

Abstract
Mutations in KCNE1, the gene encoding the β subunit of the slowly activating delayed rectifier potassium current (IKs) channel protein, may lead to the long QT syndrome (LQTS), a condition associated with enhanced arrhythmogenesis. Mice with homozygous deletion of the coding sequence of KCNE1 have inner ear defects strikingly similar to those seen in the corresponding human condition. The present study demonstrated and assessed the mechanism of ventricular arrhythmias in Langendorff-perfused whole heart preparations from homozygous KCNE1-/- mice compared to wild-type mice of the same age. The effects of programmed electrical stimulation with decremental pacing from the basal right ventricular epicardial surface upon electrogram waveforms recorded from the basal left ventricle were assessed and quantified using techniques of paced electrogram fractionation analysis for the first time in an experimental system. All KCNE1-/-(n = 10) but not wild-type (n = 14) mouse hearts empirically demonstrated marked pacing-induced ventricular arrhythmogenicity. This correlated with significant increases in electrogram dispersion, consistent with a wider spread in conduction velocities, in parallel with clinical findings from LQTS patients with potassium channel mutations. In contrast, introduction of 100 nM isoprenaline induced arrhythmogenicity in both KCNE1-/- (n = 7) and wild-type (n = 6) hearts during pacing. Furthermore, pretreatment with 1 μM nifedipine exerted a strong anti-arrhythmic effect in the KCNE1-/- hearts (n = 12) that persisted even in the presence of 100 nM isoprenaline (n = 6). Our findings associate KCNE1-/- with an arrhythmogenic phenotype that shows an increased dispersion of conduction velocities, and whose initiation is prevented by nifedipine, a finding that in turn may have therapeutic applications in conditions such as LQTS.
Sudden cardiac death (SCD), attributable to ventricular arrhythmogenesis, is responsible for over 400 000 deaths per year in the USA (Zipes & Wellens, 1998; Zheng et al. 2001). It is usually associated with heart failure in which ventricular myocytes may show a ≈40 % reduction in potassium (K+) currents (e.g. the inward rectifier potassium current (IK1) and the transient outward potassium current (ITO) (Beuckelmann et al. 1993; Tomaselli et al. 1994; Tomaselli & Marban, 1999)) compromising repolarisation of the action potential. In contrast, monogenic long QT syndromes (LQTS) account for less than 1 % of SCD but similarly result from repolarisation abnormalities, commonly as a result of reduced potassium currents (Marban, 2002). Genetically modified (GM) animals incorporating mutations designed to resemble those seen in LQTS potentially provide tractable experimental models to investigate the mechanisms of delayed repolarisation and arrhythmogenesis applicable to both conditions.
The present experiments describe the electrophysiological phenotype of Langendorff-perfused whole heart preparations from mice. Firstly, we chose a GM system in which the entire KCNE1 coding sequence was deleted (Vetter et al. 1996). KCNE1 encodes the potassium ion channel β subunit that co-assembles with KCNQ1, which encodes the potassium ion channel α subunit, to produce a channel that mediates the slowly activating delayed rectifier potassium current, IKs (Barhanin et al. 1996; Sanguinetti et al. 1996). Mutations in this gene may compromise channel function, decreasing IKs, delaying repolarisation and thereby prolonging action potential duration (Splawski et al. 1997). These mutations have been associated with a clinical subtype of the long QT syndrome, LQT5 (Splawski et al. 1997; Duggal et al. 1998). Homozygote KCNE1-/- mice exhibit circular movements with repetitive falling and nodding, characterised as Shaker/Waltzer behaviour and are deaf (Vetter et al. 1996), findings also seen in homozygote mice with targeted disruption of KCNQ1 (Vetter et al. 1996; Lee et al. 2000; Casimiro et al. 2001). Each of these findings has been attributed to absent transepithelial potassium transport in the inner ear with a resulting collapse of the potassium-rich endolymphatic space (Vetter et al. 1996; Lee et al. 2000; Casimiro et al. 2001). The deafness and inner ear defects in the mice parallel the sensorineural deficits observed in the human autosomal recessive variant of LQTS, the Jervell and Lange-Nielsen (JLN) syndrome, characterised by a particularly severe arrhythmogenic phenotype, some of whom are also known to have mutations in KCNE1 (Schulze-Bahr et al. 1997; Duggal et al. 1998).
