

Abstract
There is controversy over the extent to which glutamate released at one synapse can escape from the synaptic cleft and affect receptors at other synapses nearby, thereby compromising the synapse-specificity of information transmission. Here we show that the glial glutamate transporters GLAST and GLT-1 limit the activation of Purkinje cell AMPA receptors produced by glutamate diffusion between parallel fibre synapses in the cerebellar cortex of juvenile mice. For a single stimulus to the cerebellar molecular layer of wild-type mice, increasing the number of activated parallel fibres prolonged the parallel fibre EPSC, demonstrating an interaction between different synapses. Knocking out GLAST, or blocking GLT-1 in the absence of GLAST, prolonged the EPSC when many parallel fibres were stimulated but not when few were stimulated. When spatially separated parallel fibres were activated by granular layer stimulation, the EPSC prolongation produced by stimulating more fibres or reducing glutamate transport was greatly reduced. Thus, GLAST and GLT-1 curtail the EPSC produced by a single stimulus only when many nearby fibres are simultaneously activated. However when trains of stimuli were applied, even to a small number of parallel fibres, knocking out GLAST or blocking GLT-1 in the absence of GLAST greatly prolonged and enhanced the AMPA receptor-mediated current. These results show that glial cell glutamate transporters allow neighbouring synapses to operate more independently, and control the postsynaptic response to high frequency bursts of action potentials.
Until the mid 1990s, it was generally assumed that synapses must operate independently. Recently, however, spillover of transmitter from one synaptic release site to receptors at nearby release sites, or to extrasynaptic receptors, has been suggested to occur for glutamate at auditory, hippocampal, olfactory and cerebellar synapses (Otis et al. 1996; Kullmann et al. 1996; Isaacson, 1999; Lozovaya et al. 1999; Carter & Regehr, 2000; Arnth-Jensen et al. 2002; DiGregorio et al. 2002), and for GABA at hippocampal and cerebellar synapses (Isaacson et al. 1993; Hamann et al. 2002). Transmitter crosstalk between synapses will disrupt the specificity of synaptic transmission, and may degrade the information processing capability of the brain. For excitatory synapses, whether transmitter crosstalk compromises synaptic independence is in part determined by the density of glutamate transporters. Rapid glutamate uptake by postsynaptic neuronal transporters (Takahashi et al. 1996; Otis et al. 1997; Auger & Attwell, 2000; Diamond, 2001) or by glial transporters located near release sites (Chaudhry et al. 1995; Bergles et al. 1997; Clark & Barbour, 1997; Dzubay & Jahr, 1999) will remove transmitter and thus help to terminate the EPSC, but will also prevent glutamate diffusing to nearby synapses. Modelling studies have concluded, depending on the assumptions made, either that glutamate diffusion between boutons is likely to produce an important contribution to postsynaptic currents (Barbour & Häusser, 1997; Rusakov & Kullmann, 1998) or that crosstalk is negligible and synapses operate independently (Barbour, 2001).
Cerebellar parallel fibre synapses onto Purkinje cells are strongly wrapped by glia expressing a high density of GLAST (and to a lesser extent GLT-1) glutamate transporters (Palay & Chan-Palay, 1974; Lehre & Danbolt, 1998), suggesting that these transporters could play a major role in limiting synaptic crosstalk. Knocking out GLAST produces motor defects but has been reported to have no effect on the parallel fibre EPSC (Watase et al. 1998). Blocking glutamate uptake pharmacologically prolongs the AMPA receptor EPSC at these synapses (Barbour et al. 1994; Takahashi et al. 1995), but it is unclear whether this reflects a block of glial glutamate transporters, or of the postsynaptic neuronal glutamate transporters EAAT4 and EAAC1 (Takahashi et al. 1996; Otis et al. 1997; Auger & Attwell, 2000).
Here we have studied the effects on the parallel fibre to Purkinje cell EPSC of preventing glial glutamate uptake (either genetically or pharmacologically), as a function of the number of parallel fibres stimulated. If synapses operate independently, then the EPSC time course and its prolongation by uptake block should be the same, irrespective of how many parallel fibres are active. By contrast, if the EPSC time course and the prolongation produced by uptake block are dependent on the number of fibres stimulated, then crosstalk between synapses produced by glutamate spillover must be occurring. Experimentally, the EPSC was found to be longer when more fibres were stimulated, and block of glial glutamate uptake had a strong effect on the EPSC duration when many fibres were stimulated but not when only a few were active. These data suggest that a major role of glial glutamate transporters in the cerebellar cortex is to allow synapses to signal more independently.
METHODS
Transgenic mice
The generation of mice lacking GLAST, by disrupting exon 6 of the GLAST gene which encodes the 4th transmembrane region of the GLAST protein, has been described in detail previously (Watase et al. 1998). Knock-out (−/−) and wild-type (+/+) mice were produced by breeding heterozygote (+/−) mice which had been backcrossed nine times with C57Black/6 mice. Experiments comparing wild-type and knock-out mice were carried out as far as possible on pairs of +/+ and −/− animals from the same litter to reduce any variability that may occur between litters.
Cerebellar slices
Parasagittal cerebellar slices 200 μm thick were cut, using a vibrating slicer, from the cerebellum of P14- to 21-day-old mice (killed by cervical dislocation in accordance with the UK Animals (Scientific Procedures) Act, 1986), and stored at room temperature before use. Young, rather than adult, mice were used because the dendritic tree of their Purkinje cells is less elaborate and more voltage uniform (Llano et al. 1991). A few experiments were also carried out on slices from 15-day-old Sprague-Dawley rats, as described for Fig. 2. For cutting and storing the slices the extracellular solution was modified from that described below by the addition of 1 mM kynurenate to block glutamate receptors and thus reduce neuronal death, and the omission of GABAzine.
Figure 2. Comparison of stimulus-response data and single bouton properties in wild-type and knock-out cells.
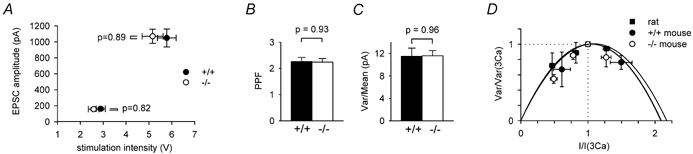
A, mean EPSC amplitude as a function of stimulation intensity in 20 wild-type (filled circles) and 15 knock-out (open circles) cells: stimuli were adjusted to produce EPSC amplitudes near 160 pA, then doubled to produce the larger responses. B, paired-pulse facilitation ratio (PPF, for stimuli separated by 5 ms) in cells from 20 wild-type and 15 GLAST knock-out cells (mean EPSC amplitudes were 162 ± 17 and 157 ± 16 pA in wild-type and knock-out cells, respectively). C, ratio of the variance to the mean of the peak of the EPSC in 29 wild-type and 43 GLAST knock-out cells (mean EPSC amplitudes were 671 ± 21 and 653 ± 18 pA in wild-type and knock-out cells, respectively). D, plots of variance versus mean of EPSC amplitudes (normalised to the values in 3 mM [Ca2+]o), with release probability altered by raising or lowering [Ca2+]o in exchange for [Mg2+]o. Experiments for 4 cells from 15-day-old rats (filled squares, shown for comparison with mouse data) and 7 cells in 17-18-day-old GLAST knock-out mice (open circles) employed [Ca2+]o = 2, 2.5, 3 and 3.5 mM, while the experiments for 2 cells in 20-day-old wild-type mice used [Ca2+]o = 2, 3 and 4 mM. Best-fit parabolae shown are fits to eqn (5) and gave the release probabilities quoted in the text.
Solutions
Normal extracellular solution contained (mM): NaCl 124, KCl 2.5, NaH2 PO4 1, NaHCO3 26, CaCl2 3, MgCl2 1, glucose 10, GABAzine 0.01 (to block GABAA receptors) bubbled with 95 % O2-5 % CO2. Normal intracellular (pipette) solution contained (mM): caesium gluconate 120, NaCl 4, Hepes 10, BAPTA 10, MgATP 4, Na4 GTP 0.5, QX314-Cl (to block voltage-gated sodium channels) 10, pH-adjusted to 7.25 with CsOH. Similar effects of uptake block on EPSC duration were seen using an internal solution containing 5 mM EGTA and 0.5 mM CaCl2 instead of BAPTA. Drugs were added by dissolving stock solutions, typically 100 mM made up in distilled water, in the extracellular solution.
Electrophysiology
Slices were transferred from the holding chamber to a bath on the stage of an Olympus BX50WI microscope, and superfused with extracellular solution at 2ml min−1. Purkinje cells were whole-cell patch-clamped to −70 mV under visual control using an Axopatch 200B (Axon Instruments). Most experiments were at room temperature (27 °C), with some experiments being carried out at 36 °C (as described in the text) to show that glutamate spillover also occurs at body temperature. Pipette resistance in external solution was typically 2-3 M Ω, and series resistance was compensated, by up to 90 %, to produce a residual series resistance lower than 1 M Ω as described in the main text. Parallel fibre inputs were stimulated, normally at 0.05 or 0.1 Hz with a pulse width of 50 μs, using a patch pipette filled with extracellular solution. The stimulus electrode was located in the molecular layer for most experiments, but for some (as stated in the text) it was located in the granular layer. Subtracting the stimulus artefact (measured in 25 μM NBQX) altered the calculated value of the time constant for the EPSC decay (from eqn (2) below) by only 1 % (increased by 0.95 ± 0.79 % in 11 cells with an EPSC amplitude of ≈200 pA). Identification of the stimulated input as being from the parallel fibres rather than the climbing fibre was verified (Konnerth et al. 1990; Perkel et al. 1990) by checking (i) that the EPSC showed a graded increase as the stimulus strength was increased and more parallel fibres were recruited (by contrast the climbing fibre EPSC shows one or occasionally two discrete step increases when the stimulus is increased), and (ii) that the second response to paired-pulse stimulation was larger (by contrast the second climbing fibre response is smaller). Synaptic currents were abolished by TTX (2 μM). They were also abolished by the AMPA/kainate receptor blocker NBQX (25 μM, Fig. 10D), demonstrating that activation of metabotropic or NMDA receptors does not contribute to the EPSC. In addition, they were abolished by 50 μM GYKI 53655, which non-competitively blocks AMPA receptors with much less effect on kainate receptors (Bleakman et al. 1996), implying that the EPSCs are produced almost entirely by AMPA receptors (single EPSCs in GLAST knock-out animals treated with 200 μM dihydrokainate as in Fig. 7 were reduced in amplitude by GYKI 53655 by 99.6 ± 0.3 % and reduced in charge transfer by 99.8 ± 0.1 % in three cells; the charge transfer produced by 10 stimuli at 200 Hz (as in Fig. 10D) was reduced by 99.7 % in one cell, data not shown).
