1991; Clancy et al. 2001) with a general consensus that a 10- to 14-day-old rat is equivalent to a term human. Term rats are therefore equivalent to preterm to extreme preterm human infants, exactly those who receive prenatal glucocorticoids in utero. A further concern is the comparison of neural cell cycle times. Ideally we would expose the rat brain cells to betamethasone for the same number of cell cycles as occurs in the human. Fortuitously for us, the cell cycle time of the developing primate (and presumably human) brain is three to five times longer than that of a rodent fetus in the third trimester of pregnancy, which is similar to the difference in betamethasone half-life (Kornack & Rakic, 1998). Ideally we would also measure betamethasone levels reaching the fetus as there are distinct differences in placental morphology and transfer between the rat and the human, but this was not carried out.
Betamethasone (Celestone Chronodose, Schering-Plough, The Netherlands) was diluted in its own buffer to a concentration of 230 μg ml−1. The exact buffer recipe used in the Celestone Chronodose suspension is a proprietary secret of Schering-Plough and may not be published but it contains the following ingredients in low concentrations: betamethasone acetate, betamethasone disodium phosphate, EDTA, H2NaPO4, Na2HPO4 and benzalkonium chloride in MilliQ water. The animals were injected in the nape of the neck with the high dose (340 μg kg−1), low dose (170 μg kg−1) or vehicle-only at 10.00 and 14.00 h on day 20 of pregnancy (E20). All the animals delivered on day 22 of pregnancy which was designated as pup age P0. For birth measurements the pups were sexed, weighed and had their crown-tail (C-T) length and head diameter measured using a digital vernier caliper within 2 h of birth. All measurements were taken by the same investigator (A.S.) to preserve consistency. During this time the dam was never left without any pups and the pups were not separated from their dam for more than 3 min to minimise the stress levels incurred.
Bioactivity check
In order to check that the betamethasone preparation was bioactive in its diluted form and in our hands we used non-pregnant adult Fisher 344 females and treated these to the same betamethasone protocol as described above (n = 3 per group). Ideally this would have been measured in the pregnant dams but blood sampling is known to be stressful and is itself an inducer of growth retardation in offspring (Drago et al. 1999). Blood samples were taken by tail bleed 1 h before the first injection and at 2, 6, 24, 44 and 72 h after the second betamethasone injection. The animals remained on ad libitum food and water in standard housing conditions throughout the experiment. Blood glucose was measured immediately using the Accu-chek blood glucose metre (Roche, USA). Further blood samples were taken into heparinised blood collection tubes (Microvette CB300, Sarstedt, Germany) kept on ice and then centrifuged at 3000 g for 5 min at 4°C and the plasma frozen down to −20°C for subsequent determination of corticosterone by radioimmunoassay assay (RIA).
Tracer method
We used a well known, convenient and previously described proliferation assay (DiCicco-Bloom et al. 1993; Tao et al. 1996, 1997; Wolf et al. 1997; Wagner et al. 1999; Cheng et al. 2001; Scheepens et al. 2003) to measure brain region specific proliferation at 1, 2 and 21 days after birth. On their assigned day the pups were injected with a single dose of 5 μCi (g body weight)−1 [3H]thymidine ([3H]thy) (25 Ci mmol−1, 1 m Ci ml−1 in 0.9 % NaCl, Amersham Pharmacia Biotech, The Netherlands) by subcutaneous injection into the nape of the neck. The infusate was pre-warmed to 34°C to prevent cooling of the pups and the volume injected did not exceed 5 μl (g body weight)−1. Following [3H]thy infusion the pups were kept at 34°C and 75-85 % humidity in a paediatric incubator. Exactly 1 h later the pups were killed by decapitation. The brains were then quickly removed and dissected using the method of Wagner et al. (1999). Regions known to be actively proliferating postnatally were taken including: the sub ventricular zone (SVZ) contained within the rostral forebrain, the entire hippocampal formation, the olfactory bulbs and the cerebellum; the rest of the brain was also taken for analysis. The microdissected regions were then weighed, quickly snap frozen in liquid nitrogen and stored at −70°C. All dissections were performed by the same investigator (A.S.) to preserve consistency. Subsequently the heart, lungs, spleen, kidney and carcass were taken and weighed.
The dissected brain regions were then placed in pre-cooled lysis buffer containing: 137 mM NaCl, 20 mM Tris-HCl (pH 8.0), 1 % NP-40, 10 % glycerol and a complete protease inhibitor tablet (Roche, The Netherlands). The samples were then homogenised using a Mini-Bead Beater (Biospec products, OK, USA) three times for 30 s each time with cooling of the samples on ice between runs. An aliquot was then taken to determine the total amount of [3H]thy which had diffused into each brain region. Another aliquot of the same size was then taken and all cellular DNA and macromolecules were precipitated using a standard trichloroacetic acid (TCA) precipitation protocol. This precipitated sample represents the total amount of [3H]thy which was incorporated into DNA during the 1 h exposure. The percentage of [3H]thy incorporated into DNA compared to the total amount of [3H]thy present in that brain region then represents the number of S-phase cells or proliferative potential of the tissue (Tao et al. 1996, 1997; Wagner et al. 1999). This measure is independent of differences in diffusion of the tracer through the blood-brain barrier (BBB) and to the total amount of tracer infused.
Liquid scintillation counting
Homogenised tissue and DNA samples were solubilised in 1 ml Soluene-350 (Packard Instruments, The Netherlands) at 45-50°C for 2-24 h or until the samples were completely dissolved. A 5 ml volume of Hionic-Flour scintillation cocktail (Packard Instruments, The Netherlands) was then added and the samples were read for 20 min on a Wallac WinSpectral 1414 liquid scintillation counter. The appropriate quench curves were produced using tritium standards added to homogenised brain tissue and subsequently used to convert the sample counts per minute to disintegrations per minute.
Blood sampling and corticosterone radioimmunoassay
Trunk blood samples were taken from the pups at post mortem after a 1 h separation from the dam, a known stressor. Therefore, the measured corticosterone does not represent basal corticosterone level; however, the P1 and P2 pups are within the stress hyporesponsive period (SHRP; Henning, 1978; Scheepens et al. 2003) and it is unlikely that their corticosterone secretion is altered due to maternal separation; however, this is not the case for the P21 pups. Further, all the pups were kept in a warm, humid (34 °C, 75-85 % humidity) and quiet paediatric incubator and generally slept during the 1 h maternal separation. All pups were handled in exactly the same way and the high-, low- and vehicle-dose pups were taken alternately to prevent the influence of time of day on corticosterone levels between groups. For the determination of the plasma corticosterone concentrations, 50 μl of plasma was extracted with 3 ml dichloromethane and vortexed for 1 min. The corticosterone was then measured directly on 1 ml dried dichloromethane and extracted for radioimmunoassay using 125I-corticosterone as tracer and an anti-corticosterone serum. The radioimmunological reaction was performed overnight at 4°C. A second antibody system was used to separate bound and unbound steroid as previously described in detail (Sulon et al. 1978).