Secondly, we adapted pacing and analysis protocols from those used in clinical studies to stratify LQTS patients for arrhythmic risk (Saumarez & Grace, 2000; Saumarez et al. 2003). There is a substantial need for translational whole-heart electrophysiology applicable to both mice and humans and our adaptation of clinical protocols allows parallels to be drawn between potential arrhythmogenic phenotypes seen in these mice and those found in patients. We aimed to establish such a correlation in order to demonstrate further that the experimental mice offer a suitable model for LQTS, and we provide a quantitative assessment of this propensity.
Thirdly, we used pharmacological agents to probe further the ionic channel properties that might give rise to such arrhythmogenesis, giving results suggestive of possible therapeutic applications using calcium channel blockade.
METHODS
Experimental animals
The mice used, first described by Vetter et al. (1996) were initially supplied by that group. All mice were inbred and of the 129/sv strain. The mice were kept in an animal house at room temperature and subjected to 12 h : 12 h light : dark cycles, fed with sterile rodent chow and had access to water at all times. Mice aged 3 to 6 months were used in all experiments. Breeding pairs of homozygotes, which displayed Shaker/Waltzer behaviour, and wild-type mice were set up and offspring were weaned and used when of the correct age.
Langendorff-perfused preparation
The experiments used a Langendorff-perfused preparation (Langendorff, 1895) as first used in dog and rabbit hearts, adapted for the murine heart. Mice were given 100 i.u. of intraperitoneal heparin to prevent coagulation, left for 10 min, and then killed by cervical dislocation in accordance with Schedule 1 of the UK Animals (Scientific Procedures) Act 1986. The heart was then carefully but rapidly excised and placed in ice-cold bicarbonate-buffered Krebs-Henseleit solution that contained (mM): 119 NaCl, 25 NaHCO3, 4 KCl, 1.2 KH2PO4, 1 MgCl2, 1.8 CaCl2, 10 glucose and 2 sodium pyruvate. The solution was bubbled with a 95 % O2-5 % CO2 gas mixture (British Oxygen Company, Manchester, UK). Tissues surrounding the heart were carefully removed leaving a small (3 to 4 mm) section of aorta. The aorta was then cannulated under the buffer surface with a 21-gauge tailor-made cannula constructed from a blunted needle attached to small length of plastic tubing which had been pre-filled with ice-cold buffer solution in order to prevent air bubbles from entering the system. The aorta was then firmly sutured to the cannula needle with fine thread and transferred to the perfusion apparatus, to which the cannula was attached and perfusion commenced in a retrograde manner via the aorta with the aforementioned bicarbonate-buffered Krebs-Henseleit solution. The perfusate was maintained at a flow rate of 2 to 2.5 ml min−1 using a peristaltic pump (Watson-Marlow Bredel pumps model 505S, Falmouth, Cornwall, UK). It was passed through 200 μm and 5 μm pore-size filters (Millipore (UK), Watford, UK) and warmed and maintained at 37 °C via a water jacket and circulator (Techne model C-85A, Cambridge, UK) before entering the aorta. The aortic valve was shut by the pressure of the perfusate entering the aorta and the perfusate therefore passed into the coronary arteries. The perfusate ultimately drained via the coronary sinus into the right atrium and then into the stumps of the inferior and superior vena cavae. Following the start of perfusion, healthy, viable hearts suitable for subsequent experimentation regained a healthy pink colouration and spontaneous rhythmic contraction with warming.
Electrophysiological procedures
Programmed electrical stimulation (PES) of the heart was then carried out using an adaptation of the corresponding clinical techniques (Saumarez & Grace, 2000; Saumarez et al. 2003); the resulting data were analysed using paced electrogram fractionation analysis (PEFA) (Saumarez & Grace, 2000; Saumarez et al. 2003).
