

Abstract
KCNQ1 K+ channels in humans are important for repolarization of cardiac action potentials and for K+ secretion in the inner ear. The pore-forming channel subunits form heteromeric complexes with small regulatory subunits of the KCNE family, in particular with KCNE1 to form channels that conduct a slow delayed rectifier K+ current, Iks. This association leads to alteration of biophysical properties, including a slowing of activation, a suppression of inactivation and an increase of the apparent single-channel conductance. In addition, inward Rb+ currents conducted by homomeric KCNQ1 channels are about threefold larger than K+ currents, whereas heteromeric KCNQ1-KCNE1 channels have smaller inward Rb+ currents compared to K+ currents. We determined inactivation properties and compared K+vs. Rb+ inward currents for channels formed by co-assembly of KCNQ1 with KCNE1, KCNE3 and KCNE5, and for homomeric KCNQ1 channels with point mutations in the pore helix S5 or S6 transmembrane domains. Several of the channels with point mutations eliminated the apparent inactivation of KCNQ1, as described previously (Seebohm et al. 2001). We found that the extent of inactivation and the ratio of Rb+/K+ currents were positively correlated. Since the effect of Rb+ on the current size has been shown previously to be related to a fast ‘flickery’ process, our results suggest that inactivation of KCNQ1 channels is related to a fast flicker of the open channel. A kinetic model incorporating two open states, no explicit inactivated state and a fast flicker that is different for the two open states is able to account for the apparent inactivation and the correlation of inactivation and large Rb+ currents. We conclude that an association between KCNQ1 and KCNE subunits or removal of inactivation by mutation of KCNQ1 stabilizes the open conformation of the pore principally by altering an interaction between the pore helix and the selectivity filter and with S5/S6 domains.
K+ channels fulfil numerous physiological roles, including repolarization of action potentials and stabilization of a negative resting membrane potential in excitable cells (Hille, 2001). Voltage-gated K+ channels are composed of a homo- or heterotetrameric assembly of pore-forming α-subunits and additional regulatory β-subunits. The α-subunits contain six transmembrane helices (S1-S6) and a membrane-embedded, but not membrane-traversing, pore segment. The fourth helical domain, S4, contains numerous positively charged, evenly spaced amino acids and is the voltage sensor that moves in response to changes in the transmembrane voltage gradient (Yellen, 2002; Jiang et al. 2003a, b). The S5-S6 domains form the ion conduction pathway (Heginbotham et al. 1994; Doyle et al. 1998) with a structure that is presumed to be very similar to that of the small, bacterial KcsA channel recently resolved by X-ray crystallography (Doyle et al. 1998) and whose conductive properties are very similar to those of voltage-gated K+ channels (LeMasurier et al. 2001).
Five different human KCNQ channels have been cloned. Mutations in specific KCNQ genes can cause cardiac arrhythmia, epilepsy or deafness (Wang et al. 1996; Biervert et al. 1998; Charlier et al. 1998; Schroeder et al. 1998; Kubisch et al. 1999; Dedek et al. 2001). KCNQ1 was first identified by a positional cloning approach to define the cause of long QT syndrome, an inherited arrhythmia (Wang et al. 1996). Heterologous expression of KCNQ1 channels induced a current with biophysical properties that did not correspond to a known cardiac K+ current (Barhanin et al. 1996; Sanguinetti et al. 1996). However, when KCNQ1 was co-expressed with KCNE1 (formerly called Isk or minK; Takumi et al. 1988), the induced current resembled the slow delayed rectifier K+ current, IKs. KCNQ1 was subsequently shown to be associated with other members of the KCNE family of small, single-transmembrane-segment proteins (Barhanin et al. 1996; Sanguinetti et al. 1996; Schroeder et al. 2000; Tinel et al. 2000; Angelo et al. 2002). The association between KCNQ1 and KCNE1 leads to a drastic change in channel properties, including an increase in current magnitude, slowed rate of activation, elimination of inactivation and an increase in single-channel conductance (Barhanin et al. 1996; Sanguinetti et al. 1996; Pusch, 1998; Sesti & Goldstein, 1998; Yang & Sigworth, 1998). Inward Rb+ currents are about threefold larger than inward K+ currents for homomeric KCNQ1 channels, but about the same size for heteromeric KCNQ1-KCNE1 channels (Pusch et al. 2000). This is in contrast to most K+ channels, which conduct Rb+ ions less well than K+ ions (Hille, 2001).