Figure 10. The effect of GLAST knock out, and of blocking GLT-1 with DHK, on the EPSC generated by trains of stimuli to the parallel fibres.
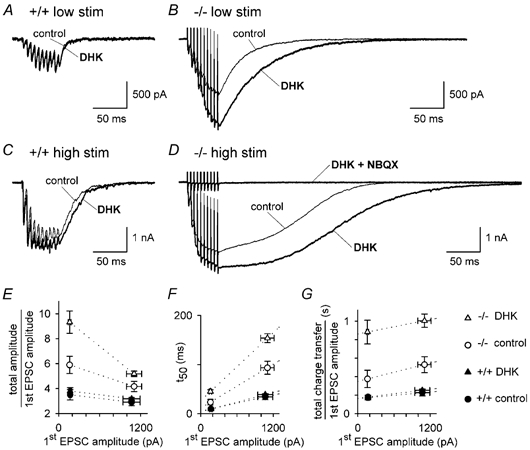
A, response of a Purkinje cell in a wild-type slice to 10 stimuli at 200 Hz in the absence and presence of 200 μM DHK. Stimulus intensity was adjusted to produce an initial EPSC of amplitude ≈170 pA (‘low stim’, mean value 168 ± 21 pA in 20 cells). B, as for A, but in a knock-out slice (mean initial EPSC amplitude 160 ± 21 pA in 15 cells). C, same cell as in A but at double the stimulation intensity (‘high stim’), producing an initial EPSC amplitude of ≈1100 pA (mean value 1068 ± 125 pA). D, same cell as in B, but at double the stimulation intensity (mean initial EPSC amplitude 1105 ± 111 pA). E, peak current amplitude evoked by the train, normalised to the amplitude of the first EPSC in the train in control conditions, for data as in A-D from 20 wild-type and 15 GLAST knock-out cells. F, time needed for the current evoked by the train to decay to half its amplitude at the end of the train. G, total charge transfer evoked by the train (integrated from the start of the train to 1 s after the train), normalised to the amplitude of the first EPSC in the train in control conditions.
Figure 7. EPSC prolongation produced by block of GLT-1 is larger when more fibres are stimulated.

A, specimen parallel fibre EPSCs of different amplitude evoked by molecular layer stimulation in the same GLAST knock-out cell. B, EPSCs in the same cell and with the same stimulus strength as A, after application of 200 μM DHK. C and D, data in A and B normalised to have the same peak. E, weighted decay time constant for different stimulation intensities, in the absence and presence of DHK, measured as in A-D and normalised to the value (4.83 ± 0.44 ms) for an EPSC amplitude near 640 pA in control solution, in 7 knock-out cells. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were as follows. For low stimulation, in control: 118 ± 12 pA, 3.69 ± 0.19 ms, 3.05 ± 2.07 pA, 22.8 ± 8.1 ms (4 out of 7 cells showed a slow component); in DHK: 76.4 ± 11.0 pA, 3.07 ± 0.41 ms, 20.4 ± 8.9 pA, 25.1 ± 10.6 ms (5 out of 7 cells showed a slow component). For medium stimulation, in control: 539 ± 44 pA, 4.09 ± 0.47 ms, 66.5 ± 35.5 pA, 17.4 ± 6.1 ms (5 out of 7 cells showed a slow component); in DHK: 482 ± 44 pA, 4.37 ± 0.68 ms, 124 ± 35 pA, 36.3 ± 7.9 ms (all cells showed a slow component). For high stimulation, in control: 1060 ± 196 pA, 4.40 ± 0.67 ms, 451 ± 187 pA, 21.4 ± 10.6 ms (6 out of 7 cells showed a slow component); in DHK: 1102 ± 61 pA, 5.91 ± 0.62 ms, 338 ± 52 pA, 50.9 ± 8.7 ms (all cells showed a slow component).
Statistics
Data are shown as means ± s.e.m. P values were obtained from two-tailed t tests. EPSCs were fitted with the sum of two exponential components (see eqn (1)), a fast component with a time constant < 6 ms, and a slow component with a time constant > 6 ms. If no slow component was present, the EPSC was fitted with a single fast exponential, the amplitude of the slow component was defined as zero when calculating a mean value for the slow component amplitude, and no time constant from that cell was included when calculating the mean value of the slow component time constant.
RESULTS
Purkinje cell properties in wild-type and GLAST knock-out mice
As reported previously, Purkinje cells in mice lacking GLAST showed anatomical and physiological properties broadly similar to those in wild-type littermates, apart from the change of EPSC properties documented in this paper and the persistence of multiple climbing fibre innervation to older ages in the knock-out (Watase et al. 1998). The overall anatomy of the cerebellum, including its size and foliation, Purkinje cell dendrite arborization, the area of the granule cell layer and the density of granule cells, the structure of parallel and climbing fibre synapses, and the parallel fibre synapse density, have been shown by a combination of light and electron microscopy not to differ significantly between the wild-type and GLAST knock-out mice and, like in wild-type mice, the parallel fibre fast EPSC is mediated solely by AMPA receptors with no NMDA receptor component (Watase et al. 1998). Western blotting of cerebellum from wild-type and knock-out mice has indicated that knocking out GLAST does not alter the total expression of the other cerebellar glutamate transporters GLT-1, EAAC1 and EAAT4 (Watase et al. 1998). To check whether knocking out GLAST had any secondary effects on the properties of Purkinje cells, we compared the input resistance and capacity current transients of Purkinje cells in knock-out and wild-type mice. These did not differ significantly. The membrane resistance at −70 mV was 75.3 ± 6.9MΩ and 73.7 ± 6.7MΩ in 11 wild-type and 9 knock-out cells, respectively (not significantly different, P = 0.87). Capacity currents evoked by 10 mV pulses were fitted with the sum of three decaying exponentials which had amplitudes (A) and time constants (τ) of A1 = 1865 ± 266 pA, τ1 = 0.12 ± 0.03 ms; A2 = 562 + 54 pA, τ2 = 5.95 ± 0.61 ms; A3 = 113 ± 43 pA, τ3 = 54.6 ± 29.9 ms in 11 wild-type cells, and A1 = 1412 ± 260 pA, τ1 = 0.14 ± 0.03 ms; A2 = 590 ± 56 pA, τ2 = 6.99 ± 0.77 ms; A3 = 67 ± 43 pA, τ3 = 105 ± 67 ms in 9 knock-out cells (not significantly different: P = 0.24, 0.51, 0.72, 0.31, 0.47 and 0.51 for comparison of A1, τ1, A2, τ2, A3 and τ3, respectively).
We analyse in detail below the properties of parallel fibre EPSCs, and show that the probability of transmitter release from parallel fibre boutons, and the peak postsynaptic current produced per bouton, were the same in wild-type and knock-out mice, suggesting no change of AMPA receptor number at each parallel fibre synapse. In addition, as shown below, the stimulus strength needed to evoke an EPSC of a given size was not altered in the knock-out, suggesting an unchanged density of parallel fibre synapses. However, applying 0.5 μM AMPA (in the presence of 1 μM TTX and 10 μM GABAzine to block action potentials and GABAA receptors) evoked an inward current at −70 mV, which was slightly but significantly larger (24 %, P = 0.011) in 13 knock-out cells than in 13 wild-type cells (mean values were 2.42 ± 0.13 and 1.96 ± 0.10 nA, respectively, measured from the plateau current reached after 3 min of AMPA application). This may reflect the presence of extra receptors associated with the multiple climbing fibre innervation persisting in the knock-out mice (Watase et al. 1998), or a greater AMPA-evoked rise of extracellular glutamate concentration in the knock-out (if depolarization of granule cells by AMPA can lead to action potential-independent release of glutamate).
Parallel fibre EPSCs are longer in GLAST knock-out mice
We initially assessed the contribution of GLAST transporters to terminating EPSCs evoked by a single stimulus applied to the parallel fibres in the molecular layer. To assess the effect of GLAST knock-out on the parallel fibre EPSC decay, which unavoidably involves a comparison between cells in slices from different animals, we had to standardize the number of parallel fibres stimulated, and to ensure that the EPSC was not distorted by series resistance filtering (since increasing the number of nearby active release sites in calyceal synapses can lead to a prolongation of the EPSC decay (Trussell et al. 1993) and the apparent duration of the parallel fibre EPSC is increased when the series resistance of the whole-cell electrode is excessive (Llano et al. 1991)). We initially started with the hypothesis (which will be justified below) that an equal EPSC amplitude implies that the same number of input parallel fibres are stimulated, and standardized the number of parallel fibres stimulated by adjusting the stimulation intensity to produce EPSCs with an amplitude of approximately 620 pA in both knock-out and wild-type mice (615 ± 10 pA (n = 47) and 624 ± 13 pA (n = 37), respectively, not significantly different, P = 0.58). Assuming that each stimulated parallel fibre which contacts the recorded cell produces on average a peak current of ≈10 pA in both wild-type and knock-out mice (as explained in detail below), this corresponds to approximately 62 parallel fibres being stimulated.
Figure 1A shows specimen EPSCs recorded at −70 mV from wild-type and knock-out mice. To quantify the duration of the EPSC, its decay (from 90 % of the peak amplitude) was fitted with the sum of two decaying exponentials (which usually gave a better fit than one exponential, although some cells showed only a fast exponential component (τ < 6 ms) as detailed in the figure legends):
| (1) |
where A1 and A2 are the amplitudes of the components with time constants τ1 and τ2, and an amplitude-weighted decay time constant was calculated as:
| (2) |
The weighted time constant is equal to the charge transfer of the EPSC normalised by the EPSC amplitude (for an instantaneously rising EPSC). It therefore provides a convenient single parameter measure of the duration of the EPSC: a current with the amplitude of the EPSC would have to flow for the duration of the weighted time constant in order to produce the same charge transfer as the EPSC (equivalently, a mono-exponentially decaying current with a time constant equal to the weighted time constant would generate the same charge transfer as a bi-exponentially decaying EPSC of the same amplitude).
Figure 1. Knocking out glial GLAST transporters prolongs parallel fibre synaptic currents.