Caspase-3 activation assay
Caspase-3-like activity was measured in the six male and six female cerebellar, SVZ and hippocampal homogenates from each of the low-dose- and vehicle-treated groups and from all ages using the method previously described in detail by Puka-Sundvall et al. (2000a,b). In short, samples of homogenate (30 μl) were mixed on a 96-well microtitre plate (Dynex, USA) with 70 μl of extraction buffer, containing (mM): 50 Tris-HCl (pH 7.3), 100 NaCl, 5 EDTA, 1 EGTA, 3 NaN3, 1 phenylmethylsulphonyl fluoride (PMSF), 1 μg ml−1 pepstatin, 2.5 μg ml−1 leupeptin, 10 μg ml−1 aprotinin and 0.2 % 3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulphonate (CHAPS). After incubation for 15 min at room temperature, 100 μl of the extraction buffer was added but without protease inhibitors and CHAPS and including 4 mM dithiothreitol (DTT) and 50 μM peptide substrate Ac-DEVD-AMC (Biomol, Germany). Cleavage of Ac-DEVD-AMC was measured at 37 °C using a Spectramax Gemini microplate fluorometer (Molecular Devices, USA) at an excitation wavelength of 380 nm and an emission wavelength of 440 nm. The cleavage was followed starting 15 min after addition of all the reagents at 2 min intervals for 2.5 h. Fluorometric measures were corrected for the wet weight of tissue added and are expressed as fluorometric units per milligram wet weight per minute. To check the specificity of the assay, an additional 5 μl of 5 mg ml−1 DEVD-CHO (Biomol, Germany), a selective caspase-3 inhibitor, was added to one sample of each region at each time point. As a result of the addition of this selective inhibitor the reaction was completely inhibited thus confirming the assay specificity for caspase-3 proteolytic activity.
Statistics
We used a standardised and randomised block design for these studies whereby each dam had one of each sex of pup used at each time point to remove any litter effects (Chapman & Stern, 1978). All statistics were evaluated using a pair wise two-way ANOVA analysis for either dose by time or dose by sex comparison. Post hoc tests were performed using the Bonferroni t test corrected for repeated measures. Differences in litter size were tested using a one-way ANOVA. Mortality was tested using the Fisher exact test. All statistics were carried out using SigmaStat software version 2.03 (SPSS Inc., USA). Statistical significance was assumed to exist at P ≤ 0.05. All data are presented as means ± standard error of the mean (s.e.m.).
RESULTS
Bioactivity check
Both the high and low dose of betamethasone caused a rapid and long-lasting, dose-dependent hyperglycaemia which continued for 24 h after the second injection (Fig. 1A) as expected. There was a significant overall effect for dose (dose: F(2,30) = 14.5, P = 0.005), a time effect (time: F(5,30) = 42.6, P > 0.001) and an interaction (dose × time: F(10,30) = 9.7, P < 0.001). Further, both high and low doses caused rapid and long-lasting hypocorticosteronaemia returning to control values by 44 h (Fig. 1B). This also showed a significant dose effect (dose: F(2,36) = 16.4, P < 0.001), time effect (time: F(5,30) = 2.5, P = 0.046) and interaction effect (dose × time: F(10,36) = 3.8, P = 0.001). The increasing level of plasma corticosterone within the vehicle group was due to the stress induced by the vehicle injections and three tail bleeds which took place on the first day.
Figure 1. Bioactivity check.

A, the betamethasone-induced hyperglycaemia in a separate set of adult, female animals. There is a significant dose, time and interaction effect (see Results). Glucose levels returned to control values by 24 h after the second (last) betamethasone injection. B, illustrates the hypocorticosteronaemia induced by both the high and low betamethasone doses in the same animals. Values represent means ± s.e.m., n = 3 per dose; symbols used for post hoc differences; * P < 0.05, ** P < 0.001 as compared to the vehicle only group.
Mortality and litter size
Of the 151 pups used for this study, four pups died and all were from betamethasone-treated dams. However, betamethasone-induced mortality was not significant (Fisher exact test). Further, one high-dose dam died during delivery and, as this is unusual, we performed a post mortem examination but found no obvious cause for death. There was no difference in litter size between the groups (litter size: F(2,12) = 0.26, P = 0.774)
Birth weight
Betamethasone caused significant dose-dependent growth retardation at birth (P0) in both the high- and low-dose groups and in both males and females (Fig. 2). There was a significant overall effect for dose (dose: F(2,151) = 27.0, P < 0.001) and for gender (sex: F(1,151) = 48.3, P > 0.001), but no interaction between the two.
Figure 2. Overview birth measures.
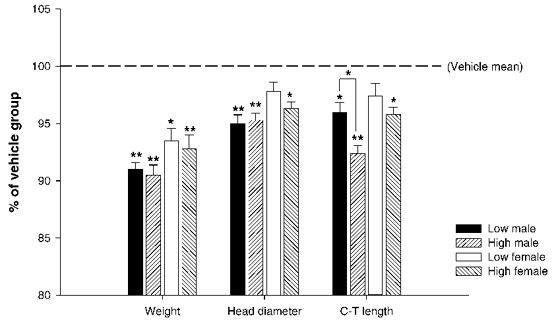
Graph showing growth retardation in both the high and low betamethasone-treated groups for all measures. Values are expressed as percentages of the vehicle-only group. Note that a true dose effect exists only for the crown-tail (C-T) length in males; there are no significant differences between the high and low groups for any other measure. An overall dose and sex effect was found for all three measures (P < 0.001), but without a significant interaction. Values represent means ± s.e.m., n = 24 to 31 per dose per sex; * P < 0.05 compared to vehicle group, ** P < 0.001 compared to vehicle group.
Crown-tail length at birth
Betamethasone caused a dose-specific retardation of C-T length in all the animals at birth (Fig. 2). The overall effects of betamethasone were: (dose: F(2,151) = 20.0, P < 0.001), (sex: F(1,151) = 21.3, P < 0.001). Post hoc analysis showed that there was also a significant dose-response with a clear difference between the low and high doses in males, with the high dose causing a further reduction in C-T length at birth.
Head diameter at birth
Head diameter at birth was also significantly reduced due to the steroid treatment (Fig. 2). The overall effects were: (dose: F(2,151) = 15.3, P < 0.001), (sex: F(1,151) = 11.0, P < 0.001). Post hoc analysis showed that the low dose within the female group did not per se cause retardation in head diameter (P = 0.167), but the high dose did (P = 0.003). Within males, both the high and low doses caused a significantly reduced head diameter at birth.