The inactivation of homomeric KCNQ1 channels is not obvious from examination of the currents elicited by depolarizing pulses, but it becomes apparent as a non-monotonic time course of tail currents elicited by membrane repolarization. While the tail currents of most voltage-gated K+ channels decay monotonically as a result of deactivation, the tail currents of KCNQ1 channels transiently increase in magnitude before deactivating. This was interpreted as a result of the necessity for most channels to recover from inactivation before closing (Pusch et al. 1998; Tristani-Firouzi & Sanguinetti, 1998). Detailed analysis of this process led to the conclusion that homomeric KCNQ1 channels possess two kinetically distinct open states and a voltage-independent, intrinsically fast inactivation process (Pusch et al. 1998). The presence of at least two open states was confirmed based on completely independent evidence (Pusch et al. 2001). Based on the finding that KCNQ1-KCNE1 tail currents decay monotonically without a transient increase, it was concluded that KCNE1 subunits prevented KCNQ1 channels from inactivating (Pusch et al. 1998, 2001; Tristani-Firouzi & Sanguinetti, 1998).
In addition to KCNE1, KCNQ1 interacts with and forms hetero-oligomers with KCNE3 and KCNE5 subunits in heterologous expression systems and in some tissues (Schroeder et al. 2000; Angelo et al. 2002). Similar to KCNE1, the other KCNE proteins drastically change the biophysical properties of the heteromultimeric channel. KCNQ1-KCNE3 currents are mostly voltage-independent (Schroeder et al. 2000), while KCNE5 mainly shifts the voltage-dependence of channel activation to more positive voltages (Angelo et al. 2002).
Many obvious questions regarding the association between KCNE subunits and KCNQ1 remain unanswered. For example, it is not clear which domains are responsible for KCNQ1-KCNE1 subunit interaction, although some evidence suggests that KCNE1 binds to S6 of KCNQ1 (Tapper & George, 2001). It is also unknown if the diverse biophysical changes induced by KCNE1 are attributable to single or multiple interactions with the KCNQ1 channel. Although mutation of a single amino acid in the pore helix or S5 domain of KCNQ1 can almost completely eliminate inactivation (Seebohm et al. 2001), it is not clear if this indicates that these residues are the same as those affected by KCNE1 to alter the inactivation properties of the KCNQ1 channel.
Here we show that the seemingly unrelated phenomena of voltage-dependent inactivation and preferential conductance of Rb+ compared to K+ of KCNQ1 channels may result from a common gating mechanism. We analysed the functional consequences of several missense mutations in the pore helix, and the S5 and S6 domains. Surprisingly, we found a tight correlation between the extent of inactivation and the ratio of Rb+/K+ inward currents. We propose that inactivation of KCNQ1 channels reflects a fast flickery process of a late open state that depends on the occupancy of the pore by Rb+ and that KCNE1 stabilizes the open state of KCNQ1 channels, perhaps through an indirect interaction with the S6 domain.
METHODS
Molecular biology
The molecular biology procedures used are the same as have been described previously (Franqueza et al. 1999; Seebohm et al. 2001; Chen et al. 2002). Site-directed mutagenesis of the human KCNQ1 gene was performed by PCR using cloned Pfu-polymerase. All constructs were confirmed by automated DNA sequencing.
Oocyte expression
Stage VI oocytes were collected from Xenopus laevis frogs anaesthetized with tricaine (0.17 %). After surgery frogs were allowed to recover and after the final oocyte collection, frogs were killed humanely. The use of frogs was approved by the University of Utah IACUC committee. Oocytes were injected with ≈ 50 nl of RNA and incubated at 18 °C for 2-5 days, as described previously (Chen et al. 2002).
Electrophysiology
Standard two-electrode voltage-clamp techniques (Stühmer, 1998) were used to record whole-cell currents in Xenopus oocytes at room temperature (23-25 °C). Data were acquired with the Clampex program of pCLAMP 8.0 software (Axon Instruments) and analysed with Clampfit (Axon Instruments), Origin 6.0 (OriginLab Corp.) and custom software. For voltage-clamp experiments, oocytes were bathed in a modified ND96 solution containing (mM): 96 NaCl, 4 KCl, 1.8 MgC12, 0.1 CaC12, 5 Hepes; pH 7.6. For the high-K+ solution, 96 mM NaCl was replaced with 96 mM KCl (resulting in a total [KCl] of 100 mM). For the high-Rb+ solution, NaCl was replaced with RbCl. Liquid junction potentials were measured (< 1 mV) and were not corrected. Errors introduced by series resistance of the oocytes were not corrected. To minimize series resistance errors we kept expression at a reasonably low level (generally < 10 μA for the relevant tail currents that were fitted with the bi-exponential function). Assuming a series resistance of 0.5 kΩ for a ‘normal’ oocyte, a 10 μA current gives rise to a 5 mV error. Because the tail current kinetics of KCNQ1 are not extremely voltage dependent (Pusch et al. 1998), this error was judged to be acceptable.