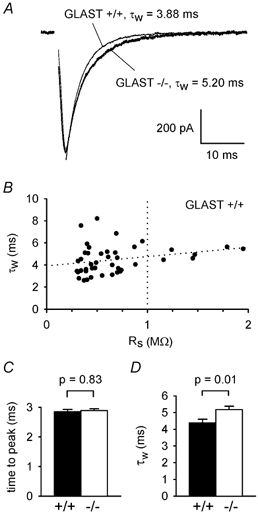
A, specimen EPSCs at −70 mV evoked in Purkinje cells by parallel fibre stimulation in the molecular layer, adjusted to produce the same EPSC amplitude in a wild-type cell (+/+) and a cell from a mouse with GLAST knocked out (−/−). Stimulus artefacts removed for clarity. Smooth lines through the EPSC decays are fits of eqn (1), with weighted time constants (τw, from eqn (2)) as shown. B, weighted decay time constant as a function of the series resistance remaining after ≈90 % compensation, RS, in 43 cells from wild-type mice. Almost-horizontal dotted line is a linear regression through the data. Vertical dotted line is the upper limit of 1MΩ which we set for acceptable values of RS. C and D, mean (± s.e.m.) values of time from the start of the stimulus to the EPSC peak (C), and τw (D), for EPSCs with amplitudes around 620 pA in 37 wild-type and 47 knock-out cells. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) for the data in D were: in wild-type cells, 542 ± 16 pA, 4.06 ± 0.22 ms, 21.3 ± 12.1 pA, 31.1 ± 11.2 ms (13 out of 37 cells showed a slow component; for the other 24 cells Aslow was set to 0 and no τslow value was included in the average); and in knock-out cells: 486 ± 14 pA, 4.34 ± 0.19 ms, 59.6 ± 12.0 pA, 19.7 ± 2.8 ms (35 out of 47 cells showed a slow component).
In wild-type animals the weighted time constant increased slowly with the residual series resistance (Rs) remaining after 90 % compensation (Fig. 1B), so for comparing wild-type and knock-out animals we selected cells with a residual Rs of less than 1MΩ (mean values were 0.53 ± 0.03MΩ for wild-type cells and 0.54 ± 0.03MΩ for knock-out cells in Fig. 1C and D, not significantly different, P = 0.7). There was no difference in the time to peak of the 620 pA amplitude EPSC between the wild-type and knock-out cells (P = 0.83, Fig. 1C), but the weighted decay time constant in 47 cells in the GLAST knock-out mice was 18 % longer than that found for 37 cells in the wild-type mice (mean values were 4.38 ± 0.22 and 5.17 ± 0.21 ms in wild-type and knock-out mice, respectively, P = 0.01, Fig. 1D). The contributions to this difference, of changes in the faster and slower exponential components of the decay, will be assessed in detail below.
Stimulus-response curve, release probability and single fibre EPSC amplitude are unaltered in the GLAST KO
In this section we assess whether knocking out GLAST has any effect on the number of parallel fibres activated by stimulation, or on the single bouton release probability and single bouton current, since these parameters could in principle affect the size and shape of the EPSC. We also estimate the current produced by stimulation of a single input parallel fibre, in order to convert EPSC size to an approximate number of input fibres activated.
The strength of stimulus needed to evoke a parallel fibre EPSC of a given size was not significantly different in the wild-type and the knock-out mice, either for small or for large EPSCs (Fig. 2A). To evoke an EPSC of 160 pA amplitude, the stimulus strength needed for wild-type cells was 2.98 ± 0.20 V (in 20 cells; the amplitude was 162 ± 17 pA) and for knock-out cells was 2.58 ± 0.23 V (in 15 cells; the amplitude was 157 ± 16 pA, P = 0.82 compared to the wild-type amplitude). These stimulus strengths are not significantly different (P = 0.32). These stimulus strengths were then doubled, and the resulting EPSC size was measured to investigate possible changes in the shape of the stimulus-response curve. The resulting EPSC amplitudes (1049 ± 112 pA in wild-type cells and 1070 ± 89 pA in knock-out cells) did not differ significantly (P = 0.89).
Changes of transmitter release probability are generally reflected in changes of paired-pulse facilitation of the EPSC (Oleskevich et al. 2000). As already reported by Watase et al. (1998), GLAST knock-out had no effect on paired-pulse facilitation evoked at parallel fibre synapses with pulses separated by 5 ms (Fig. 2B). In 20 wild-type cells and 15 cells in slices from knock-out mice the ratios of the amplitude of the second EPSC to that of the first EPSC were 2.26 ± 0.16 and 2.24 ± 0.14, respectively (not significantly different, P = 0.93), suggesting that knocking out GLAST had no effect on the release probability from the parallel fibres.
To estimate the postsynaptic current produced by activation of a single parallel fibre, and check whether this was altered when GLAST was knocked out, we measured the ratio of the variance of the EPSC peak current to its mean value. Assuming that there is approximately one synaptic release site from each parallel fibre that contacts the recorded Purkinje cell (Palay & Chan-Palay, 1974; Napper & Harvey, 1988b), all of which, for simplicity, either release or fail to release glutamate with the same release probability p, the ratio of the variance (var) of the EPSC amplitude to its mean is given by
| (3) |
where i is the peak current produced when release does not fail (Sigworth, 1980; Meyer et al. 2001). For EPSCs of mean amplitude around 660 pA (similar to those in Fig. 1) produced by molecular layer stimulation, the value of this ratio (over ≈20 EPSCs, from time epochs selected for stability of the mean EPSC amplitude to avoid adding variance due to movement of the stimulating electrode or response run-down) was 11.50 ± 1.46 and 11.59 ± 0.94 pA, respectively, in 29 wild-type and 43 knock-out cells (Fig. 2C: not significantly different, P = 0.96). Increasing the intensity of stimulation to evoke EPSCs of about 1670 pA in amplitude (as used below) decreased the var/mean ratio by 2 ± 10 % (pooled data from five wild-type and seven knock-out cells; not significant, P = 0.86), and stimulation in the granular layer (as used below) gave a value of var/mean which was 11 ± 12 % lower than that measured for molecular layer stimulation in the same cell (pooled data from six wild-type and nine knock-out cells; not significantly different, P = 0.34). Since the paired-pulse facilitation experiments above indicate that the value of p is not altered in GLAST knock-out mice, the lack of change in the knock-out of var/mean in eqn (3) implies that there is also no change in i, the current produced when glutamate is released from a single fibre. Thus, since the mean EPSC amplitude is given by:
| (4) |
where N is the number of activated parallel fibres making synapses onto the recorded cell, and p and i are the same in wild-type and knock-out animals; comparing EPSCs of the same peak amplitude in wild-type and knock-out mice implies that the comparison is being made with the same number of input parallel fibres activated.
It is useful to have an approximate estimate of the number of input parallel fibres contributing to an EPSC of a certain magnitude. To obtain this, it is necessary to know the release probability, p. We therefore measured the variance and mean of the EPSC at different [Ca2+]o (substituted for Mg2+), and plotted the data (Fig. 2D) normalised to their values in 3 mM [Ca2+]o/1 mM [Mg2+]o (i.e. the concentrations used for the rest of the experiments). If the same number of fibres are stimulated in all conditions (and a subscript 3 means the value of a parameter in 3 mM [Ca2+]o/1 mM [Mg2+]o), eqns (3) and (4) give the predicted form of the graph in Fig. 2D as:
| (5) |
From the fits of eqn (5) to the data in Fig. 2D, we estimated p3 = 0.48 for two cells in 20-day-old wild-type mice, p3 = 0.46 for seven cells in 17-18-day-old GLAST knock-out mice, and (for comparison) p3 = 0.48 for four Purkinje cells in 15-day-old rat brain slices. Using these values of release probability, the variance/mean values derived from a large number of cells in Fig. 2C give mean single fibre EPSC amplitudes, pi, calculated as (var/mean)(p/(1 - p)), of 10.6 ± 1.3 and 9.9 ± 0.8 pA, respectively, for the wild-type and knock-out mice, which are not significantly different. (A reservation of the approach in Fig. 2D is that surface charge effects could result in fewer parallel fibres being stimulated when [Ca2+]o is raised; this would decrease the variance at high [Ca2+]o, and cause an overestimate of the values of p3 obtained from Fig. 2D, and hence an overestimate of pi). In the remainder of the text we take a rounded value of pi = 10 pA to provide an estimate of the number of activated input fibres contributing to a measured EPSC amplitude (and thus 20 pA as an estimate of the current produced when an activated bouton does release glutamate); however, the particular value assumed for this parameter will not alter any of the major conclusions reached in the paper.
Stimulating more parallel fibres prolongs the EPSC more in the GLAST knock-out
The prolongation of the EPSC with GLAST knock-out in Fig. 1A could reflect direct removal of glutamate from the synaptic cleft by GLAST transporters, or could reflect a role for GLAST in preventing glutamate diffusing between synapses and interacting with receptors that are normally activated by glutamate released from another bouton. Interaction between the effects of glutamate released from neighbouring synaptic boutons is more likely to occur when more parallel fibres are simultaneously active. Figure 3A and B shows specimen EPSCs recorded from Purkinje cells in a wild-type and in a GLAST knock-out cerebellar slice, when the stimulus strength was altered to evoke peak EPSC amplitudes of around 130, 620 and 1670 pA, corresponding to stimulation of approximately 13, 62 and 167 parallel fibres which contact the recorded cell. Normalising these responses to the peak amplitude, to allow comparison of their decays (Fig. 3C and D), shows that in both the wild-type and the knock-out cells the weighted time constant of the decay increases with the number of stimulated fibres. This indicates an interaction between synapses, since if synapses behaved independently τw would be independent of the number of active synapses. Furthermore, Fig. 3C and D show that this interaction is larger in the knock-out. Similar behaviour was seen in nine wild-type and 19 knock-out cells. Mean data from these cells are plotted in Fig. 3E, with the τw values obtained at a peak EPSC amplitude of around 620 pA normalised to 1 for the wild-type mice, and normalised to 1.18 for the knock-out mice (i.e. normalised to reproduce the relative values of 5.17 and 4.38 ms shown in Fig. 1D that were obtained from a larger sample of cells; normalization is used to remove scatter in the absolute values of τw in different cells, allowing more accurate assessment of the fractional change of τw at different EPSC sizes).
Figure 3. EPSC prolongation by GLAST knock out is larger when more parallel fibres are stimulated.