In the present experiments, paired platinum stimulating and recording electrodes of 1 mm inter-pole spacing were positioned on the basal epicardial surfaces of the right and left ventricles, respectively. Hearts were initially paced for 20 min at 10 Hz before beginning PES. The stimulus amplitude was set at three times the excitation threshold and the heart paced using 2 ms square-wave stimuli (Grass S48 stimulator, Grass-Telefactor, UK, Slough, UK). Stimulation protocols began by applying standard pacing stimuli at frequencies of 8 or 10 Hz for 200 ms. Following this initial short pacing period, a drive train of eight paced beats (S1) again at 8 or 10 Hz was followed by an extrastimulus (S2) every ninth beat initially at an S1S2 interval equal to the pacing interval and reduced by 1 ms with each nine-beat cycle until ventricular refractoriness was reached, whereupon the S2 stimulus elicited no electrogram. Frequencies of 8 and 10 Hz corresponding to physiological whole-animal heart rates were used (Papadatos et al. 2002) and protocols using 8 Hz in addition to those using 10 Hz were carried out on each heart. The resulting bipolar electrograms (BEGs) were monitored by the recording electrode whose signals were amplified, filtered (band-pass filter 30 Hz to 1 kHz)(Gould 2400S, Gould-Nicolet Technologies, Ilford, Essex, UK) and digitised using an analog-to-digital converter (CED 1401plus, Cambridge Electronic Design, Cambridge, UK).
Analysis of the electrogram waveforms used Spike II software (Cambridge Electronic Design, Cambridge, UK) and specific electrogram features were extracted and analysed on PC in a similar way to that performed in clinical studies using PEFA (Saumarez & Grace, 2000; Saumarez et al. 2003). In resulting ventricular conduction curves, which were constructed from sequences not resulting in arrhythmias, electrogram latencies following the S2 stimulus of successive peaks and troughs on the electrogram were plotted against the interval between the S1 and S2 stimuli (S1S2 interval) at that point. The electrogram duration (EGD) was taken as the time difference between the arrival of the first and the last peak or trough on the electrogram. The same filters were used in all experiments in order to get a consistent, empirical representation of the EGD. Results are all expressed as means ± S.E.M. comparing different groups of experimental animals using ANOVA (SPSS software).
Pharmacological agents
All drugs were purchased from Sigma-Aldrich, Poole, UK. Isoprenaline was initially dissolved in distilled water to make stock concentrations of 1 mM and nifedipine was dissolved in 96 % ethanol to make a similar stock concentration with final drug concentrations achieved by dilution with buffer solution. Nifedipine stock solutions were kept refrigerated at 4 °C and were kept wrapped in foil to prevent light degradation. Isoprenaline stock solutions were stored at -20 °C.
RESULTS
Our experimental data were derived from recording BEGs from hearts from wild-type (WT) and KCNE1-/- mice, initially establishing their arrhythmogenic phenotype. The procedures were then repeated in hearts following administration of several cardiotropic agents. Experimental caesium (Cs+) block of potassium currents provokes early afterdepolarisations (EADs) and arrhythmias in vivo (Levine et al. 1985) and GM mice with potassium channel gene mutations might therefore exhibit similar arrhythmogenic tendencies.
The initial series of experiments was based on 24 mice (14 WT: seven male and seven female; 10 KCNE1-/-: five male and five female). These compared the characteristics of electrograms between WT and KCNE1-/- mice using PES in the absence of pharmacological agents. Subsequent data from the second experimental series, which involved the use of pharmacological agents, used 23 mice (11 WT: five male and six female, 12 KCNE1-/-: six male and six female).
Perfused hearts from KCNE1-/- mice show an arrhythmic tendency
PES induced sustained ventricular tachycardia (VT) (mean cycle length 49.8 ± 5 ms, n = 10) in all KCNE1-/- hearts. The arrhythmia persisted for 30 s despite continuing stimulation, by which time the pacing sequence had ended; the arrhythmia was then pace terminated. In any given pacing sequence, VT invariably began immediately following an S2 extrastimulus delivered at an S1S2 interval that approached the ventricular effective refractory interval (VERP). In contrast, the same PES protocols consistently failed to induce VT in WT hearts (n = 14). Figure 1 shows typical experimental traces during PES of extracellular voltage plotted against time (drive train at 8 Hz) from WT (top panel) and KCNE1-/- hearts (lower panel). The single vertical markers at the base of each trace indicate the timings of the S1 stimuli with double vertical lines indicating the timing of S2 stimuli. Both traces show the same point in the PES sequence at which there is shown a succession of three S1 stimulus artifacts closely followed by the resulting electrograms (S1EG). This is followed by an extrastimulus artifact (S2) and its resulting electrogram (S2EG) succeeded by a further five S1 stimulus artifacts and electrograms seen after these in the top panel recorded from a WT heart. In the lower panel from a KCNE1-/- heart, VT has been initiated by the extrastimulus and is sustained, being unaffected by five further S1 stimuli.