Data analysis
The relative Rb+/K+ conductance, GRb/GK, was determined as the ratio of the peak inward tail current at -120 mV after a 1 s prepulse to +40 mV measured from the same oocyte bathed in the high-Rb+ followed by the high-K+ solution. The degree of KCNQ1 or KCNQ1-KCNE channel inactivation for oocytes bathed in the high-K+ solution was assessed by analysis of tail currents that were fitted to a bi-exponential equation of the form:
| (1) |
with a slow time constant τs and a fast time constant τf, and their respective amplitudes, as and af, and a steady-state current, a0. The faster time constant of this double exponential relaxation was of the same order of magnitude (i.e. between about 10 and 100 ms) for all mutants and for KCNQ1-KCNE1 heteromers. In addition, the slower time constant was similar for all constructs. A larger fast component, af, indicates the presence of a more pronounced ‘hook’ of the tail current, and the ratio af/as is a parameter that depends strongly on the degree of inactivation (Pusch et al. 1998). For a simple model, as the one described in scheme (1) of Discussion, the degree of steady-state inactivation (i.e. the quantity pI/(pO2 + pI) = λ/(λ + μ), where λ and μ are the rate constants of inactivation) can be obtained from the parameters of the double-exponential tail-current fit provided that the tail potential is sufficiently negative ensuring that the backward rate constants δ and β are much larger than the corresponding forward rate constants δ and α:
| (2) |
Here, the ratio of the absolute values of the fast and slow components, af/as, are ‘corrected’ by the ratio of the time constants, τf/τs. For small τf/τs, the two measures coincide. For some mutants, the quantity calculated by eqn (2) was slightly smaller than zero. In this case it was set to zero (see Fig. 5). For KCNQ1-KCNE3 heteromers that are apparently voltage-independent, the analysis described above is of course questionable, because the gating scheme (1) is clearly not applicable. The data concerning this channel have therefore to be considered with caution.
Scheme 1.
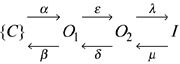
Figure 5. Predictions of a kinetic scheme for KCNQ1 channels with two closed and two open states.
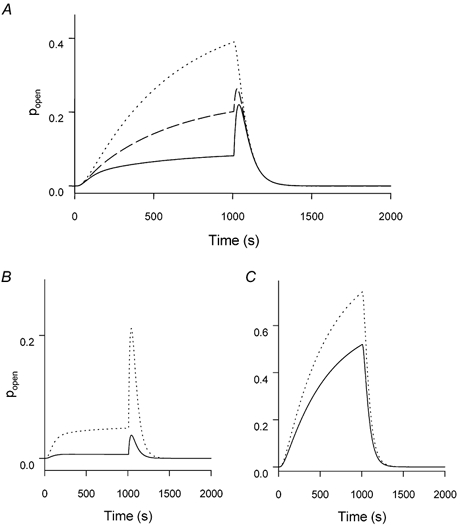
The flicker process was simulated as described in Methods and the effect of varying the three parameters KC, KO1 and KO2 are shown. In all panels the response is shown (i.e. the sum of the probability of states O1 and O2) to a voltage pulse to 50 mV of 1 s duration followed by a tail pulse to -120 mV (holding potential -80 mV). A, simulations for KC =KO1 = 1 and KO2 = 1 (dotted line), KO2 = 3 (dashed line) and KO2 = 10 (continuous line) that illustrate how a stronger flicker-closed probability of O2 leads to apparent inactivation. B, simulation of homomeric KCNQ1 channels in the presence of high extracellular [K+] (continuous line) or [Rb+] (dotted line) with KO2/KO1 = 20; KO1 =KC, and KC = 10 for K+ and KC = 1 for Rb+. The effect of Rb+ is to decrease the flicker-closed probability simultaneously for all states. C, simulation of heteromeric KCNQ1-KCNE1 channels or a non-inactivating mutant of KCNQ1 in high K+ (continuous line) or Rb+ (dotted line) with KO2/KO1 = 1; KO1 = 0.05 ×KC, and KC = 10 for K+ and KC = 1 for Rb+. The effect of Rb+ is to decrease the flicker-closed probability simultaneously for all states by a factor of 10. The difference compared to B is a decreased open state flicker, in particular for the second open state.
Simulations
The kinetic scheme (2) described in Discussion was simulated using custom software (available on website: www.ge.cnr.it/ICB/conti-moran-pusch/programs-pusch/software-mik.htm). Simulations were performed using two closed states with the rate constants between the flicker open states as described in Pusch et al. (1998):
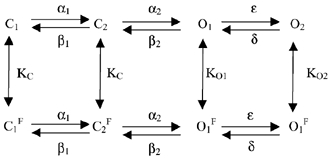 |
with (rates in s-1, θ = VF/(RT)):
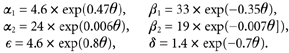 |
Flicker-closed states (superscript F) and flicker-open states (no superscript) are in fast equilibrium described by the three constants KC, KO1, and KO2. These are the only parameters that were varied for the simulations shown in Fig. 5. The simulations were performed using a holding potential of -80 mV, a prepulse to 50 mV for 1 s followed by a ‘tail’ pulse to -120 mV for 1 s. The open probability plotted in Fig. 5 is the sum of the probabilities to stay in states O1 and O2.