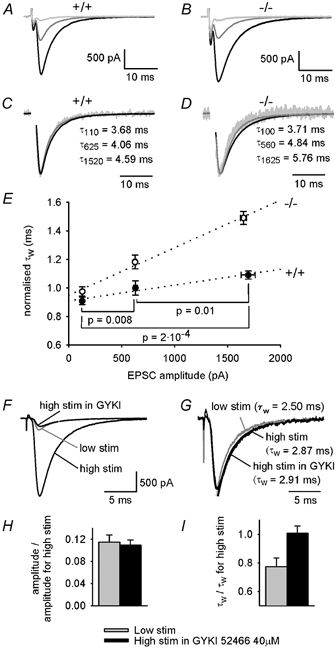
A, EPSCs with different amplitudes (produced by molecular layer stimulation of different intensities) in a specimen wild-type (+/+) cell. B, as A, but for a cell from a GLAST knock-out mouse (−/-). C and D, data in A and B normalised to have the same peak, with values of weighted decay time constant indicated (subscript indicates amplitude of EPSC in pA). E, mean (± s.e.m.) weighted decay time constants measured at 3 EPSC amplitudes in each of 9 wild-type and 19 knock-out cells, and rescaled to have the values 1 and 1.18 for 620 pA EPSCs in wild-type and knock-out cells, respectively, to reproduce the relative τw values in the much larger sample of cells in Fig. 1D (the actual ratio of τw values for the −/− and +/+ cells studied here was 1.15). The amplitudes and time constants of the two exponential components from which these data were calculated are included in the data in Fig. 4 legend. EPSC amplitudes could not be set exactly to the same three desired values (approximately 130, 620 and 1670 pA) in each cell; horizontal error bars show the scatter around the mean values. Dotted lines are linear regressions, the slopes of which differ significantly (P = 1.2 × 10−5). Horizontal lines indicate the +/+ data compared for the p values given. F-I, the observed variation of τw with EPSC amplitude is not due to changes in series resistance voltage errors and voltage non-uniformity. F, EPSC evoked in a wild-type cell with a strong stimulus in the absence and presence of GYKI 52466 (40 μM), and evoked by a weaker stimulus (grey trace) that produces an amplitude similar to that seen with the strong stimulus in GYKI 52466. G, the data in F normalised to the same peak and fitted with double exponential decays. H, the amplitude reduction produced by reducing the stimulus strength (grey) is similar (P = 0.73) to that produced by GYKI 52466 (black) in 11 wild-type cells. I, a significant fractional reduction of τw is produced by reducing the stimulus strength (grey, P = 0.004 comparing low and high stimulus strength) but not by applying GYKI 52466 (black, P = 0.88 comparing the absence and presence of GYKI) in the same 11 wild-type cells. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were, for reduced stimulus strength: 173 ± 20 pA, 2.92 ± 0.31 ms, 5.1 ± 2.9 pA, 7.46 ± 1.27 ms (3 out of 11 cells showed a slow component); and for GYKI 52466: 159 ± 17 pA, 3.14 ± 0.31 ms, 16.3 ± 7.7 pA, 25.1 ± 13.9 ms (7 out of 11 cells showed a slow component).
The lines through the points in Fig. 3E show that knocking out GLAST has a relatively small effect on the EPSC decay when a small number of parallel fibres are active: the weighted decay time constant is increased by only 7 % for an EPSC amplitude of ≈130 pA, and extrapolation to an EPSC amplitude of 20 pA suggests no significant difference when only one fibre is releasing glutamate onto the recorded cell. However GLAST knock-out has a large effect when many fibres are active: the weighted τ is increased by 37 % when the EPSC amplitude is ≈1670 pA. This dependence of the prolongation on the number of fibres activated implies a greater interaction between synapses when GLAST is absent, and excludes a compensatory change of postsynaptic receptor properties in the GLAST knock-out as the cause of the EPSC prolongation. If an increase in receptor affinity, or in the number of receptors around a synapse, were responsible for the EPSC prolongation seen in the knock-out, and contrary to our conclusion there was no interaction between synapses, then the EPSC prolongation would be observed even when a single input fibre was activated. Experimentally, however, this prolongation is not seen when a single bouton releases glutamate but only occurs when many fibres are active.
We wanted to check whether any of the small changes of EPSC duration seen in wild-type mice when altering the EPSC amplitude (Fig. 3E) were due to changes in voltage non-uniformity or series resistance voltage errors produced when the amplitude of the EPSC was altered (cf. Takahashi et al. 1995). To do this, we compared the effect on τw of decreasing the EPSC amplitude from about 1800 to 200 pA either by decreasing the number of fibres stimulated, or by applying the non-competitive AMPA receptor blocker GYKI 52466 (40 μM, Bleakman et al. 1996) while keeping the stimulus strength constant (Fig. 3F-I). Decreasing the number of fibres stimulated decreased τw by 23 ± 6 % in 11 cells (P = 0.004, Fig. 3F, G, and I), similar to the +/+ data shown in Fig. 3E, while decreasing the amplitude by the same factor (Fig. 3H) with GYKI 52466 (Fig. 3F) produced no significant change of τw in the same cells (increased by 0.8 ± 5.0 %, P = 0.88, Fig. 3G and I). We conclude that the effect on τw of changing the number of fibres stimulated is not produced by changes in voltage non-uniformity or series resistance voltage errors.
The increase in EPSC duration, which is produced by GLAST knock-out at all but the lowest stimulus intensities, was the result of an increased charge transfer by the slower exponential component of the EPSC decay, with no change of the charge transfer by the initial faster exponential component in eqn (1) (Fig. 4). The increased charge transfer therefore presumably reflects a slow tail of extracellular glutamate concentration resulting from the decrease in glutamate uptake rate. The amplitudes and time constants of the fast and slow exponential components from which these charge transfers were calculated are given in the legend to Fig. 4. These show that, when GLAST is knocked out, the increase in the fraction of charge transfer by the slower exponential component is due largely to an increase in the amplitude of the slower component relative to that of the faster component; when GLAST is knocked out, for 130 pA EPSCs the ratio (amplitude of the slow component)/(amplitude of fast component) increases from 2.2 to 3.0 %, for 620 pA EPSCs it increases from 3.9 % to 12.2 %, and for 1670 pA EPSCs it increases from 10.8 % to 33 %. In addition, in both the wild-type and knock-out mice, part of the increase in τw with increased stimulation strength reflects a significant increase in the time constant of the decay of the fast exponential component (≈20 % when the EPSC amplitude is increased from 130 to 1670 pA), as can be seen directly from the EPSC data in Fig. 3C and D.
Figure 4. Knocking out GLAST selectively increases charge transfer by the slower decaying component of the EPSC at large EPSC amplitudes.
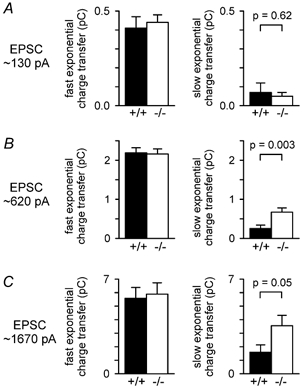
A-C, charge transfer by the faster exponential component of eqn (1) (left panel), and by the slower exponential component of eqn (1) (right panel), for EPSCs with amplitudes around 130 pA (A), 620 pA (B) and 1670 pA (C) (A and C, same cells as in Fig. 3E; B, all cells from Figs 3E and 1D). The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) for the cells of Fig. 3E were as follows. In wild-type cells: for 130 pA EPSCs 117 ± 8 pA, 3.45 ± 0.45 ms, 2.63 ± 1.82 pA, 45.9 ± 20.2 ms (3 out of 9 cells showed a slow component); for 620 pA EPSCs 542 ± 16 pA, 4.06 ± 0.22 ms, 21.3 ± 12.1 pA, 31.1 ± 11.2 ms (13 out of 37 cells showed a slow component); for 1670 pA EPSCs 1352 ± 41 pA, 4.06 ± 0.49 ms, 146 ± 71 pA, 28.8 ± 16.3 ms (6 out of 9 cells showed a slow component). In knock-out cells: for 130 pA EPSCs 114 ± 5 pA, 3.89 ± 0.26 ms, 3.37 ± 1.74 pA, 18.01 ± 5.98 ms (6 out of 19 cells showed a slow component); for 620 pA EPSCs 486 ± 14 pA, 4.34 ± 0.19 ms, 59.6 ± 12.0 pA, 19.2 ± 2.8 ms (35 out of 47 cells showed a slow component); for 1670 pA EPSCs 1125 ± 102 pA, 4.70 ± 0.48 ms, 374.1 ± 98.3 pA, 19.8 ± 4.5 ms (17 out of 19 cells showed a slow component).
Granular layer stimulation in GLAST KO mice yields a faster decaying EPSC
Stimulating parallel fibres with an electrode in the molecular layer, as in the experiments described above, may activate a narrow beam of fibres that are more adjacent than the fibres activated during the normal function of the cerebellum, and may thus promote interaction between the effects of glutamate released from different boutons. To try to produce a more natural pattern of activation, we used an electrode in the granule cell layer to stimulate ascending granule cell axons, in order to activate synapses that might be more spread out in the Purkinje cell dendritic tree. In these experiments the stimulus strength was initially adjusted to produce an EPSC amplitude of around 600 pA. In wild-type mice, granular layer stimulation produced an EPSC with a weighted decay time constant not significantly different from that produced in the same cell by stimulation in the molecular layer (Fig. 5A and C, six cells). However, for GLAST knock-out mice, granular layer stimulation evoked an EPSC with a significantly smaller weighted decay time constant than that for similar size EPSCs produced by stimulation in the molecular layer (Fig. 5B and C, 12 cells), and this shortening of τw was due to less charge transfer by the slower exponential component of the EPSC (Fig. 5D and E), presumably reflecting a faster decay of extracellular glutamate concentration. With GLAST absent, the weighted decay time constant seen when stimulating in the granular layer was similar in value to that seen for stimulation of only a few afferents in the molecular layer, and was independent of the size of the EPSC that was produced (Fig. 5F).
Figure 5. Granular layer stimulation evokes briefer parallel fibre EPSCs than molecular layer stimulation in GLAST knock-out mice.