Postnatal somatic growth
Somatic growth was clearly inhibited by prenatal steroid treatment during the first 21 days of postnatal life (Table 1). At P1 there was an (sex non-specific) overall reduction in body weight related to the dose (dose: F(2,42) = 10.5, P < 0.001), as for head diameter (dose: F(2,42) = 7.7, P = 0.001) and C-T length (dose: F(2,42) = 10.1, P < 0.001). At P2 this effect continued with overall dose effects for body weight (dose: F(2,42) = 12.5, P < 0.001) and head diameter (dose: F(2,42) = 14.3, P < 0.001). No difference was found for the C-T length at P2. At P21 there was still an overall dose-related reduction in body weight (dose: F(2,46) = 7.5, P = 0.001) and head diameter (dose: F(2,46) = 14.3, P < 0.001). C-T length measures were not taken at P21 and sex by dose interactions were not found for any somatic measure at any time point.
Table 1.
Somatic growth
| Sex and dose | P1 | P2 | P21 | ||
|---|---|---|---|---|---|
| Body weight (g) | Male | Vehicle | 5.2 ± 0.14 | 5.8 ± 0.12 | 33.5 ± 1.2 |
| Low | 4.6 ± 0.17* | 5.5 ± 0.11 | 31.1 ± 0.7 | ||
| High | 4.6 ± 0.08* | 5.3 ± 0.18 | 28.4 ± 0.5* | ||
| Female | Vehicle | 4.8 ± 0.13 | 5.4 ± 0.08 | 29.7 ± 0.9 | |
| Low | 4.5 ± 0.14 | 4.7 ± 0.24* | 29.1 ± 1.1 | ||
| High | 4.5 ± 0.12 | 4.5 ± 0.16** | 27.9 ± 0.5 | ||
| Dose effect | P < 0.001 | P < 0.001 | — | ||
| Sex effect | P = 0.015 | P < 0.0001 | — | ||
| Head diameter (mm) | Male | Vehicle | 9.2 ± 0.19 | 9.8 ± 0.14 | 16.1 ± 0.1 |
| Low | 8.6 ± 0.17* | 9.2 ± 0.17* | 15.9 ± 0.1 | ||
| High | 8.5 ± 0.12* | 8.9 ± 0.16* | 15.2 ± 0.1*† | ||
| Female | Vehicle | 8.8 ± 0.17 | 9.5 ± 0.19 | 15.6 ± 0.1 | |
| Low | 8.5 ± 0.21 | 8.8 ± 0.16* | 15.6 ± 0.2 | ||
| High | 8.2 ± 0.07* | 8.7 ± 0.13* | 14.9 ± 0.2*† | ||
| Dose effect | P = 0.001 | P < 0.001 | — | ||
| Sex effect | P = 0.025 | P = 0.03 | — | ||
| Crown–tail length (mm) | Male | Vehicle | 43.1 ± 0.8 | 43.7 ± 0.8 | — |
| Low | 40.1 ± 0.4* | 43.6 ± 0.5 | — | ||
| High | 39.6 ± 0.7** | 43.3 ± 0.6 | — | ||
| Female | Vehicle | 40.7 ± 0.4 | 42.9 ± 0.4 | — | |
| Low | 39.5 ± 0.5 | 41.4 ± 0.9 | — | ||
| High | 39.0 ± 0.6 | 40.8 ± 0.5 | — | ||
| Dose effect | P < 0.001 | n.s. | — | ||
| Sex effect | P = 0.018 | P < 0.001 | — | ||
Effect of prenatal betamethasone on somatic growth parameters at postnatal days (P) 1, 2 and 21, (means ±s.e.m., n = 7–10 per sex per dose per time point). Symbols used for post hoc differences
P < 0.05 compared to vehicle
P < 0.001 compared to vehicle
P < 0.05 between low and high groups. No significant interactions (sex × dose) were found.
Relative organ weights
All relative organ weights (percentage of body weight) were reduced by the betamethasone treatment at P1 with exception of the heart (Table 2). By the second day of postnatal life, heart weights were actually significantly higher in the group treated with the high dose of betamethasone compared to the group treated with vehicle. Furthermore, a significant sex by dose interaction was found for the kidney at P2. There was a significant overall dose effect for the liver at all time points.
Table 2.
Relative organ weights
| Sex and dose | P1 | P2 | P21 | ||
|---|---|---|---|---|---|
| Heart | Male | Vehicle | 0.57 ± 0.03 | 0.63 ± 0.03 | 0.74 ± 0.02 |
| Low | 0.59 ± 0.03 | 0.72 ± 0.03 | 0.80 ± 0.02 | ||
| High | 0.63 ± 0.03 | 0.76 ± 0.03* | 0.81 ± 0.03 | ||
| Female | Vehicle | 0.58 ± 0.03 | 0.63 ± 0.03 | 0.80 ± 0.02 | |
| Low | 0.62 ± 0.03 | 0.74 ± 0.03 | 0.77 ± 0.02 | ||
| High | 0.60 ± 0.03 | 0.68 ± 0.03* | 0.77 ± 0.02 | ||
| Dose effect | n.s | P = 0.002 | n.s. | ||
| Sex effect | n.s. | n.s. | n.s. | ||
| Lung | Male | Vehicle | 0.18 ± 0.07 | 2.15 ± 0.09 | 1.07 ± 0.05 |
| Low | 0.19 ± 0.07 | 2.05 ± 0.08 | 1.13 ± 0.04 | ||
| High | 0.16 ± 0.08 | 2.02 ± 0.10 | 1.11 ± 0.05 | ||
| Female | Vehicle | 0.18 ± 0.07 | 2.06 ± 0.09 | 1.13 ± 0.05 | |
| Low | 0.18 ± 0.07 | 1.98 ± 0.10 | 1.09 ± 0.05 | ||
| High | 0.19 ± 0.07 | 1.85 ± 0.09 | 1.13 ± 0.04 | ||
| Dose effect | n.s. | n.s. | n.s. | ||
| Sex effect | n.s. | n.s. | n.s. | ||
| Liver | Male | Vehicle | 4.15 ± 0.09 | 3.86 ± 0.12 | 3.63 ± 0.05 |
| Low | 3.81 ± 0.09* | 3.94 ± 0.10 | 3.56 ± 0.04 | ||
| High | 3.67 ± 0.10* | 3.95 ± 0.13 | 3.44 ± 0.06* | ||
| Female | Vehicle | 4.22 ± 0.09 | 3.79 ± 0.12 | 3.62 ± 0.05 | |
| Low | 3.83 ± 0.09* | 4.29 ± 0.13* | 3.58 ± 0.05 | ||
| High | 3.72 ± 0.09** | 3.82 ± 0.12* | 3.53 ± 0.04 | ||
| Doses effect | P < 0.001 | P = 0.04 | P = 0.025 | ||
| Sex effect | n.s. | n.s. | n.s. | ||
| Spleen | Male | Vehicle | 0.18 ± 0.01 | 0.25 ± 0.01 | 0.35 ± 0.01 |
| Low | 0.16 ± 0.01 | 0.26 ± 0.01 | 0.33 ± 0.01 | ||
| High | 0.15 ± 0.01 | 0.26 ± 0.02 | 0.32 ± 0.01 | ||
| Female | Vehicle | 0.16 ± 0.01 | 0.21 ± 0.01 | 0.33 ± 0.01 | |
| Low | 0.16 ± 0.01 | 0.21 ± 0.02 | 0.33 ± 0.01 | ||
| High | 0.17 ± 0.01 | 0.18 ± 0.01 | 0.33 ± 0.01 | ||
| Dose effect | n.s. | n.s. | n.s. | ||
| Sex effect | n.s. | P < 0.001 | n.s. | ||
| Kidney | Male | Vehicle | 0.86 ± 0.02 | 0.95 ± 0.02 | 1.14 ± 0.01 |
| Low | 0.83 ± 0.02 | 0.95 ± 0.02 | 1.15 ± 0.01 | ||
| High | 0.79 ± 0.03 | 0.99 ± 0.02 | 1.14 ± 0.01 | ||
| Female | Vehicle | 0.85 ± 0.02 | 0.94 ± 0.02 | 1.20 ± 0.01 | |
| Low | 0.86 ± 0.02 | 0.98 ± 0.02 | 1.19 ± 0.01 | ||
| High | 0.88 ± 0.02 | 0.92 ± 0.02 | 1.20 ± 0.01 | ||
| Dose effect | n.s. | n.s. | n.s. | ||
| Sex effect | n.s. | n.s. | P < 0.001 | ||
Effect of prenatal betamethasone on organ weights (percentage of body weight) (means ± s.e.m., n = 7–10 per sex per dose per time point). Symbols used for post hoc differences
P < 0.05 compared to vehicle
P < 0.001 compared to vehicle. No significant interactions (sex × dose) were found, except for the P2 kidney (P = 0.045).