Figure 1. Ventricular tachycardia in KCNE1-/- hearts.

Representative traces from isolated perfused mouse hearts during PES, pacing at 8 Hz as indicated below the traces, taken from WT (A) and KCNE1-/- hearts (B) showing induction of ventricular tachycardia (VT) following an S2 stimulus (delivered at a 30 ms coupling interval) in the KCNE1-/- but not in the WT hearts at the same point in the pacing sequence. The single vertical markers at the base of each trace indicate the timings of the S1 stimuli with double vertical lines indicating the timing of S2 stimuli. BEG, bipolar electrogram; S1, drive train stimulus; S1EG, S1 electrogram; S2, interpolated extrastimulus; S2EG, S2 electrogram.
Paced electrogram fractionation analysis (PEFA) can be applied to the experimental results
The initial experiments thus established an arrhythmogenic tendency in KCNE1-/- in contrast to WT hearts. Application of PEFA allowed the derivation of ventricular conduction curves that demonstrated specific electrogram features hitherto correlated with an arrhythmogenic phenotype in previous invasive clinical electrophysiological studies in over 500 patients (Saumarez & Grace, 2000; Saumarez et al. 2003). Figure 2 exemplifies conduction curves from data acquired using PES from Langendorff-perfused mouse hearts. Figure 2A shows typical experimental electrogram traces derived at the beginning of the PES sequence (pacing at 8 Hz), taken from a WT heart, and plots extracellular voltage against time. It consists of a pacing (S1) stimulus artifact followed by the resulting electrogram, and an extrastimulus (S2) artifact followed by an electrogram. The peaks and troughs on the electrogram following S2 are represented by a, b, c and d. The conduction curve in the Fig. 2B plots the latencies of these individual components of electrograms following S2 against the S1S2 interval at which the electrograms were obtained, thereby highlighting the consequences of reducing the S1S2 interval. The conduction curve also provides a number of additional parameters, in parallel with results from clinical studies (Saumarez & Grace, 2000; Saumarez et al. 2003), which can be compared between murine hearts. First, there is the S1S2 interval corresponding to the VERP (e in Fig. 2B). Secondly, the increase in dispersion of conduction velocities is reflected by a corresponding increase in electrogram duration (EGD) during PES. The resulting fractional increase in EGD is calculated by normalising the EGD following the S2 extrastimulus just prior to the VERP for the heart to the EGD following S2 at the beginning of the sequence, i.e. (d1 - a1)/(d - a) in Fig. 2B, and thereby corrects for small variations in electrogram morphology owing to electrode position. Thirdly, the critical S1S2 is the point at which the electrogram latencies in the conduction curve significantly start to increase (> 5 % above first recorded latencies in sequence)(f in Fig. 2B). Comparisons of the conduction velocity dispersion and the critical S1S2 in clinical studies of hypertrophic cardiomyopathy and LQTS have enabled an apparent prediction of arrhythmia risk (Saumarez & Grace, 2000; Saumarez et al. 2003), which is now currently being tested in prospective clinical trials (Saumarez et al. 2003).
Figure 2. Application of paced electrogram fractionation analysis (PEFA) to experimental electrogram traces.