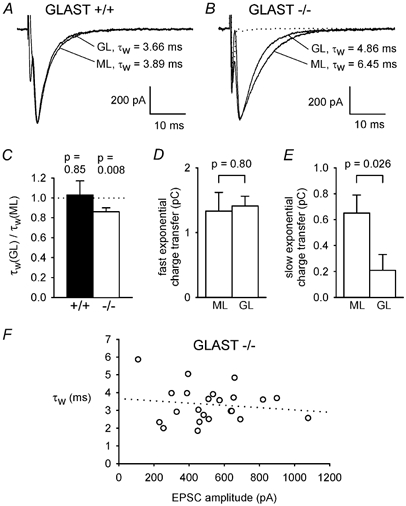
A, specimen parallel fibre EPSCs of similar amplitude recorded in the same wild-type cell (+/+) when stimulating the granule cell axons either in the molecular layer (ML) or the granular layer (GL), with weighted decay time constants (τw) indicated. B, as for A but in a GLAST knock-out cell (−/-). C, ratio of τw for GL and ML stimulation in 6 wild-type and 12 knock-out cells. P values are for t tests comparing data with unity. Amplitudes of EPSCs were 653 ± 72 pA for GL and 660 ± 52 pA for ML stimulation in the +/+ cells, and 578 ± 68 pA for GL and 578 ± 70 pA for ML stimulation in the −/− cells (not significantly different: P = 0.94 for +/+ and 0.998 for −/−). D and E, charge transfer by the faster exponential component (D), and by the slower exponential component of eqn (1) (E), for ML and GL stimulation in the 12 knock-out cells. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were: for ML stimulation, 467 ± 58 pA, 3.34 ± 0.41 ms, 47.5 ± 18.1 pA, 9.12 ± 1.90 ms (7 out of 12 cells showed a slow component); and for GL stimulation: 484 ± 55 pA, 2.99 ± 0.20 ms, 19.9 ± 14.1 pA, 14.5 ± 2.8 ms (4 out of 12 cells showed a slow component). F, τw as a function of EPSC amplitude for GL stimulation in 23 knock-out cells (includes some cells stimulated only in the GL layer). This experiment (unlike that of Fig. 3E) required inclusion of data from different cells because the range of EPSC amplitudes which can be generated by altering the intensity of GL stimulation is much smaller than for ML stimulation.
The effect of blocking GLT-1 in wild-type and GLAST knock-out mice
The glutamate analogue dihydrokainate (DHK) is a non-transported blocker of GLT-1 transporters (Arriza et al. 1994; Levy et al. 1998). For an initial EPSC amplitude of around 660 pA, i.e. stimulating ≈66 input parallel fibres in the molecular layer, in Purkinje cells from wild-type mice DHK (200 μM) had no effect on the parallel fibre EPSC amplitude, time to peak or weighted decay time constant (Fig. 6A and D-F), presumably because GLT-1 contributes a relatively minor fraction of the glutamate transport activity in the molecular layer (GLT-1 is expressed at only ≈1/6 the density of GLAST in the molecular layer; Lehre & Danbolt, 1998). By contrast, in Purkinje cells from GLAST knock-out mice, DHK reversibly prolonged the EPSC decay (Fig. 6B and F) and slightly increased its amplitude (presumably because the peak glutamate concentration is increased; Fig. 6B and D). This suggests that when GLAST is not functioning GLT-1 provides a significant fraction of the uptake present, and blocking it slows glutamate removal enough to prolong the EPSC. As for the effect of the GLAST knock-out, the EPSC prolongation arose from an increase in the charge transfer of the slower exponential component of the EPSC, with no significant change in the charge transfer by the faster component (Fig. 6G and H). Interestingly DHK also prolonged the time to peak of the EPSC (Fig. 6B inset and 6E); the possible reason for this is considered in the Discussion. When stimulating in the granular layer in GLAST knock-out mice the prolongation of the EPSC by DHK was less than that seen for stimulation in the molecular layer (Fig. 6C and F), consistent with less spillover interaction occurring when the stimulated synapses are more separated.
Figure 6. Block of GLT-1 transporters prolongs parallel fibre EPSCs in the GLAST knock-out.
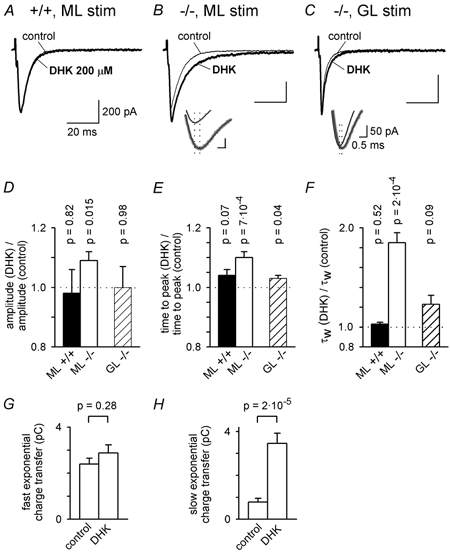
A, parallel fibre EPSCs evoked by molecular layer (ML) stimulation in a wild-type (+/+) Purkinje cell in the absence (control) and presence of 200 μM dihydrokainate (DHK). B, as for A, but in a GLAST knock-out (−/-) cell. Inset shows time at the peak of the EPSCs (vertical dotted lines); smooth black curves through grey data are multi-exponential fits to define the peak better and the thicker grey trace is in DHK. C, as for B, but with stimulation in the granular layer (GL). D-F, mean values of EPSC amplitude (D), time from the start of the stimulus to the peak of the EPSC (E), and weighted decay time constant (F) in DHK relative to the value in the same cell in control solution, for 6 wild-type cells stimulated in the ML (control EPSC amplitude was 666 ± 33 pA), 18 knock-out cells stimulated in the ML (control EPSC amplitude was 608 ± 12 pA), and 4 knock-out cells stimulated in the GL (control EPSC amplitude was 555 ± 64 pA, not significantly different from ML stimulation, P = 0.47). P values are for t tests comparing data with unity. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were as follows. For ML stimulation in wild-type, in control: 597 ± 33 pA, 4.80 ± 0.15, 18.5 ± 15.8 pA, 23.0 ± 10.5 ms (2 out of 6 cells showed a slow component); in DHK: 588 ± 61 pA, 4.87 ± 0.21 ms, 17.3 ± 10.9 pA, 18.3 ± 3.2 ms (2 out of 6 cells showed a slow component). For ML stimulation in knock-out cells, in control: 502 ± 28 pA, 4.54 ± 0.38 ms, 71.0 ± 25.2 pA, 24.4 ± 5.3 ms (16 out of 18 cells showed a slow component); in DHK: 499 ± 21 pA, 5.63 ± 0.59 ms, 93.7 ± 15.7 pA, 43.0 ± 4.4 ms (all 18 cells showed a slow component). For GL stimulation in knock-out cells, in control: 441 ± 115 pA, 2.99 ± 0.48 ms, 46.7 ± 21.9 pA, 8.80 ± 0.74 ms (2 out of 4 cells showed a slow component); in DHK: 502 ± 66 pA, 3.34 ± 0.58 ms, 33.9 ± 22.4 pA, 36.9 ± 13.1 ms (3 out of 4 cells showed a slow component). G and H, charge transfer by the faster (G) and slower (H) exponential components of eqn (1), for ML stimulation in the 18 knock-out cells in the absence (control) and presence of DHK.
As for the effect of GLAST knock-out (Fig. 3E), the effect of blocking GLT-1 was smaller when only a small number of parallel fibres were active (a 34 % increase of τw for an EPSC amplitude of ≈130 pA, P = 0.016), but was much larger when many fibres were stimulated (a 2.4-fold prolongation for an EPSC amplitude of ≈1610 pA, P = 2 × 10−5, Fig. 7). The similarity of this effect of blocking GLT-1, for which comparisons can be made within the same cell, with the effect of knocking out GLAST is consistent with the difference between the GLAST knock-out and wild-type cells in Fig. 3E being a result of preventing uptake rather than of some non-specific effect of the knock-out.
Effect of temperature
Spillover of glutamate between synapses in the hippocampus is reported to be less important at physiological temperatures than at room temperature (Asztély et al. 1997). We therefore tested whether at 37 °C inhibiting uptake still prolonged the EPSC evoked by stimulating a large number of parallel fibres.
In wild-type mice, increasing the temperature from our normal experimental temperature (27 °C) to 37 °C decreased the time from the stimulus to the peak of the EPSC by a factor of 1.37 (Fig. 8A and B), decreased the weighted decay time constant by a factor of 1.29 (Fig. 8C), and increased the EPSC amplitude by a factor of 1.42 (Fig. 8D), qualitatively similar to data reported for chick calyceal synapses (Zhang & Trussell, 1994). The last factor was used to estimate the number of parallel fibres stimulated, as described below, by assuming that the increase in EPSC amplitude is not due to fibres becoming more easily excited at higher temperature. (This assumption is not crucial; if it were wrong then, in the experiments below, fewer fibres would have been stimulated than we estimate, and significant spillover would be occurring with fewer active fibres). In order to compare EPSC decays in the presence of different amounts of spillover in the same cell (rather than comparing between cells from wild-type and knock-out mice, which requires studying many more cells), we studied the effect of DHK (200 μM) on the decay of the EPSC evoked by stimulation in the molecular layer. In seven wild-type cells DHK had no effect on the EPSC at 35-37 °C (data not shown), as in Fig. 6 at 27 °C. In cells from mice lacking GLAST, increasing the EPSC amplitude from ≈365 to ≈1215 pA at 35-37 °C (equivalent to amplitudes of ≈257 and ≈856 pA at 27 °C (Fig. 8D), and thus reflecting stimulation of ≈26 and ≈86 input parallel fibres) the EPSC weighted decay time constant increased by 20 % (Fig. 8E, −/− control data), similar to the 19 % increase seen in Fig. 3E (−/- data) when increasing the EPSC size from 257 to 856 pA at 27 °C. For EPSCs of ≈1215 pA, the weighted decay time constant was prolonged by 97 % when DHK was added (Fig. 8E), similar to the prolongation seen for an EPSC amplitude of 856 pA at 27 °C (99 % from Fig. 7E), and less prolongation was seen when fewer parallel fibres were stimulated (Fig. 8E). As at 27 °C, the increase of τw at high stimulus strengths was due to more charge transfer by the slower exponential component of the EPSC decay (Fig. 8F and G). Thus, when many fibres are releasing glutamate, blocking uptake increases glutamate spillover and interactions between synapses even at physiological temperatures.