Brain and brain region weights
Steroid treatment caused overall retardation in all brain region weights measured at P1 (Table 3). By P2 there was continued brain growth retardation for all the regions measured. Specific overall sex differences were also found for all regions at P2 but without any significant interaction between dose and gender. The structure most affected at P1 and P2 was the cerebellum. At 21 days of age we found a significant dose-related growth retardation for the whole brain only. Dose by gender interactions were not found.
Table 3.
Brain region weights
| Sex and dose | P1 | P2 | P21 | ||
|---|---|---|---|---|---|
| Cerebellum | Male | Vehicle | 7.8 ± 0.5 | 10.5 ± 0.4 | 167 ± 4.0 |
| Low | 6.6 ± 0.2* | 8.8 ± 0.4* | 167 ± 3.7 | ||
| High | 6.5 ± 0.2* | 9.2 ± 0.5 | 165 ± 3.0 | ||
| Female | Vehicle | 7.3 ± 0.4 | 9.2 ± 0.2 | 161 ± 1.3 | |
| Low | 6.5 ± 0.2 | 8.5 ± 0.6 | 162 ± 2.4 | ||
| High | 5.7 ± 0.4* | 7.9 ± 0.3 | 159 ± 1.8 | ||
| Dose effect | P < 0.001 | P = 0.005 | n.s. | ||
| Sex effect | n.s. | P = 0.006 | P = 0.031 | ||
| Hippocampus | Male | Vehicle | 11.1 ± 0.4 | 14.4 ± 0.5 | 98.2 ± 3.1 |
| Low | 10.1 ± 0.5 | 12.7 ± 0.9 | 94.4 ± 1.9 | ||
| High | 9.7 ± 0.8 | 13.3 ± 0.9 | 93.4 ± 2.5 | ||
| Female | Vehicle | 11.1 ± 0.7 | 12.5 ± 0.5 | 95.3 ± 1.7 | |
| Low | 9.9 ± 0.3 | 12.0 ± 0.6 | 91.1 ± 2.8 | ||
| High | 9.6 ± 0.7 | 10.4 ± 0.7 | 90.1 ± 1.7 | ||
| Dose effect | P = 0.003 | P = 0.033 | n.s. | ||
| Sex effect | n.s. | P < 0.001 | n.s. | ||
| Olfactory bulb | Male | Vehicle | 7.4 ± 0.4 | 9.3 ± 0.4 | 60.0 ± 1.4 |
| Low | 6.5 ± 0.2 | 8.7 ± 0.2 | 60.3 ± 2.6 | ||
| High | 6.3 ± 0.2* | 9.2 ± 0.3 | 60.2 ± 2.1 | ||
| Female | Vehicle | 6.9 ± 0.2 | 8.8 ± 0.2 | 59.8 ± 1.4 | |
| Low | 6.2 ± 0.2 | 7.8 ± 0.4 | 56.5 ± 2.4 | ||
| High | 6.6 ± 0.3 | 7.2 ± 0.3* | 55.8 ± 1.1 | ||
| Dose effect | P = 0.014 | P = 0.010 | n.s. | ||
| Sex effect | n.s. | P < 0.001 | n.s. | ||
| Whole brian | Male | Vehicle | 219 ± 7.1 | 261 ± 6.9 | 1358 ± 7.2 |
| Low | 195 ± 4.8* | 244 ± 5.4 | 1334 ± 11.4 | ||
| High | 195 ± 2.7* | 245 ± 6.9 | 1302 ± 15.2* | ||
| Female | Vehicle | 209 ± 5.1 | 241 ± 1.8 | 1288 ± 7.0 | |
| Low | 191 ± 3.7 | 223 ± 9.3 | 1282 ± 17.2 | ||
| High | 193 ± 6.4 | 216 ± 4.2* | 1271 ± 9.9 | ||
| Dose effect | P < 0.001 | P = 0.004 | P = 0.015 | ||
| Sex effect | n.s. | P < 0.001 | P < 0.001 | ||
Effects of prenatal betamethasone on brain region weights (mg), (means ± s.e.m., n = 7–10 per sex per dose per time point). Symbols used for posts hoc differnces
P < 0.05 compared to vehicle
P < 0.001 compared to vehicle. No significant interactions (sex × dose) were found.
Brain region proliferation
Brain region specific proliferation was strikingly reduced at P1 due to the steroid treatment (Fig. 3A). Significant overall dose effects were found for all regions except the olfactory bulb (OB), with no differences between the sexes and no sex by dose interactions. This effect is completely reversed by P2 with no significant overall dose effect for any brain region. Interestingly, a significant overall dose and sex interaction effect was found for all regions (P < 0.011) except the cerebellum at P2. At this time point, proliferation within the male brain was significantly increased as a result of the betamethasone treatment whereas the females continued showing significantly reduced proliferation rates within the rest sample and whole brain. By P21 the females and males were once again similar with no significant dose and gender interactions found. An overall dose effect was found only for the P21 cerebellum of the treated groups, which continued to show elevated proliferation rates as compared to the vehicle-treated group.
Figure 3. Overview brain region proliferation.