Scheme 2.
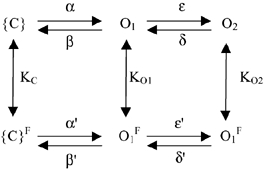
The scheme described above obviously violates microscopic reversibility (if KC is different from KO1 and/or KO1 is different from KO2). For simplicity, this was ignored because it would introduce several additional parameters. Thus, the simulations shown in Fig. 5 are at most qualitative. However, trial simulations in which the bias introduced by different values of K was fully assigned to the rightward transitions of flicker closed states in order to fulfil microscopic reversibility were very similar to the unconstrained situation (data not shown).
Molecular modelling
The KcsA structure (Doyle et al. 1998) was retrieved from the NCBI Protein Data Bank (1BL8). A three-dimensional structural model of the S5/H5/S6 domains of KCNQ1 was constructed based on 46 % homology and 36 % amino acid identity. The KCNQ1 homology model was generated using Swiss-Model (http://www.expasy.org/swissmod/SWISS-MODEL.html).
RESULTS
Inward tail currents through homomeric KCNQ1 channels are strongly dependent on the predominant species of extracellular alkali cation. Tail currents were measured at -60 mV in a single oocyte bathed sequentially with modified ND96 solutions containing either 100 mM NaCl, 96 mM KCl or 96 mM RbCl. As usual for K+-selective channels, Na+ was almost impermeant (Fig. 1A). In contrast, and as reported previously (Pusch et al. 2000), inward currents were about threefold larger in cells bathed in Rb+ (Fig. 1C) compared to K+ (Fig. 1B).
Figure 1. Effect of high extracellular concentrations of Na+, K+ or Rb+ on KCNQ1 channel currents.
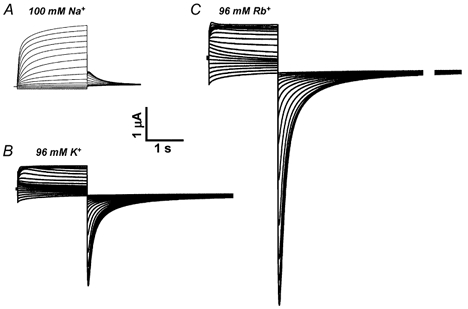
Human KCNQ1 was expressed in Xenopus oocytes and currents measured in extracellular solutions containing 100 mM NaCl (A), 96 mM KCl (B) or 96 mM RbCl (C). Currents were elicited from a holding potential of -80 mV by pulses applied in 10 mV increments to potentials ranging from -100 mV to +50 mV. These pulses were always followed by a pulse to -60 mV to elicit tail currents.
Channel conductance was strongly altered by co-expression of KCNQ1 with different KCNE subunits. Like homomeric KCNQ1 channels, all heteromeric channels had a similar discrimination vs. Na+ ions (Fig. 2, top panels). However, unlike KCNQ1 channels, both KCNE1 (Fig. 2A) and KCNE3 (Fig. 2B) prevented the increase in inward Rb+ compared to K+ currents. Only KCNQ1-KCNE5 channels (Fig. 2C) retained the Rb+ effect observed for KCNQ1 homomeric channels. In addition, however, KCNQ1- KCNE3, and even more so KCNQ1-KCNE5 channels exhibited a dramatic increase of inward and outward conductance in high-K+ and high-Rb+ solutions. Similar to what is commonly observed for KCNQ1-KCNE3 channels (Schroeder et al. 2000), high K+ also led to a partial loss of voltage-dependent gating of KCNQ1-KCNE5 channels. These findings indicate that the permeant cation has a significant effect on the gating properties of homomeric and heteromeric KCNQ1 channels and that the relative Rb+ conductance, GRb/GK, is modulated by co-expression with KCNE β-subunits.
Figure 2. Effect of high extracellular concentrations of Na+, K+ or Rb+ on KCNQ1-KCNE channel currents.
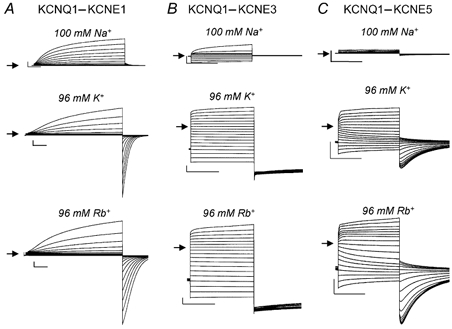
KCNQ1 was co-expressed with KCNE1 (A), KCNE3 (B) or KCNE5 (C) in Xenopus oocytes and currents measured in the solution indicated. Currents were elicited from a holding potential of -80 mV by pulses applied in 10 mV increments to potentials ranging from -100 mV to +50 mV. Tail currents were elicited by stepping to -60 mV. The horizontal scale bars indicate 1 s and the vertical bars indicate 1 μA.