Figure 8. Synaptic crosstalk occurs at physiological temperature.

A, specimen parallel fibre EPSCs evoked by molecular layer stimulation in a wild-type Purkinje cell at 27 °C and 37 °C. B-D, temperature dependence of the time from the start of the stimulus to the peak of the EPSC (B), the EPSC weighted decay time constant (C), and the EPSC amplitude normalised to its value at 27 °C (D), in 10 wild-type Purkinje cells. Mean amplitude of EPSC at 27 °C was 2320 ± 290 pA, and temperature dependence of amplitude did not depend significantly on EPSC amplitude at 27 °C for amplitudes between 1300 and 3400 pA (not shown). E, EPSC weighted decay time constant as a function of EPSC amplitude at 35-37 °C, in 6 GLAST knock-out cells (−/-) in the absence and presence of DHK (200 μM). From D, the amplitudes of EPSC studied here (≈365 and ≈1215 pA) correspond to amplitudes of ≈257 and ≈856 pA at 27 °C. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were as follows. For low stimulation, in control: 269 ± 54 pA, 2.45 ± 0.44 ms, 51.8 ± 48.8 pA, 9.96 ± 3.90 ms (2 out of 6 cells showed a slow component); in DHK: 307 ± 54 pA, 2.98 ± 0.45 ms, 24.1 ± 8.4 pA, 14.5 ± 2.6 ms (5 out of 6 cells showed a slow component). For high stimulation, in control: 982 ± 165 pA, 2.90 ± 0.45 ms, 142 ± 118 pA, 23.6 ± 5.7 ms (5 out of 6 cells showed a slow component); in DHK: 908 ± 99 pA, 3.87 ± 0.50 ms, 132 ± 29 pA, 34.8 ± 3.5 ms (all cells showed a slow component). F, charge transfer by the faster and slower exponential components of eqn (1), for the data in E at an EPSC amplitude of ≈365 pA. G, as F, but for the data in E with an EPSC amplitude of ≈1215 pA. H and I, in wild-type cells at 36 °C τw varies with EPSC amplitude, and this is not due to changes in series resistance voltage errors and voltage non-uniformity. H, the fraction of EPSC amplitude remaining is not significantly different (P = 0.17) when stimulation strength is reduced (grey) from strong (producing an EPSC amplitude of ≈ 1560 pA) to weak (≈320 pA amplitude), or when GYKI 52466 (40 μM) is applied during strong stimulation (black), in 5 wild-type cells at 36 °C. I, a significant fractional reduction of τw is produced by reducing the stimulus strength (grey, P = 0.004 comparing low and high stimulus strength) but not by applying GYKI 52466 (black, P = 0.75 comparing the absence and presence of GYKI) in the same 5 wild-type cells. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were: for reduced stimulus strength, 293 ± 49 pA, 1.93 ± 0.08 ms, 1.69 ± 1.69 pA, 22.8 ms (1 out of 5 cells showed a slow component); and for GYKI 52466, 389 ± 36 pA, 2.26 ± 0.09 ms, 1.85 ± 0.84 pA, 29.0 ± 9.4 ms (3 out of 5 cells showed a slow component).
In wild-type mice, increasing the number of fibres stimulated also increased the weighted time constant at 37 °C. Changing from strong to weak stimulation, to lower the EPSC amplitude from ≈1560 to ≈320 pA, decreased the weighted time constant by 19 % (P = 0.004; Fig. 8H and I). Applying GYKI 52466 (40 μM) to the same cells, to produce a similar decrease of EPSC amplitude during strong stimulation, had no effect on the weighted time constant (P = 0.75; Fig. 8H and I), ruling out an alteration of series resistance voltage error or voltage non-uniformity as the cause of the alteration of time constant seen when altering the number of fibres stimulated (cf. Fig. 3F-I). Thus, even without GLAST knocked out, synapses interact with each other at 37 °C when many nearby fibres are active.
The effect of inhibiting all glutamate transporters
In addition to GLAST and GLT-1, there is also significant glutamate uptake by neuronal glutamate transporters (EAAT4 and EAAC1) in Purkinje cells (Takahashi et al. 1996; Otis et al. 1997; Furuta et al. 1997; Tanaka et al. 1997; Dehnes et al. 1998; Auger & Attwell, 2000). The glutamate analogue DL-threo-β-benzyloxyaspartate (TBOA) is reported to block all Na+-dependent glutamate transporters without being transported itself (Shimamoto et al. 1998; Auger & Attwell, 2000) so, to investigate the effect of blocking a major fraction of the glutamate transporters present, particularly in wild-type mice, which lack any compensatory adaptations that might be present in the GLAST knock-out, we superfused TBOA (200 μM) in addition to DHK (200 μM), in slices from wild-type or GLAST knock-out mice at 27 °C. After a few minutes superfusion of TBOA and DHK, cells in GLAST knock-out slices developed a large inward current (presumably generated by glutamate accumulation) and often subsequently died; the data described below were recorded before this inward current developed.
The weighted decay time constant of the parallel fibre EPSC evoked by stimulating in the molecular layer (selected to have an amplitude of ≈640 pA) was prolonged more by DHK + TBOA (Fig. 9A and B) than by DHK alone (Fig. 6), in Purkinje cells both from wild-type and from GLAST knock-out mice. In wild-type cells DHK + TBOA prolonged the EPSC 3.1-fold (Fig. 9F) compared with no prolongation by DHK alone (Fig. 6F, significantly different from DHK + TBOA, P = 0.011), while in GLAST knock-out cells DHK + TBOA prolonged the EPSC 3.4-fold (Fig. 9F) compared to a 1.9-fold prolongation with DHK alone (Fig. 6F, significantly different from DHK + TBOA, P = 0.017). When stimulating in the granular layer in knock-out mice (Fig. 9C), the prolongation produced by DHK + TBOA (2.4-fold: Fig. 9F) was also greater (P = 0.003) than that seen with DHK alone (1.2-fold: Fig. 6F). These data are consistent with transporters in addition to GLAST and GLT-1, presumably EAAT4 and/or EAAC1 in Purkinje cells, playing a role in terminating the parallel fibre EPSC, but could also reflect a more complete block of GLT-1 when TBOA is present in addition to DHK. With both granular and molecular layer stimulation the prolongation of the EPSC was due to a selective increase in the charge transfer of the slower exponential component of the EPSC decay (Fig. 9G and H), and the time to peak of the EPSC was slightly increased (Fig. 9E). For GLAST knock-out mice, DHK + TBOA produced a small but non-significant decrease in EPSC amplitude (Fig. 9D).
Figure 9. The effect of blocking glutamate transporters with a combination of TBOA, DHK and GLAST knock out.
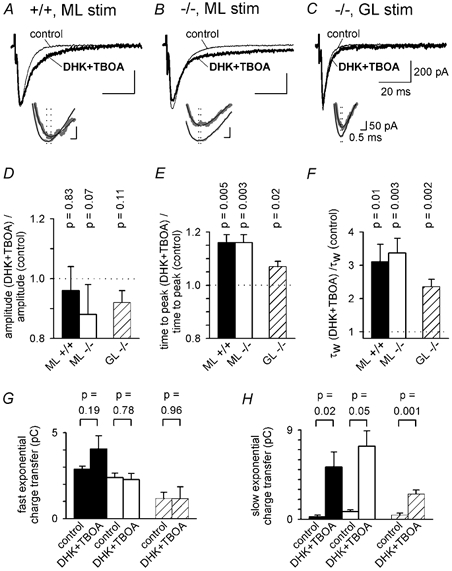
A, parallel fibre EPSCs evoked by molecular layer (ML) stimulation in a wild-type (+/+) Purkinje cell in the absence (control) and presence of 200 μM DHK + 200 μM TBOA. Inset shows time at peak of the EPSCs (vertical dotted lines); smooth black curves through grey data are multi-exponential fits to define the peak better and the thicker trace is in DHK + TBOA. B, As for A, but in a GLAST knock-out (−/-) cell. C, as for B, but with stimulation in the granular layer (GL). D-F, mean values of EPSC amplitude (D), time from the start of the stimulus to the peak of the EPSC (E), and weighted decay time constant (F) in DHK + TBOA relative to the value in the same cell in control solution, for 6 wild-type cells stimulated in the ML (control EPSC amplitude was 666 ± 33 pA), 6 knock-out cells stimulated in the ML (control EPSC amplitude was 615 ± 46 pA, P = 0.39 compared with wild-type), and 6 knock-out cells stimulated in the GL (control EPSC amplitude was 483 ± 50 pA, P = 0.08 compared with ML stimulation). P values are for t tests comparing data with unity. G and H, charge transfer by the faster (G) and slower (H) exponential components of eqn (1), in the absence and presence of DHK + TBOA, for the cells of D-F (black bars: ML +/+; white bars: ML −/−; shaded bars: GL −/−). P values are for paired t tests comparing data with and without DHK + TBOA. The amplitudes and time constants of the fast and slow exponential components of the EPSC decay (in the order Afast, τfast, Aslow, τslow) were as follows. For ML stimulation in wild-type, in control: 597 ± 33 pA, 4.80 ± 0.15 ms, 18.6 ± 15.8 pA, 23.0 ± 10.5 ms (2 out of 6 cells showed a slow component); in DHK + TBOA: 450 ± 34 pA, 8.92 ± 1.57 ms, 121 ± 26 pA, 42.6 ± 5.6 ms (all cells showed a slow component). For ML stimulation in knock-out, in control: 511 ± 26 pA, 5.20 ± 0.62 ms, 30.3 ± 14.1 pA, 33.6 ± 12.2 ms (all cells showed a slow component); in DHK + TBOA: 377 ± 61 pA, 6.53 ± 1.14 ms, 97.8 ± 11.9 pA, 79.9 ± 21.0 ms (all cells showed a slow component). For GL stimulation in knock-out, in control: 389 ± 77 pA, 2.91 ± 0.32 ms, 34.3 ± 21.3 pA, 17.3 ± 8.4 ms (3 out of 6 cells showed a slow component); in DHK + TBOA: 383 ± 37 pA, 3.43 ± 0.31 ms, 28.4 ± 3.6 pA, 87.6 ± 9.8 ms (all cells showed a slow component).
In the presence of TBOA + DHK the EPSC weighted decay time constant was not significantly different in wild-type and knock-out mice (P = 0.31). This, and the selective effect on the slower exponential component of the EPSC decay, of both TBOA + DHK in the wild-type mouse and of knocking out GLAST, is consistent with the effects of GLAST knock-out reflecting a removal of transporters and prolonging the glutamate transient rather than reflecting a compensatory effect on the properties of glutamate receptors.