Successive electrograms following S2 are used to generate conduction curves of latency vs. S1S2 interval with subsequent derivation of VERP (e) and critical S1S2 (f), the point at which electrogram latencies in the conduction curve significantly start to increase (> 5 % above first recorded latencies following S2 in sequence), and electrogram duration (EGD), taken as the time difference between the arrival of the first and the last peak or trough on the electrogram, e.g. calculated by subtracting a from d or a1 from d1. The figure shows electrogram traces taken from a WT heart (A) in which the latencies of the peaks and troughs in the electrogram following S2 (a, b, c, d in A) are plotted against the corresponding S1S2 interval at which they were obtained (B). This is repeated for electrograms following S2 at shortening S1S2 intervals until VERP is reached, when no electrogram is elicited, resulting in the generation of the conduction curve.
The arrhythmic tendency in KCNE1-/- hearts parallels a significant increase in EGD during PES
The arrhythmic tendencies shown in Fig. 1 correlated with patterns of changes in EGD demonstrated by PEFA analysis. Thus KCNE1-/- hearts showed a highly significant increase in EGD following S2 at short S1S2 intervals compared to WT. Figure 3 shows typical electrograms from WT (A) and KCNE1-/- hearts (B) during PES (pacing at 8 Hz) as the S1S2 interval approaches VERP. In both cases the S1 stimulus triggers a reference electrogram that provides a control for the electrogram elicited by the extrastimulus (S2). Figure 3C and D show the derived conduction curves for WT and KCNE1-/- hearts, respectively. The EGD following the extrastimulus (S2) is clearly more prolonged in the KCNE1-/- heart. Therefore the corresponding conduction curve from the KCNE1-/- heart shows a greater spread compared to that derived from the WT heart. Table 1 summarises an analysis of this increase in EGD in the KCNE1-/- hearts during PES when compared to WT under these conditions. In contrast, the critical S1S2 or VERP in the hearts of WT and KCNE1-/- mice were not different.
Figure 3. Greater increase in EGD following S2 in KCNE1-/- hearts.
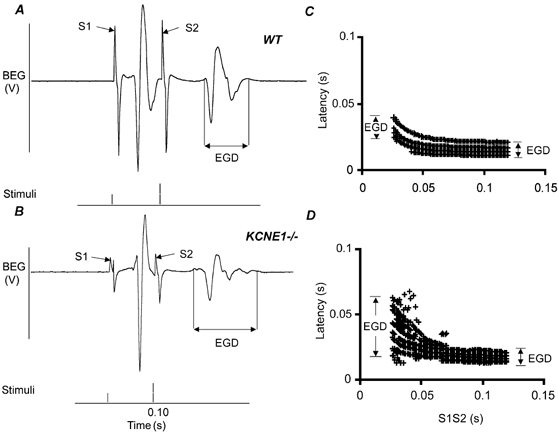
Expanded BEG traces from WT (A) and KCNE1-/- hearts (B) undergoing PES, pacing at 8 Hz, with corresponding ventricular conduction curves (C and D, respectively). The EGD is increased in KCNE1-/- hearts at shorter S1S2 intervals following an S2 stimulus (B) with a greater spread seen as a result in the conduction curve (D) compared to WT (A and C).
Table 1.
Two-way ANOVA examining varous parameters in KCNE1-/-and WT hearts in the presence and absence of nifedipine
| WT | KCNE1-/- | Level of significance | |||||
|---|---|---|---|---|---|---|---|
| Parameter | Control (n = 14) | Nifedipine (n = 11) | Control (n = 10) | Nifedipine (n = 12) | Between mice | Nifedipine vs. control | Effect of interaction |
| VERP | 30.4 ± 2.6 | 31.3 ± 1.23 | 28.8 ± 1.4 | 35.8 ± 3.1 | n.s. | n.s. | n.s. |
| Critical S1S2 | 78.1 ± 3.3 | 73.0 ± 2.4 | 79.2 ± 1.7 | 86.2 ± 1.7 | n.s. | n.s. | n.s. |
| EGD increase | 1.28 ± 0.06 | 1.21 ± 0.07 | 2.09 ± 0.17 | 2.11 ± 0.22 | P < 0.001 | n.s. | n.s. |
All data are presented as means ±s.e.m. The effect of the interaction between the type of mouse and nifedipine is tested to see if the two types of mice respond differently to the drug.