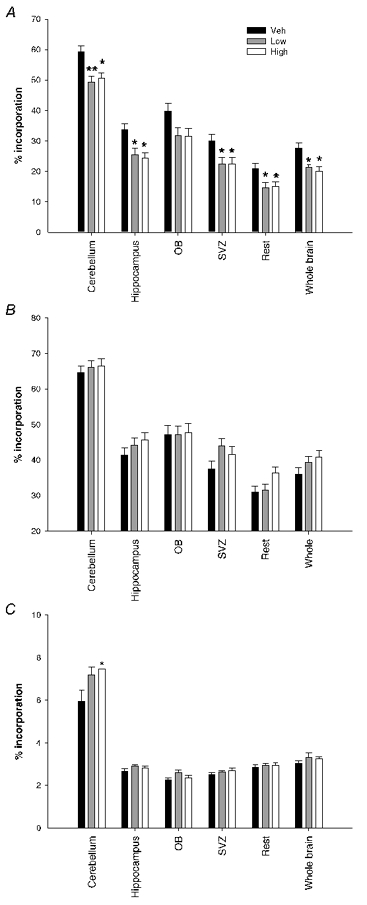
Graph showing the effect of prenatal betamethasone on postnatal brain region proliferation in both males and females combined. Betamethasone treatment caused significant decreases in proliferation at P1 in all brain regions (dose effect P < 0.05)except for the olfactory bulb (A). B, by P2 this effect is reversed with no significant differences overall but with clear sex effects. At P2 the males showed significantly increased brain cell proliferation whereas the female brain continued to have inhibited proliferation due to the treatment. C, at P21 the cerebellum is still proliferating at increased levels due to the prenatal betamethasone treatment (cerebellum overall dose effect P = 0.024). Values represent means ± s.e.m., n = 12–16 per dose per time; * P < 0.05 compared to vehicle, ** P < 0.001 compared to vehicle.
BBB permeability
Permeability of the BBB to [3H]thy at P1 was significantly increased by around 13 % by both betamethasone doses (dose: F(2,40) = 4.6, P = 0.016). This difference was not apparent at P2 or P21 (data not shown). Over all three time points we found an overall effect for dose (dose: F(2,132) = 4.1, P = 0.019) and for time (time: F(2,132) = 55.9, P < 0.001).
Corticosterone levels
Prenatal betamethasone had no effect on postnatal corticosterone values at any time point measured (Fig. 4) and there were also no significant sex by dose interactions. P1 and P2 values were low as compared to P21 because of the stress hyporesponsive period which lasts until around P15 in the rat (Scheepens et al. 2003).
Figure 4. Pup plasma corticosterone.

Effect of prenatal betamethasone on postnatal plasma corticosterone levels. No significant changes were seen between the groups at any age. Values represent means ± s.e.m., n = 12–16 per dose per time point.
Caspase-3 activation
Caspase-3 activation within the hippocampus, SVZ and cerebellum was not affected by the low dose of prenatal betamethasone (Fig. 5). We found no difference between the sexes or between the low-dose- and vehicle-treated groups, but there was a clear and significant time effect for all the regions reflecting the known decrease in developmental apoptosis occurring in the postnatal rat brain.
Figure 5. Brain region caspase-3 activation.

The low dose of prenatal betamethasone did not alter caspase-3 activation in any brain region tested at any age. There was no difference between the sexes but there was a significant decrease over time in all regions as expected. Values represent means ± s.e.m., n = 12 per time point.
DISCUSSION
By giving 170 μg kg−1 betamethasone (low-dose group) twice within 4 h to pregnant Fisher 344 rats, we have attempted to replicate a human clinical situation of a single course of prenatal glucocorticoids (two injections of 12 mg, 12 h apart; a strategy that is still being used in several institutions, though most institutions use a dose regimen of two doses of 12 mg given 24 h apart) in terms of dose, timing of the doses, the number of brain cell cycles exposed to betamethasone and the stage of fetal neurodevelopment. This is the first rodent study in which all these points have been carefully considered.
Firstly we confirmed that our treatment protocol could induce acute hyperglycaemia and hypocorticosteronaemia in the adult rat as is seen clinically (McKenna et al. 2000; Shelton et al. 2002). Subsequent analysis of pups at birth indicated that moderate growth retardation, as measured by birth weight, head diameter and C-T length, was induced by both betamethasone doses. There was no difference in the degree of growth retardation caused by the high and low doses except for C-T length in males. Somatic growth inhibition in terms of body weight and head diameter continued to be significant at P1 and P2 with only head diameter still being reduced at P21 in both sexes. These general growth retarding effects of prenatal betamethasone are likely to be due to decreased circulating levels of growth hormone (GH) and its main effector, insulin-like growth factor-1 (IGF-1), a known consequence of prenatal betamethasone (Ballard et al. 1980; Mosier et al. 1987). The reduction in head circumference we found at birth, following a single course of prenatal betamethasone, (4.3 % loss, vehicle vs. low-dose group) is the same as that reported for the human neonate following a single course (4 % loss, n = 16 322; French et al. 1999 and 3.3 % loss, n = 477; Thorp et al. 2002). Further, a reduced head circumference at birth has been shown to be clearly associated with learning problems in school-age children (Stathis et al. 1999). Repeated prenatal steroid treatment in the human clinic has also been shown to convey long-term psychomotor and neuromotor disturbances at 2 years of age (Yeh et al. 1998; Esplin et al. 2000), ‘problem child behaviour’ at 3 years of age (French et al. 1998) and a moderate decline in visual memory and visual closure at 6 years of age (MacArthur et al. 1982). One clinical study has shown that babies born after repeated courses of betamethasone had decreased cortical complexity and decreased cortical surface area (Modi et al. 2001), although others have brought these results into question (Dammann & Matthews, 2001).
Brain region weights were reduced at P1 and P2 with the cerebellum being most affected in agreement with this structure containing the highest levels of glucocorticoid receptors within the neonatal brain (Pavlik & Buresova, 1984). By P21 a significantly reduced total brain weight was only found in males. Interestingly, sex differences in the severity of side effects caused by prenatal betamethasone have also been found in the human clinic with males appearing to be more affected than females (Yeh et al. 1998).
In maternal food restriction and chronic hypoxia during pregnancy experiments resulting in growth restriction, the brain and heart are usually unaffected (Jacobs et al. 1988; Freed & Herington, 1989). The results from this experiment show that brain weight is significantly reduced as it is in the human clinic (Thorp et al. 2002), whereas relative heart weight (percentage of body weight) is actually significantly increased at postnatal day 2. We hypothesise that the brain is affected primarily due to its high content of glucocorticoid receptors (Matthews, 2000) and the relative ease with which betamethasone enters the fetal brain (Nakano et al. 1981). Increased heart weight may be related to one of the known clinical side effects of betamethasone treatment namely, transient hypertrophic cardiomyopathy at birth (Yunis et al. 1999). Alternatively this may illustrate the combined effects of hypertrophy and growth restriction on the heart.