Effect of GLAST knock-out and block of GLT-1 on the response to trains of stimuli
In vivo, in response to peripheral input, the parallel fibres can fire action potentials in bursts of up to 400 Hz (Eccles et al. 1966). High frequency release of glutamate from boutons is more likely to lead to glutamate spillover and an enhanced role for glutamate transporters. We compared the response of Purkinje cells in wild-type and GLAST knock-out mice to 10 parallel fibre stimuli applied at 200 Hz, and tested the effect of adding DHK to block GLT-1. The stimulus amplitude was set to produce an EPSC amplitude of either ≈170 or ≈1100 pA in response to a single stimulus, i.e. reflecting stimulation of about 17 or 110 input parallel fibres, respectively. During the train there may be an increase in the number of fibres which are stimulated (Merrill et al. 1978), and facilitation of release probability during the train of stimuli (Dittman et al. 2000) will increase the number of stimulated fibres which release glutamate.
In wild-type mice the response to the stimuli summated during the train due to the facilitation of glutamate release and increase in number of stimulated fibres mentioned above, and blocking GLT-1 had little effect on the response (Fig. 10A and C). In mice lacking GLAST, both for the weaker (Fig. 10B) and the stronger stimuli (Fig. 10D), the current response to the stimulus train was larger (Fig. 10E, compare control data for −/− and +/+), the current decayed much more slowly after the train (Fig. 10F), the charge transfer produced by the train was approximately doubled (Fig. 10G), and blocking GLT-1 increased these changes further. Extrapolating the data in Fig. 10E-G back to an initial amplitude of 20 pA, corresponding to glutamate being released from a single parallel fibre, knocking out GLAST is predicted to increase by a factor of 1.7 the amplitude of the maximum current produced by the train, to increase 2.3-fold the time for the current to decay to 50 % of its value at the end of the train, and to increase 2.2-fold the charge transfer produced by the train, and subsequent block of GLT-1 is predicted to produce a further 1.6-, 2.2- and 2.4-fold increase of these parameters. Thus for high frequency trains, even when small numbers of parallel fibres are stimulated, blocking uptake by GLAST, or by GLAST and GLT-1, has a dramatic effect on the resulting EPSC.
The decrease in the ratio of the peak current produced by the train to the amplitude of the first EPSC in the train, which is seen even for wild-type cells in Fig. 10E on increasing the stimulus strength, is not expected if synapses operated independently. It may reflect saturation or desensitization of AMPA receptors, or activation of presynaptic receptors inhibiting glutamate release, by the large extracellular glutamate concentration which is produced when a large number of parallel fibres are stimulated at high frequency and glutamate diffuses between synapses.
Even with high frequency stimulation at the higher intensity, no EPSC component produced by metabotropic receptors was seen: the EPSC was entirely blockable by NBQX (Fig. 10D), possibly because of the presence of intracellular QX314 which blocks G protein-mediated signalling, or the low [Ca]i maintained by the BAPTA in our intracellular solution. Thus, the prolonged EPSC in Fig. 10B and D is entirely produced by AMPA receptors.
DISCUSSION
Anatomical features relevant to parallel fibre synaptic crosstalk
The density of synapses in the parallel fibre layer is high, with a mean separation of about 1.1 μm, and 17 synapses per linear μm of the Purkinje cell processes (Napper & Harvey, 1988a,b). The strong ensheathment of parallel fibre synapses by glia expressing a high density of GLAST (and to a lesser extent GLT-1) glutamate transporters (Palay & Chan-Palay, 1974; Lehre & Danbolt, 1998; Xu-Friedman et al. 2001) may have evolved to reduce the strong synaptic crosstalk which would otherwise seem inevitable for such a short diffusion time between synapses (≈1 ms for 1 μm). Here we have investigated how well uptake of synaptically released glutamate by the ensheathing glial cells (Bergles et al. 1997; Clark & Barbour, 1997) succeeds in preventing synaptic crosstalk. Previous work showed that spillover of glutamate occurs between parallel fibre synapses when many boutons are stimulated at high frequency (Carter & Regehr, 2000). Our work extends this finding by demonstrating that spillover leads to activation of AMPA receptors following stimulation of nearby parallel fibres with a single stimulus, and shows that the AMPA receptor EPSC produced by high frequency activation of a single fibre is strongly shaped by transporter action.
Glial glutamate transporters reduce crosstalk between parallel fibres
If cerebellar parallel fibre synapses operated independently then information could be preserved during synaptic transmission and stored independently at different synapses. In this case, the waveform of the AMPA receptor EPSC evoked by a single stimulus to the parallel fibres, and any EPSC prolongation produced by blocking glutamate uptake, should be independent of the number of fibres stimulated. Experimentally, however, the EPSC duration increases with the number of fibres activated by molecular layer stimulation when uptake is functioning normally, both at 27 °C (Fig. 3E, +/+ data) and at 36 °C (Fig. 8I), showing that when many nearby synapses are active they interact with each other. Furthermore, genetic or pharmacological block of the glial glutamate transporters GLAST and GLT-1 prolongs the EPSC when many parallel fibres are active, even at 36 °C, but has little effect when only a few fibres are active (Figs. 3E, 7E and 8E). The strong dependence of this prolongation on the number of active fibres implies that with uptake blocked there is an increased interaction between different synapses, presumably as a result of glutamate diffusing between synapses and increasing the activation of AMPA receptors. Thus, the main role of GLAST and GLT-1 during synaptic transmission is to prevent diffusion of glutamate between excitatory synapses.
Since our data demonstrate that there is an interaction between synapses when more parallel fibres are releasing glutamate, glutamate released from one presynaptic site must be affecting receptors that are also influenced by glutamate released from another site. However, we cannot say whether the receptors that are sensing glutamate from two release sites are within the synaptic cleft or are extrasynaptic.
Our data suggest that, for single stimuli, GLAST and GLT-1 do not shape the AMPA receptor EPSC produced at a single active bouton, and diffusion of glutamate away from the synapse presumably terminates the EPSC. Desensitization cannot be the main cause of the decay, since when more fibres are stimulated and more glutamate is released the fast component of the decay is slowed (Fig. 3C and D and Fig. 4 legend), presumably as a result of reduced diffusion when the [glutamate]o rises around nearby synapses, whereas a raised [glutamate]o would be expected to cause faster desensitization.
At high stimulation strengths, when GLAST is knocked out the charge transfer by the single stimulus EPSC is increased by ≈40 % (Fig. 3E). The fact that much of this extra charge transfer increase occurs late in the EPSC does not reduce its importance. At this synapse a single parallel fibre action potential is insufficient to produce a postsynaptic action potential: temporal and spatial summation of about 50 individual parallel fibre EPSCs is necessary to produce a Purkinje cell action potential (Barbour, 1993), and a prolongation of each EPSC will reduce the amount of presynaptic activity needed to make the Purkinje cell fire an action potential.
For high frequency trains of action potentials, the much greater glutamate release occurring results in transporters shaping the EPSC dramatically even when a single bouton is active (Fig. 10), demonstrating the potential for information flow through the cerebellar cortex to be altered by modulation of the activity of glial glutamate transporters. Some of this effect of transporters may result from them preventing activation of facilitatory or inhibitory presynaptic glutamate receptors (Casado et al. 2000; Daniel & Crepel, 2001; Delaney & Jahr, 2002) by the accumulation of glutamate during a train.
Watase et al. (1998) observed no effect of GLAST knock-out on the parallel fibre EPSC decay. A possible cause of this discrepancy is the dependence of the decay time constant on the number of parallel fibres stimulated (Fig. 3E). Since the time constant was not studied as a function of EPSC amplitude by Watase et al. (1998) it is possible that extra variability introduced by the different numbers of fibres active could obscure the prolongation of the EPSC which we observe when GLAST is knocked out.
The role of neuronal transporters
Our experiments have focussed on the role of glial glutamate transporters, but Purkinje cells also express the neuronal transporters EAAC1 and EAAT4 postsynaptically (Furuta et al. 1997; Dehnes et al. 1998) and in hippocampus postsynaptic transporters can limit glutamate spillover between synapses (Diamond, 2001). Since, at the climbing fibre synapse, transporters in Purkinje cells rapidly remove a significant fraction of synaptically released glutamate (Otis et al. 1997; Auger & Attwell, 2000), it is likely that neuronal transporters also help to limit glutamate spillover between parallel fibre synapses.
Glutamate crosstalk prolongs AMPA receptor mediated EPSCs
Previous work has suggested that glutamate released from active synapses is able to diffuse to other nearby synapses and activate NMDA receptors (Kullmann et al. 1996; Asztély et al. 1997; Carter & Regehr, 2000). The fact that such spillover can also activate AMPA receptors at nearby synapses (this paper; Carter & Regehr, 2000), despite their lower affinity for glutamate, is consistent with simulations by Barbour (2001) which also predicted that uptake block would have a much larger effect on spillover EPSCs than on conventional (non-spillover) EPSCs, and is consistent with the demonstration of spillover-mediated AMPA EPSCs at the cerebellar mossy fibre-to-granule synapse (DiGregorio et al. 2002).
The increase in the duration of the parallel fibre EPSC when more parallel fibres are stimulated (Fig. 3E) is consistent with studies on other synapses (calyceal synapse: Trussell et al. 1993; parallel fibre-to-stellate cell synapse: Carter & Regehr, 2000; Schaffer collateral CA3-CA1 synapse: Arnth-Jensen et al. 2002). Diffusion of glutamate between synaptic release sites (or to extrasynaptic receptors which sense glutamate released from more than one bouton) can produce this effect in three distinct ways (Trussell et al. 1993; Takahashi et al. 1995; DiGregorio et al. 2002). First, with uptake inhibited or a large number of fibres activated, glutamate may diffuse to receptors on the recorded cell from release sites which do not form a synapse onto the recorded cell, and the greater diffusion distance will produce a slower rising and falling glutamate transient. Consistent with this, we found that strong uptake block (applying DHK in the GLAST knock-out, or applying TBOA + DHK in the wild-type or the knock-out: Figs. 6 and 9) prolonged the time to the peak of the EPSC. Second, even if the extra release sites recruited at high stimulus strengths form direct synapses onto the recorded cell, non-linearity of the dose-response curve of AMPA receptors can result in the larger glutamate transient which is produced by spillover from more synapses leading to a longer EPSC (Sarantis et al. 1993). Finally, saturation of uptake at the higher glutamate concentration produced by spillover when release occurs from many boutons can lead to a slower removal of glutamate and a longer EPSC.