Recently, two amino acids of KCNQ1 were identified as major determinants of the inactivation process (Seebohm et al. 2001). Mutations of either G272C in the S5 domain or V307L in the H5 domain almost completely abolished the characteristic ‘hook’ in the tail currents and other kinetic features that are indicative of the inactivation process (Seebohm et al. 2001; Fig. 3; compare currents of the mutants in high K+ with those of the wild-type (inset)). We investigated the effect of these mutations on the relative Rb+ conductance of homomeric KCNQ1 channels. Either mutation prevented the large increase of inward tail currents observed for wild-type KCNQ1 channels when the extracellular solution was changed from high K+ to high Rb+ (compare Fig. 1 and Fig. 3). This result suggests that a common mechanism is responsible for inactivation and the GRb/GK characteristic of wild-type KCNQ1 channels.
Figure 3. Mutant KCNQ1 channels that lack inactivation conduct K+ and Rb+ equally.
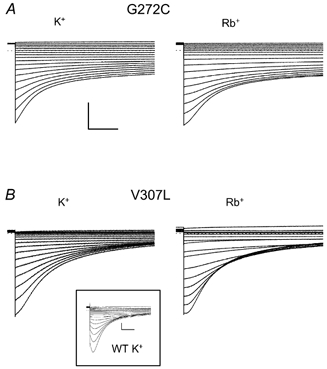
G272C KCNQ1 (A) and V307L KCNQ1 (B) channels were expressed in oocytes and tail currents were measured when the oocytes were sequentially bathed in solutions containing either 96 mM KCl or 96 mM RbCl. Tail currents were measured at potentials ranging from -120 mV to +50 mV after a 2 s pulse to +40 mV. The inset shows for comparison tail currents of wild-type (WT) KCNQ1 in high K+. The horizontal scale bars indicate 1 s, the vertical bars indicate 1 μA.
To investigate in more detail a possible link between high Rb+ conductance and inactivation of KCNQ1, we characterized several more point mutations in H5, S5 and S6. Missense mutations were introduced near position V307 in H5, at positions 272 and 273 in S5 and in positions 332, 336 and 337 in S6. The residues in the transmembrane segment S5 and the pore helix were chosen because in KCNQ1 the mutated residues were shown to influence the degree of inactivation (Seebohm et al. 2001). Furthermore, the residues L273 in S5 and V307 in the pore helix were predicted to be in close proximity and likely to interact (Seebohm et al. 2001). Interaction of the transmembrane segments S5 and S6 and the pore helix had been suggested as the structural basis for effects on open-state stability (the fast voltage-independent pore gate) in the Kv2.1 and exp-2 channels (Liu & Joho, 1998; Espinosa et al. 2001). Mutation of a residue in S6 of Kv2.1 not only altered the open-state stability, but also the ratio of Rb+vs. K+ conductance (Liu & Joho, 1998). These reports led us to speculate that an interaction of the S6 transmembrane segment with the pore helix might affect KCNQ1 inactivation. Based on a KCNQ1 homology model (see Discussion), residues 332, 336 and 337 in S6 could interact with the pore helix and were therefore chosen for further mutagenesis. The following mutations did not yield functional expression: L273A, L273Q, T309S, V307A, V307S and T311A.
All mutations in functionally expressing channels affected both the extent of inactivation and GRb/GK. A scatter plot of the degree of inactivation vs. GRb/GK (see Methods for a definition of these parameters) is presented in Fig. 4A. It can be seen that the two properties appear to be tightly correlated. Typical current traces for some of the mutant channels are shown in Fig. 4B. Mutants with a larger GRb/GK exhibited a larger degree of inactivation. The wild-type heteromeric channels (filled diamond in Fig. 4A) exhibit a similar correlation, even though the lack of apparent voltage independence of KCNQ1-KCNE3 heteromers makes the inactivation data for this channel questionable (see Methods). Using Pearson's formula, a linear correlation coefficient of r = 0.79 was calculated for the data shown in Fig. 4A, and the probability that the two parameters are uncorrelated is P = 5.1 × 10−5. This clearly demonstrates a tight link between the two seemingly independent channel properties.
Figure 4. Correlation between inactivation and GRb/GK of WT and mutant KCNQ1 and WT KCNQ1-KCNE channels.