Brain cell proliferation was strongly reduced by both betamethasone doses at P1 and a dose effect was equally apparent in both sexes. By P2 this was reversed in male pups only, with specific sex by dose interactions found for all brain structures except the cerebellum and OB. At this time the neonatal male brain appeared to be undergoing rebound or catch-up proliferation with significantly more cell proliferation in the betamethasone-treated groups. This was not seen in female pups that continued to show significantly reduced proliferation due to the steroid treatment. Catch-up of corticosteroid-treated fetuses has also been reported for its anti-myelination effect within the neonatal sheep brain (Quinlivan et al. 1999). Further, within the P2 males we see an inverted dose effect with more proliferation in the high-dose as compared to the low-dose-treated groups. Interestingly, although the males have an earlier rebound in proliferation this does not compensate for total brain weight at P21, which remains significantly reduced due to the steroid treatment in males but not females. Whether the betamethasone-induced increase in brain cell proliferation is beneficial or not, remains to be determined.
It is important to note that this measure of proliferation is a ‘snapshot’ of 30-40 min worth of neural proliferation (the approximate time [3H]thy is able to be incorporated into DNA; Boswald et al. 1990). Thus the significant reduction in proliferation seen at P1 has possibly started within hours of the first betamethasone injection at E20, 4 days earlier. The sum of the anti-proliferative effects of prenatal betamethasone on the brain is therefore substantial. At P21, proliferation within the cerebellum of both males and females was significantly increased by the steroid treatment, indicating that the females also show some rebound proliferation but with a greater delay than is seen in males. The precise mechanisms behind the anti-proliferative effect of betamethasone on the brain are unknown but possibly involve the downregulation in expression of the trophic growth factors: brain-derived neurotrophic factor (BDNF), IGF-1 and basic fibroblast growth factor (bFGF). All three of these factors are known regulators of neural proliferation, migration and differentiation and all are reduced by steroid treatment (Adamo et al. 1988; Schaaf et al. 1997; Molteni et al. 2001). Furthermore, administration of any one of these three factors has been shown to specifically increase neurogenesis (Wagner et al. 1999; Aberg et al. 2000; Pencea et al. 2001).
These data show that brain cell proliferation is inhibited for up to 4 days after a single course of prenatal betamethasone in the rat. Thus when repeated courses of prenatal betamethasone are given clinically, neural proliferation is likely to be inhibited for the entire length of treatment.
Betamethasone treatment significantly increased BBB permeability to [3H]thy, as expected (Stonestreet et al. 2000). The proliferation assay used here is independent of BBB permeability as it is based on the proportion of [3H]thy incorporated. These results clearly illustrate that proliferation assays using thymidine analogues which do not correct for BBB permeability, such as BrdU, are inappropriate in experiments where BBB integrity may be compromised including after steroid treatment, stress, epilepsy and especially after brain injury or intracerebroventricular infusions.
Pup plasma corticosterone values were unchanged by the treatment as is seen in the human clinic (Terrone et al. 1997). This indicates that no long-term changes in basal corticosterone secretion have been induced and further, that the significant reduction in brain cell proliferation seen is not due to altered endogenous glucocorticoid levels. Whether stress-induced HPA activity is affected by the treatment remains unknown.
Lastly, we measured caspase-3 activation, an accepted measure of apoptosis (Puka-Sundvall et al. 2000a,b), within the hippocampal, SVZ and cerebellar samples of the low-dose- and vehicle-treated groups. No changes in caspase-3 activation were seen although we did see the developmental decrease in apoptosis during early postnatal life as shown previously (Blaschke et al. 1998). These data show no correlation between brain cell proliferation and apoptosis within the juvenile rat brain.
In summary, we show for the first time that a clinically relevant single course (two injections within 24 h) of prenatal betamethasone induces moderate, sexually divergent, somatic growth retardation in the rat which is almost completely resolved by 21 days of age. Further, this treatment causes substantial and long-term changes in brain cell proliferation but not in caspase-3-mediated apoptosis, during what is a critically important stage of neural development. We also show for the first time that some catch-up recovery of the anti-proliferative effects of prenatal betamethasone on brain cells occurs and that this happens earlier in males than in females. Given these results we do not recommend this particular strategy and re-iterate the need for further clinical trials on the effectiveness of a single course of prenatal betamethasone on lung maturation in preterm infants, at lower concentrations than the widely used 24 mg over 24 h. Further, pregnant women receiving betamethasone who fail to deliver within 1 week are often given a second, third or even fourth course of glucocorticoids even though clinical evidence abounds that this is no more effective than a single dose and carries many detrimental side effects for both mother and fetus (Banks et al. 1999; Guinn et al. 2001; Vermillion et al. 2001). In conclusion, we show that a single course of prenatal betamethasone does cause major neural side effects and should not be as readily prescribed to pregnant women threatening to deliver preterm as it has been to date.
Acknowledgments
The authors would like to thank Mrs Heleen Huyten for her invaluable advice and help with the scintillation counting protocol and quench curve preparation, and Ms Wilma van de Berg for her assistance with the caspase-3 assay. We also thank Professor Harry Steinbusch for his financial support and for the use of his laboratory.