An estimate of the minimum distance over which synaptic crosstalk occurs
A significant increase in EPSC duration is seen in wild-type mice when ≈100 input parallel fibres are simultaneously actived by a single stimulus (Fig. 3E). If the Purkinje cell dendritic tree receives 105 parallel fibre inputs over an area of (approximately) 100 μm × 100 μm, then 100 parallel fibres would occupy an area of ≈10 μm2 corresponding to a circle of radius 1.8 μm. Thus, if all the stimulated input fibres are close together, spillover can occur between synapses roughly 1.8 μm apart (if the 100 fibres are more separated, then spillover must occur over a greater distance). The time needed to diffuse a distance x = 1.8 μm can be estimated from the diffusion equation as t = x2/2(D/λ2), where the diffusion coefficient D = 7.6 × 10−10 m2 s−1 (the value for glutamine: Longsworth, 1953) and the tortuosity factor λ = 1.55 (Nicholson & Phillips, 1981) which is 5.1 ms. This is consistent with spillover having an effect on the EPSC decay ≈5 ms after the EPSC peak (Fig. 1 and Fig. 3). By contrast, Carter & Regehr (2000) observed spillover of glutamate over tens of microns when transmitter release was evoked by trains of stimuli.
Does synaptic crosstalk improve signal to noise ratio?
Glutamate spillover degrades the specificity of information transmission but, by allowing synaptic transmission to employ a larger effective number of release sites or to activate a larger number of postsynaptic receptors (which fluctuate in their opening and closing), it may increase the signal to noise ratio (DiGregorio et al. 2002). Barbour (1993) estimated that ≈50 parallel fibres need to be simultaneously active to evoke a Purkinje cell action potential. If these inputs were very close together, Fig. 3E indicates that, in wild-type mice, spillover would increase the weighted decay time constant and the charge transfer, and hence the number of channels opened or the effective number of release sites, by only 6 % (+/+ data for an EPSC amplitude of 50 × 10 pA = 500 pA, compared to the value extrapolated for a very small EPSC). Since the signal to noise ratio is expected to be proportional to the square root of the total number of channels opened, we conclude that spillover will produce an insignificant increase (3 %) of signal to noise ratio. Some synaptic crosstalk may occur, therefore, not as a design feature, but simply because it is not feasible to have a sufficient density of glutamate transporters to prevent all leakage of glutamate out of the synaptic cleft.
The role of glial glutamate transporters in vivo
As our experiments were done on relatively young animals, we cannot be sure that the results obtained will also reflect the properties of mature parallel fibre synapses. In addition, the importance of glial glutamate transporters for limiting synaptic crosstalk in vivo will depend both on the rate of firing of parallel fibres and on how close together simultaneously active parallel fibres are. For single stimuli we observed much less spillover-evoked EPSC prolongation when stimulating granule cell axons in the granular layer than when using molecular layer stimulation, presumably because the same number of activated synapses are spread over a larger area of the dendritic tree. Thus, physiological patterns of low frequency parallel fibre activity may produce significantly less interaction between parallel fibre synapses than does the experimental application of a strong stimulus to the molecular layer. For high frequency trains of action potentials however, even in a single parallel fibre, the activity of glial transporters has a major effect on the EPSC charge transfer (Fig. 10).
Acknowledgments
Supported by a Marie-Curie fellowship to P.M., the European Union, the Wellcome Trust and a Wolfson-Royal Society Award to D.A. We thank Prof. Kohichi Tanaka for supplying the knock-out mice and Hélène Marie, Stephen McGuiness and Manuela Lahne for genotyping them, and thank Philippe Isope, Angus Silver and Alasdair Gibb for helpful discussion.
REFERENCES
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztély F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Auger C, Attwell D. Fast removal of synaptic glutamate by postsynaptic transporters. Neuron. 2000;28:547–558. doi: 10.1016/s0896-6273(00)00132-x. [DOI] [PubMed] [Google Scholar]
- Barbour B. Synaptic currents evoked in Purkinje cells by stimulating individual granule cells. Neuron. 1993;11:759–769. doi: 10.1016/0896-6273(93)90085-6. [DOI] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci. 2001;20:7969–7984. doi: 10.1523/JNEUROSCI.21-20-07969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Häusser B. Intersynaptic diffusion of neurotransmitter. Trends Neurosci. 1997;20:377–384. doi: 10.1016/s0166-2236(96)20050-5. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in Bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci U S A. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakman D, Ballyk BA, Schoepp DD, Palmer AJ, Bath CP, Sharpe EF, Woolley ML, Bufton HR, Kamboj RK, Tarnawa I, Lodge D. Activity of 2,3-benzodiazepines at native rat and recombinant human glutamate receptors in vitro: stereospecificity and selectivity profiles. Neuropharmacology. 1996;12:1689–1702. doi: 10.1016/s0028-3908(96)00156-6. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Dieudonne S, Ascher P. Presynaptic N-methyl-D-aspartate receptors at the parallel fibre-Purkinje cell synapse. Proc Natl Acad Sci U S A. 2000;97:11593–11597. doi: 10.1073/pnas.200354297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, Van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Clark BA, Barbour B. Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. J Physiol. 1997;502:335–350. doi: 10.1111/j.1469-7793.1997.335bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel H, Crepel F. Control of Ca2+ influx by cannabinoid and metabotropic glutamate receptors in rat cerebellar cortex requires K+ channels. J Physiol. 2001;537:793–800. doi: 10.1111/j.1469-7793.2001.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci. 1998;18:3606–3619. doi: 10.1523/JNEUROSCI.18-10-03606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney AJ, Jahr CE. Kainate receptors differentially regulate release at two parallel fibre synapses. Neuron. 2002;36:475–482. doi: 10.1016/s0896-6273(02)01008-5. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digregorio D, Nusser Z, Silver R. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 2002;35:521–533. doi: 10.1016/s0896-6273(02)00787-0. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Llinas R, Sasaki K. The mossy fiber-granule cell relay in the cerebellum and its inhibition by Golgi cells. Exp Brain Res. 1966;1:82–101. doi: 10.1007/BF00235211. [DOI] [PubMed] [Google Scholar]
- Furuta A, Martin LJ, Lin CL, Dykes-Hoberg M, Rothstein J. Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience. 1997;81:1031–1042. doi: 10.1016/s0306-4522(97)00252-2. [DOI] [PubMed] [Google Scholar]
- Hamann M, Rossi D, Attwell D. Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron. 2002;33:625–633. doi: 10.1016/s0896-6273(02)00593-7. [DOI] [PubMed] [Google Scholar]
- Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/s0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Erdemli G, Asztély F. LTP of AMPA and NMDA receptor-mediated signals evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Warr O, Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol. 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longsworth LG. Diffusion measurements at 25 °C of aqueous solutions of amino acids, peptides and sugars. J Am Chem Soc. 1953;75:5705–5709. [Google Scholar]
- Lozovaya NA, Kopanitsa MV, Boychuk YA, Krishtal OA. Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-D-aspartate receptors in the rat hippocampus. Neuroscience. 1999;91:1321–1330. doi: 10.1016/s0306-4522(98)00638-1. [DOI] [PubMed] [Google Scholar]
- Merrill EG, Wall PD, Yaksh TL. Properties of two unmyelinated fibre tracts of the central nervous system lateral Lissauer tract, and parallel fibres of the cerebellum. J Physiol. 1978;284:127–145. doi: 10.1113/jphysiol.1978.sp012531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of Held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napper RMA, Harvey RJ. Quantitative study of the Purkinje cell dendritic spines in the rat cerebellum. J Comp Neurol. 1988a;274:158–167. doi: 10.1002/cne.902740203. [DOI] [PubMed] [Google Scholar]
- Napper RMA, Harvey RJ. Number of parallel fibre synapses on an individual Purkinje cell in the cerebellum of the rat. J Comp Neurol. 1988b;274:168–177. doi: 10.1002/cne.902740204. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Phillips JM. Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol. 1981;321:225–257. doi: 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. J Physiol. 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, Jahr CE. Postsynaptic glutamate transport at the climbing fiber-Purkinje cell synapse. Science. 1997;277:1515–1518. doi: 10.1126/science.277.5331.1515. [DOI] [PubMed] [Google Scholar]
- Otis TS, Yuh-Cherng W, Trussell LO. Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci. 1996;16:1634–1644. doi: 10.1523/JNEUROSCI.16-05-01634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar Cortex: Cytology and Organisation. Berlin: Springer; 1974. [Google Scholar]
- Perkel DJ, Hestrin S, Sah P, Nicoll RA. Excitatory synaptic currents in Purkinje cells. Proc R Soc Lond B Biol Sci. 1990;241:116–121. doi: 10.1098/rspb.1990.0074. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarantis M, Ballerini L, Miller B, Silver RA, Edwards M, Attwell D. Glutamate uptake from the synaptic cleft does not shape the decay of the non-NMDA component of the synaptic current. Neuron. 1993;11:541–549. doi: 10.1016/0896-6273(93)90158-n. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kovalchuk Y, Attwell D. Pre- and post-synaptic determinants of EPSC waveform at cerebellar climbing fibre and parallel fibre to Purkinje cell synapses. J Neurosci. 1995;15:5693–5707. doi: 10.1523/JNEUROSCI.15-08-05693.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Sarantis M, Attwell D. Postsynaptic glutamate uptake in rat cerebellar Purkinje cells. J Physiol. 1996;497:523–530. doi: 10.1113/jphysiol.1996.sp021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Ichikawa R, Watanabe M, Yanaka K, Inoue Y. Extra-junctional localization of glutamate transporter EAAT4 at excitatory Purkinje cell synapses. NeuroReport. 1997;8:2461–2464. doi: 10.1097/00001756-199707280-00010. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Harris KM, Regehr WG. Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J Neurosci. 2001;21:6666–6672. doi: 10.1523/JNEUROSCI.21-17-06666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Trussell LO. Voltage clamp analysis of excitatory synaptic transmission in the avian nucleus magnocellularis. J Physiol. 1994;480:123–136. doi: 10.1113/jphysiol.1994.sp020346. [DOI] [PMC free article] [PubMed] [Google Scholar]