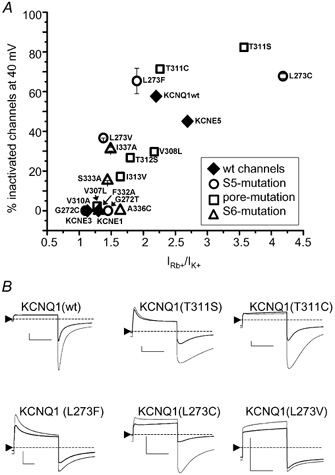
A, plot of the degree of channel inactivation at +40 mV vs. GRb/GK for KCNQ1 and KCNQ1-KCNE channels as indicated. At least four oocytes were analysed for each data point. B, representative current traces for WT and five mutant KCNQ1 channels elicited by a 2 s pulse to +40 mV and a return to -120 mV. Currents recorded in high K+ (thick trace) are overlayed with those recorded from the same oocyte bathed in high Rb+ (thin trace). The zero-current level is indicated by the dotted line. The horizontal scale bars indicate 1 s, the vertical bars indicate 1 μA.
DISCUSSION
We investigated two apparently unrelated properties of the KCNQ1 K+ channel: inactivation, a gating behaviour normally attributed to a conformational change of the channel protein, and the relative Rb+ conductance, GRb/GK, which is generally considered to be a property of the pore. Surprisingly, these two properties appeared to be tightly correlated as single-point mutations in the S5, S6 and H5 regions that reduced or increased the GRb/GK and reduced or increased the degree of inactivation, respectively. Association of KCNQ1 with members of the KCNE family also altered both properties in parallel. These findings suggest that inactivation and preferential Rb+ conductance could be consequences of a common mechanism.
Pusch et al. (2000) have shown that the large GRb/GK is determined by the dependence of a fast flickery process of open channels on the ion occupancy of the pore. This fast flicker very likely represents an instability of a fast voltage independent pore gate. Rb+-occupied pores are more stable, while K+-occupied pores have a larger ‘flicker-closed’ probability. Inactivation, on the other hand, was reported to be a fast, but intrinsically voltage-independent process (Pusch et al. 1998; Tristani-Firouzi & Sanguinetti, 1998). A kinetic scheme of the following form had been proposed in order to quantitatively describe inactivation of KCNQ1:
Here {C} denotes a set of closed states, O1 and O2 are two kinetically distinct open states, and I is a non-conducting inactivated state. The most important properties of the model are the presence of at least two open states, and that the rate-constants λ and μ are voltage independent (Pusch et al. 1998). The presence of at least two open states has been independently confirmed based on the finding that an open pore block by intracellular Na+ is not very pronounced if channels are opened only briefly, not allowing them to significantly enter the open state O2. A stronger block is seen after longer depolarizing pulses, which drive more channels into the Na+-sensitive state O2 (Pusch et al. 2001).
A simple interpretation of the correlation between inactivation and GRb/GK could be that the inactivation process itself reflects the fast flickery process that is associated with the large Rb+ conductance. However, such a model is inconsistent with the observation that Rb+ only slightly affects the two time constants of inward tail currents (Pusch et al. 2000). A slightly more complicated extension of scheme (1) is given by:
Here states with a superscript ‘F’ are flicker-closed states that are in rapid equilibrium with their respective flicker-open states, an equilibrium described by the respective constant, Ks for state s (K = pflicker-closed/pflicker-open). Apparent inactivation of channels arises if KO2 > KO1. This is shown in Fig. 5A, which illustrates the predictions of scheme (2) with two closed states, rate constants α, β, δ and δ as described in Methods, and KC = KO1 = 1, and KO2 = 1 (dotted line), or KO2 = 3 (dashed line), or KO2 = 10 (continuous line). Apart from the almost fivefold smaller open probability at the end of the +50 mV test pulse, the kinetics of the tail currents show the typical biphasic time course if KO2 = 10 × KO1, but not so if KO2 = KO1. The predictions with KO2 = 3 are intermediate (dashed line). Can scheme (2) account also for the correlation between inactivation and GRb/GK? A possible explanation of how Rb+ might differentially affect the magnitude of inward tail currents in strongly inactivating channels compared to poorly inactivating channels is illustrated in Fig. 5B and C, respectively.
A phenotype that corresponds qualitatively to that of wild-type KCNQ1 (i.e. relatively strong inactivation evidenced by a pronounced ‘hook’ in the tail conductance and large GRb/GK is shown in Fig. 5B, while a simulation with little inactivation is shown in Fig. 5C. In both cases, Rb+ (dotted line) is assumed to decrease the flicker block overall by a factor of 10 compared to K+ (continuous line; i.e. Ks(Rb+) = 0.1 × Ks(K+) for all states, s; see legend for all parameters used for the simulation). The basic difference between the simulations in Fig. 5B and C is the different overall degree of open channel flicker block. In the case of a strong flicker closed probability, Rb+, which stabilizes the flicker open state, has a much stronger effect on the current amplitude.
While these simulations are only qualitative, they demonstrate, first, that the apparent inactivation of KCNQ1 could well be the kinetic outcome of a flicker process that is different for the two open states and second, the possibility that the flicker process can underlie the tight correlation between inactivation and GRb/GK. It would be interesting to investigate the relationship between flicker and inactivation at the single-channel level. This is hampered, however, by the small single-channel conductance and the high-frequency components of the flicker process (Pusch, 1998, 2000; Sesti & Goldstein, 1998; Yang & Sigworth, 1998). Thus, a quantitative analysis of the flicker process is not feasible at the single-channel level. In addition, noise analysis is of limited practical value as it provides only relatively rough estimates of the underlying process (Pusch et al. 2000).