REFERENCES
- Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamo M, Werner H, Farnsworth W, Roberts CT, Jr, Raizada M, Leroith D. Dexamethasone reduces steady state insulin-like growth factor I messenger ribonucleic acid levels in rat neuronal and glial cells in primary culture. Endocrinology. 1988;123:2565–2570. doi: 10.1210/endo-123-5-2565. [DOI] [PubMed] [Google Scholar]
- Antonow-Schlorke I, Schwab M, Li C, Nathanielsz PW. Glucocorticoid exposure at the dose used clinically alters cytoskeletal proteins and presynaptic terminals in the fetal baboon brain. J Physiol. 2003;547:117–123. doi: 10.1113/jphysiol.2002.025700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard PL, Ballard RA. Scientific basis and therapeutic regimens for use of antenatal glucocorticoids. Am J Obstet Gynecol. 1995;173:254–262. doi: 10.1016/0002-9378(95)90210-4. [DOI] [PubMed] [Google Scholar]
- Ballard PL, Gluckman PD, Liggins GC, Kaplan SL, Grumbach MM. Steroid and growth hormone levels in premature infants after prenatal betamethasone therapy to prevent respiratory distress syndrome. Pediatr Res. 1980;14:122–127. doi: 10.1203/00006450-198002000-00011. [DOI] [PubMed] [Google Scholar]
- Banks BA, Cnaan A, Morgan MA, Parer JT, Merrill JD, Ballard PL, Ballard RA. Multiple courses of antenatal corticosteroids and outcome of premature neonates. North American Thyrotropin-Releasing Hormone Study Group. Am J Obstet Gynecol. 1999;181:709–717. doi: 10.1016/s0002-9378(99)70517-x. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396:39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Boswald M, Harasim S, Maurer-Schultze B. Tracer dose and availability time of thymidine and bromodeoxyuridine: application of bromodeoxyuridine in cell kinetic studies. Cell Tissue Kinet. 1990;23:169–181. doi: 10.1111/j.1365-2184.1990.tb01113.x. [DOI] [PubMed] [Google Scholar]
- Brabham T, Phelka A, Zimmer C, Nash A, Lopez JF, Vazquez DM. Effects of prenatal dexamethasone on spatial learning and response to stress is influenced by maternal factors. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1899–1909. doi: 10.1152/ajpregu.2000.279.5.R1899. [DOI] [PubMed] [Google Scholar]
- Chapman RH, Stern JM. Maternal stress and pituitary-adrenal manipulations during pregnancy in rats: effects on morphology and sexual behavior of male offspring. J Comp Physiol Psychol. 1978;92:1074–1083. doi: 10.1037/h0077509. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Tao Y, Black IB, DiCicco-Bloom E. A single peripheral injection of basic fibroblast growth factor (bFGF) stimulates granule cell production and increases cerebellar growth in newborn rats. J Neurobiol. 2001;46:220–229. doi: 10.1002/1097-4695(20010215)46:3<220::aid-neu1004>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Crowley PA. Antenatal corticosteroid therapy: a meta-analysis of the randomized trials, 1972 to (1994) Am J Obstet Gynecol. 1995;173:322–335. doi: 10.1016/0002-9378(95)90222-8. [DOI] [PubMed] [Google Scholar]
- Dammann O, Matthews SG. Repeated antenatal glucocorticoid exposure and the developing brain. Pediatr Res. 2001;50:563–564. doi: 10.1203/00006450-200111000-00004. [DOI] [PubMed] [Google Scholar]
- Dicicco-Bloom E, Friedman WJ, Black IB. NT-3 stimulates sympathetic neuroblast proliferation by promoting precursor survival. Neuron. 1993;11:1101–1111. doi: 10.1016/0896-6273(93)90223-e. [DOI] [PubMed] [Google Scholar]
- Drago F, Di Leo F, Giardina L. Prenatal stress induces body weight deficit and behavioural alterations in rats: the effect of diazepam. Eur Neuropsychopharmacol. 1999;9:239–245. doi: 10.1016/s0924-977x(98)00032-7. [DOI] [PubMed] [Google Scholar]
- Dunlop SA, Archer MA, Quinlivan JA, Beazley LD, Newnham JP. Repeated prenatal corticosteroids delay myelination in the ovine central nervous system. J Maternal Fetal Med. 1997;6:309–313. doi: 10.1002/(SICI)1520-6661(199711/12)6:6<309::AID-MFM1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Esplin MS, Fausett MB, Smith S, Oshiro BT, Porter TF, Branch DW, Varner MW. Multiple courses of antenatal steroids are associated with a delay in long-term psychomotor development in children with birth weights < 1500 grams. Am J Obstet Gynecol. 2000;182:S24. Abstract 27. [Google Scholar]
- Freed KA, Herington AC. Insulin-like growth factor-I and its autocrine role in growth of MCF-7 human breast cancer cells in culture. J Mol Endocrinol. 1989;3:183–190. doi: 10.1677/jme.0.0030183. [DOI] [PubMed] [Google Scholar]
- French NP, Hagan R, Evans SF, Godfrey M, Newnham JP. Repeated antenatal corticosteroids; behaviour outcomes in a regional population of very preterm infants. Pediatr Res. 1998;43 Abstract no. 1252. [Google Scholar]
- French NP, Hagan R, Evans SF, Godfrey M, Newnham JP. Repeated antenatal corticosteroids: size at birth and subsequent development. Am J Obstet Gynecol. 1999;180:114–121. doi: 10.1016/s0002-9378(99)70160-2. [DOI] [PubMed] [Google Scholar]
- Fuentes-Pardo B, Hernandez-Falcon J, Velazquez PN, Romano MC. Role of corticosterone on the development of passive electrical properties of cultured chick embryo neurons. J Dev Physiol. 1990;13:67–73. [PubMed] [Google Scholar]
- Guinn DA, Atkinson MW, Sullivan L, Lee M, MacGregor S, Parilla BV, Davies J, Hanlon-Lundberg K, Simpson L, Stone J, Wing D, Ogasawara K, Muraskas J. Single vs. weekly courses of antenatal corticosteroids for women at risk of preterm delivery: A randomized controlled trial. JAMA. 2001;286:1581–1587. doi: 10.1001/jama.286.13.1581. [DOI] [PubMed] [Google Scholar]
- Henning SJ. Plasma concentrations of total and free corticosterone during development in the rat. Am J Physiol. 1978;235:E451–456. doi: 10.1152/ajpendo.1978.235.5.E451. [DOI] [PubMed] [Google Scholar]
- Huang WL, Beazley LD, Quinlivan JA, Evans SF, Newnham JP, Dunlop SA. Effect of corticosteroids on brain growth in fetal sheep. Obstet Gynecol. 1999;94:213–218. doi: 10.1016/s0029-7844(99)00265-3. [DOI] [PubMed] [Google Scholar]
- Jacobs R, Robinson JS, Owens JA, Falconer J, Webster ME. The effect of prolonged hypobaric hypoxia on growth of fetal sheep. J Dev Physiol. 1988;10:97–112. [PubMed] [Google Scholar]
- Kornack DR, Rakic P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci U S A. 1998;95:1242–1246. doi: 10.1073/pnas.95.3.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laborde JB, Hansen DK, Young JF, Sheehan DM, Holson RR. Prenatal dexamethasone exposure in rats: effects of dose, age at exposure, and drug-induced hypophagia on malformations and fetal organ weights. Fundam Appl Toxicol. 1992;19:545–554. doi: 10.1016/0272-0590(92)90093-w. [DOI] [PubMed] [Google Scholar]
- Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. [PubMed] [Google Scholar]