The association between KCNQ1 and KCNE1 abolishes inactivation and decreases GRb/GK, as do several point mutations in the homomeric channel (in particular G272C or T in S5, V307L in H5 and F332A or A336C in S6). A common mechanism might underlie these effects. This has implications concerning the way in which KCNE1 might interact with KCNQ1. First of all, it is likely that Rb+ stabilizes the flicker-open state because the selectivity filter in the open state has a different pore occupancy in high Rb+ compared to high K+ (Morais-Cabral et al. 2001). Ion and water content of the selectivity filter was described by the occupancy of four distinct sites (Morais-Cabral et al. 2001; Fig. 6): in high K+, positions 1-4 are about evenly occupied in KcsA, whereas in high Rb+, position 2 is less favoured than are positions 1, 3 and 4. This feature probably generates an energy barrier for the conduction of Rb+. Consistent with this idea, KcsA conducts more ions in high K+ than in high Rb+ (Morais-Cabral et al. 2001).
Figure 6. Position of mutated residues.
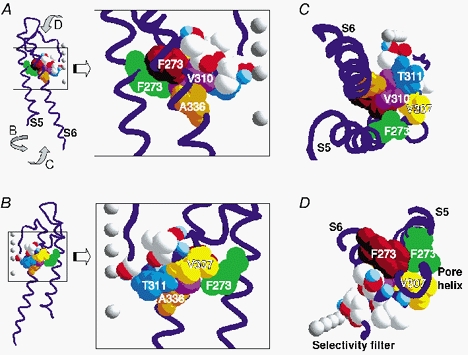
Homology model of the S5-S6 domains of a single KCNQ1 subunit. The backbone is shown in blue and selected residues are represented in space-fill mode. The residues in S6 (brown = F332, orange = A336) that are close to residues of the pore helix (yellow = V307, pink = V310) and the S5 helical segment (green = F273) are shown. In addition, amino acid T311 (blue) and the other residues (CPK colouring) studied are shown. The grey balls represent the favoured positions of K+ or Rb+ in the selectivity filter and the central cavity at high ionic concentrations (Morais-Cabral et al. 2001). The favoured position of K+ or Rb+ in the occupied selectivity filter is numbered according to Morais-Cabral et al. (2001). A, the model is shown with transmembrane segment S6 in front and the selectivity filter to the right. The enlargement of boxed area in A shows the area of interest and the numbering of selected residues. B, the model is shown turned around the central axis by 180 °. Enlargement depicts residues of S5 and the pore helix. C, the model shown from the inner membrane side. D, the backbone and residues shown from outside of the cell membrane. For clarity the outer S5-, H5- and H5-S6-linkers are not shown.
The crystal structure of KcsA predicts that the selectivity filter contacts the pore helix, which is linked to S5 via S6 (Fig. 6). Mutations at residues T311 (S5) and L273 (pore-helix) produce particularly large effects on both GRb/GK and inactivation. For example, T311S drastically increases inactivation and GRb/GK and mutating L273 to various residues leads to both increases and decreases of these properties. However, the changes don't seem to be clearly correlated with a particular physicochemical property of the side-chain. These two residues may have a crucial role in KCNQ1 function, which is underscored by the finding that both were found to be mutated in forms of long QT1 syndrome (Wang et al. 1996; Shalaby et al. 1997; Saarinen et al. 1998; Splawski et al. 2000; Seebohm et al. 2001). This is also in agreement with the observation that mutants T311A, L273A and L273Q did not yield functional expression. The three-dimensional homology model suggests an interaction between L273 and V307 of the pore helix (Fig. 6; Seebohm et al. 2001), which could affect the position of T311. Also, at position 307, two mutations prevented functional expression (V307C and V307S). L273 could interact with the pore helix and the selectivity filter, influencing ion conduction and KCNQ1 inactivation. Interestingly, the KCNQ1 272 homologous residue in the Kv2.1 K+ channel (G421) may interact with Y480, a residue in S6 (Kv2.1-Y480 is homologous to KCNQ1-I337) to stabilize the open state via a further interaction of the S6 transmembrane domain with the pore helix (Liu & Joho, 1998). A further coupling of the inactivation process to the activation machinery could result from interaction of V310 with the S6 segment (Fig. 6).
Cys 331 within S6 could be crosslinked to a cysteine engineered into KCNE1, demonstrating that KCNE1 is interacting with S6 (Tapper & George, 2001). We speculate that KCNE1 could influence concerted interactions between S5, S6, the pore helix and the selectivity filter. Such an influence is likely to be indirect since the side-chain of the putatively directly interacting Cys 331 is pointing away from the pore towards the outside of the channel protein in our homology model.