- MacArthur BA, Howie RN, Dezoete JA, Elkins J. School progress and cognitive development of 6-year-old children whose mothers were treated antenatally with betamethasone. Pediatrics. 1982;70:99–105. [PubMed] [Google Scholar]
- McKenna DS, Wittber GM, Nagaraja HN, Samuels P. The effects of repeat doses of antenatal corticosteroids on maternal adrenal function. Am J Obstet Gynecol. 2000;183:669–673. doi: 10.1067/mob.2000.106755. [DOI] [PubMed] [Google Scholar]
- Matthews SG. Antenatal glucocorticoids and programming of the developing CNS. Pediatr Res. 2000;47:291–300. doi: 10.1203/00006450-200003000-00003. [DOI] [PubMed] [Google Scholar]
- Modi N, Lewis H, Al-Naqeeb N, Ajayi-Obe M, Dore CJ, Rutherford M. The effects of repeated antenatal glucocorticoid therapy on the developing brain. Pediatr Res. 2001;50:581–585. doi: 10.1203/00006450-200111000-00008. [DOI] [PubMed] [Google Scholar]
- Molteni R, Fumagalli F, Magnaghi V, Roceri M, Gennarelli M, Racagni G, Melcangi RC, Riva MA. Modulation of fibroblast growth factor-2 by stress and corticosteroids: from developmental events to adult brain plasticity. Brain Res Brain Res Rev. 2001;37:249–258. doi: 10.1016/s0165-0173(01)00128-x. [DOI] [PubMed] [Google Scholar]
- Mosier HD, Jr, Spencer EM, Dearden LC, Jansons RA. The effect of glucocorticoids on plasma insulin-like growth factor I concentration in the rat fetus. Pediatr Res. 1987;22:92–95. doi: 10.1203/00006450-198707000-00021. [DOI] [PubMed] [Google Scholar]
- Nakano M, Takashima A, Nishiuchi M, Takeuchi M, Doteuchi M, Yamada H. Placental transfer and distribution of betamethasone 17,21-dipropionate in various regions of the brain of pregnant rats and mice (author's translation) Nippon Yakurigaku Zasshi. 1981;77:165–176. [PubMed] [Google Scholar]
- Pavlik A, Buresova M. The neonatal cerebellum: the highest level of glucocorticoid receptors in the brain. Brain Res. 1984;314:13–20. doi: 10.1016/0165-3806(84)90171-8. [DOI] [PubMed] [Google Scholar]
- Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci. 2001;21:6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puka-Sundvall M, Hallin U, Zhu C, Wang X, Karlsson JO, Blomgren K, Hagberg H. NMDA blockade attenuates caspase-3 activation and DNA fragmentation after neonatal hypoxia-ischemia. Neuroreport. 2000a;11:2833–2836. doi: 10.1097/00001756-200009110-00002. [DOI] [PubMed] [Google Scholar]
- Puka-Sundvall M, Wallin C, Gilland E, Hallin U, Wang X, Sandberg M, Karlsson J, Blomgren K, Hagberg H. Impairment of mitochondrial respiration after cerebral hypoxia-ischemia in immature rats: relationship to activation of caspase-3 and neuronal injury. Brain Res Dev Brain Res. 2000b;125:43–50. doi: 10.1016/s0165-3806(00)00111-5. [DOI] [PubMed] [Google Scholar]
- Quinlivan JA, Dunlop SA, Newnham JP, Evans SF, Beazley LD. Repeated, but not single, maternal administartion of corticostreroids delays myelination in the brain of fetal sheep. Prenat Neonatal Med. 1999;4:47–55. [Google Scholar]
- Romijn HJ, Hofman MA, Gramsbergen A. At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby. Early Hum Dev. 1991;26:61–67. doi: 10.1016/0378-3782(91)90044-4. [DOI] [PubMed] [Google Scholar]
- Scheepens AW, Piersma MJ, Van de Berg WD, Blanco CE. A delayed increase in hippocampal proliferation following global asphyxia in the neonatal rat. Brain Res Dev Brain Res. 2003;142:67–76. doi: 10.1016/s0165-3806(03)00032-4. [DOI] [PubMed] [Google Scholar]
- Shelton SD, Boggess KA, Smith T, Herbert WN. Effect of betamethasone on maternal glucose. J Matern Fetal Neonatal Med. 2002;12:191–195. doi: 10.1080/jmf.12.3.191.195. [DOI] [PubMed] [Google Scholar]
- Stathis SL, O'Callaghan M, Harvey J, Rogers Y. Head circumference in ELBW babies is associated with learning difficulties and cognition but not ADHD in the school-aged child. Dev Med Child Neurol. 1999;41:375–380. doi: 10.1017/s0012162299000833. [DOI] [PubMed] [Google Scholar]
- Stonestreet BS, Sadowska GB, McKnight AJ, Patlak C, Petersson KH. Exogenous and endogenous corticosteroids modulate blood-brain barrier development in the ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2000;279:R468–477. doi: 10.1152/ajpregu.2000.279.2.R468. [DOI] [PubMed] [Google Scholar]
- Sulon J, Demey-Ponsart L, Beauduin P, Sodoyez JC. Radioimmunoassay of corticosterone, cortisol and cortisone: their application to human cord and maternal plasma. J Steroid Biochem. 1978;9:671–676. doi: 10.1016/0022-4731(78)90180-2. [DOI] [PubMed] [Google Scholar]
- Tamvakopoulos CS, Neugebauer JM, Donnelly M, Griffin PR. Analysis of betamethasone in rat plasma using automated solid-phase extraction coupled with liquid chromatography-tandem mass spectrometry. Determination of plasma concentrations in rat following oral and intravenous administration. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776:161–168. doi: 10.1016/s1570-0232(02)00271-4. [DOI] [PubMed] [Google Scholar]
- Tao Y, Black IB, DiCicco-Bloom E. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF) J Comp Neurol. 1996;376:653–663. doi: 10.1002/(SICI)1096-9861(19961223)376:4<653::AID-CNE11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Tao Y, Black IB, DiCicco-Bloom E. In vivo neurogenesis is inhibited by neutralizing antibodies to basic fibroblast growth factor. J Neurobiol. 1997;33:289–296. [PubMed] [Google Scholar]
- Terrone DA, Smith LG, Jr, Wolf EJ, Uzbay LA, Sun S, Miller RC. Neonatal effects and serum cortisol levels after multiple courses of maternal corticosteroids. Obstet Gynecol. 1997;90:819–823. doi: 10.1016/S0029-7844(97)00427-4. [DOI] [PubMed] [Google Scholar]
- Thorp JA, Jones PG, Knox E, Clark RH. Does antenatal corticosteroid therapy affect birth weight and head circumference. Obstet Gynecol. 2002;99:101–108. doi: 10.1016/s0029-7844(01)01656-8. [DOI] [PubMed] [Google Scholar]
- Velazquez PN, Romano MC. Corticosterone therapy during gestation: effects on the development of rat cerebellum. Int J Dev Neurosci. 1987;5:189–194. doi: 10.1016/0736-5748(87)90029-3. [DOI] [PubMed] [Google Scholar]
- Vermillion ST, Soper DE, Newman RB. Is betamethasone effective longer than 7 days after treatment. Obstet Gynecol. 2001;97:491–493. doi: 10.1016/s0029-7844(00)01178-9. [DOI] [PubMed] [Google Scholar]
- Wolf E, Wagner JP, Black IB, DiCicco-Bloom E. Cerebellar granule cells elaborate neurites before mitosis. Brain Res Dev Brain Res. 1997;102:305–308. doi: 10.1016/s0165-3806(97)00111-9. [DOI] [PubMed] [Google Scholar]
- Yeh TF, Lin YJ, Huang CC, Chen YJ, Lin CH, Lin HC, Hsieh WS, Lien YJ. Early dexamethasone therapy in preterm infants: a follow-up study. Pediatrics. 1998;101:1168–1169. doi: 10.1542/peds.101.5.e7. [DOI] [PubMed] [Google Scholar]
- Yunis KA, Bitar FF, Hayek P, Mroueh SM, Mikati M. Transient hypertrophic cardiomyopathy in the newborn following multiple doses of antenatal corticosteroids. Am J Perinatol. 1999;16:17–21. doi: 10.1055/s-2007-993830. [DOI] [PubMed] [Google Scholar]