The flicker closing may underlie some, but not all of the effects exerted by the association of KCNQ1 with KCNE subunits. In particular, the almost complete lack of voltage-dependent gating induced by KCNE3 is probably due to a different mechanism. Apart from a specific interest for KCNQ1 channels, the flickery process that is so strongly affected by Rb+ may be of general importance for the mechanisms of gating and conductance of all voltage-gated K+ channels.
Acknowledgments
The support by Telethon-Italy (grant 1079), grant HL55236 from NHLBI/NIH and DFG-fellowship SE-1077 are gratefully acknowledged. We thank T. Jespersen for providing the hKCNE5 cDNA.
REFERENCES
- Angelo K, Jespersen T, Grunnet M, Nielsen MS, Klaerke DA, Olesen SP. KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys J. 2002;83:1997–2006. doi: 10.1016/S0006-3495(02)73961-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Chen J, Seebohm G, Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A. 2001;98:12272–12277. doi: 10.1073/pnas.211431298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Espinosa F, Fleischhauer R, McMahon A, Joho RH. Dynamic interaction of S5 and S6 during voltage-controlled gating in a potassium channel. J Gen Physiol. 2001;118:157–170. doi: 10.1085/jgp.118.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueza L, Lin M, Shen J, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J Biol Chem. 1999;274:21063–21070. doi: 10.1074/jbc.274.30.21063. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland, MA, USA: Sinauer; 2001. [Google Scholar]
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003a;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003b;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- LeMasurier M, Heginbotham L, Miller C. KcsA: it's a potassium channel. J Gen Physiol. 2001;118:303–314. doi: 10.1085/jgp.118.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Joho RH. A side chain in S6 influences both open-state stability and ion permeation in a voltage-gated K+ channel. Pflugers Arch. 1998;435:654–661. doi: 10.1007/s004240050566. [DOI] [PubMed] [Google Scholar]
- Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- Pusch M. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with minK. Pflugers Arch. 1998;437:172–174. doi: 10.1007/s004240050765. [DOI] [PubMed] [Google Scholar]
- Pusch M, Bertorello L, Conti F. Gating and flickery block differentially affected by rubidium in homomeric KCNQ1 and heteromeric KCNQ1/KCNE1 potassium channels. Biophys J. 2000;78:211–226. doi: 10.1016/S0006-3495(00)76586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ferrera L, Friedrich T. Two open states and rate-limiting gating steps revealed by intracellular Na+ block of human KCNQ1 and KCNQ1/KCNE1 K+ channels. J Physiol. 2001;533:135–143. doi: 10.1111/j.1469-7793.2001.0135b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Magrassi R, Wollnik B, Conti F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys J. 1998;75:785–792. doi: 10.1016/S0006-3495(98)77568-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarinen K, Swan H, Kainulainen K, Toivonen L, Viitasalo M, Kontula K. Molecular genetics of the long QT syndrome: two novel mutations of the KVLQT1 gene and phenotypic expression of the mutant gene in a large kindred. Hum Mutat. 1998;11:158–165. doi: 10.1002/(SICI)1098-1004(1998)11:2<158::AID-HUMU9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, Jentsch TJ. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–199. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- Seebohm G, Scherer CR, Busch AE, Lerche C. Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J Biol Chem. 2001;276:13600–13605. doi: 10.1074/jbc.M008373200. [DOI] [PubMed] [Google Scholar]
- Sesti F, Goldstein SA. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J Gen Physiol. 1998;112:651–663. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby FY, Levesque PC, Yang WP, Little WA, Conder ML, Jenkins-West T, Blanar MA. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation. 1997;96:1733–1736. doi: 10.1161/01.cir.96.6.1733. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- St, ühmer W. Electrophysiologic recordings from Xenopus oocytes. Methods Enzymol. 1998;293:280–300. doi: 10.1016/s0076-6879(98)93019-1. [DOI] [PubMed] [Google Scholar]
- Takumi T, Ohkubo H, Nakanishi S. Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science. 1988;242:1042–1045. doi: 10.1126/science.3194754. [DOI] [PubMed] [Google Scholar]
- Tapper AR, George AL., Jr Location and orientation of minK within the I(Ks) potassium channel complex. J Biol Chem. 2001;276:38249–38254. doi: 10.1074/jbc.M103956200. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000;19:6326–6330. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristani-Firouzi M, Sanguinetti MC. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J Physiol. 1998;510:37–45. doi: 10.1111/j.1469-7793.1998.037bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Vanraay TJ, Shen J, Timothy KW, Vincent GM, De Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Yang Y, Sigworth FJ. Single-channel properties of IKs potassium channels. J Gen Physiol. 1998;112:665–678. doi: 10.1085/jgp.112.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]