Arrhythmic tendency can be stratified using PEFA in KCNE1-/- and WT hearts
Previous clinical reports that have plotted the increase in EGD against critical S1S2 in different patients have aggregated results from LQTS patients demonstrated to be at high risk of ventricular fibrillation in the top, right quadrant of a resulting scattergram (Saumarez & Grace, 2000; Saumarez et al. 2003). Figure 4 shows a similar plot of the increase in EGD against the critical S1S2 in our experimental system during PES. Such plots show that data from KCNE1-/- hearts clearly differ from results from the WT mice in clustering nearer the top right quadrant of the scattergram, in common with the clinical findings (Saumarez & Grace, 2000; Saumarez et al. 2003).
Figure 4. Stratification of arrhythmogenic risk using PEFA in murine hearts.

Scattergram showing a greater increase in EGD in KCNE1-/- hearts during PES when compared to WT. In contrast, the critical S1S2 value is not significantly different.
Nifedipine pretreatment suppresses ventricular arrhythmias in KCNE1-/- hearts
The findings above thus clearly establish that the arrhythmogenic phenotype of KCNE1-/- hearts correlates with the corresponding clinical observations (Saumarez & Grace, 2000; Saumarez et al. 2003). This need not be unexpected: a loss of potassium channel function might well be arrhythmogenic in both situations (Roden et al. 2002). Conversely, increased calcium entry through L-type calcium channels has also been implicated in the initiation of arrhythmias (Roden & Anderson, 2000) and led to our exploration of the effect of calcium channel blockade in our proarrhythmic mouse model. Our specific choice of the dihydropyridine L-type calcium channel blocker nifedipine was prompted by its specific action on L-type calcium channels with little effect on potassium channels (Zhang et al. 1999). Doses of nifedipine of up to 50 μM do not block the rapidly activating delayed rectifier potassium channel (IKr), and a dose of 1 μM was used in all experiments. In contrast the benzothiazipine diltiazem, the phenylalkylamine verapamil and the diarylaminopropylamine bepridil all also block IKr, although higher doses are required for diltiazem, thus resulting in an effect which could mimic LQT2 (Chouabe et al. 1998; Zhang et al. 1999).
Figure 5 shows typical electrogram traces from both WT and KCNE1-/- hearts obtained at the same point in the PES sequence (pacing at 10 Hz). It demonstrates that pretreatment with 1 μM nifedipine prevented ventricular arrhythmias in the KCNE1-/- hearts during PES. The traces from WT hearts whether spared (e.g. Fig. 5A; n = 14) or subjected to nifedipine pretreatment (Fig. 5C; n = 11) demonstrated that application of extrastimuli (S2) during PES was without arrhythmic effect. In contrast, Fig. 5B confirms induction of ventricular tachycardia following similar extrastimuli in KCNE1-/- hearts (n = 10) that was prevented by nifedipine pretreatment (Fig. 5D; n = 12). PEFA detected no significant effect of nifedipine upon the VERP, critical S1S2 or increase in electrogram duration during PES in either WT or KCNE1-/- hearts (see Table 1).
Figure 5. Ventricular arrhythmias induced by extrastimuli are suppressed in the KCNE1-/- mouse heart by pretreatment with nifedipine.

Recordings from a WT heart (A) and following pretreatment with 1 μM nifedipine (C). Ventricular arrhythmias generated in a KCNE1-/- heart (B) but not following pretreatment with 1 μM nifedipine (D). All traces are taken from the same point in the pacing sequence. Pacing is at 10 Hz.
Isoprenaline induces VT in both WT and KCNE1-/- hearts
β-Adrenergic stimulation is known to increase L-type calcium currents and predispose to ventricular arrhythmias in LQTS (Schwartz et al. 2001) and, accordingly, we explored the effects of the β-adrenergic agonist isoprenaline, alone and in combination with calcium channel blockade, in the KCNE1-/- hearts. Isoprenaline has been used in pharmacological models of LQTS at a dose of 100 nM in experiments studying single cardiomyocytes (Shimizu & Antzelevitch, 2000a). In one such series that looked at a LQT1 model using the IKs blocker chromanol293B, and thus also providing a potential model for LQT5, 100 nM isoprenaline prolonged action potential duration and produced torsade de pointes (Shimizu & Antzelevitch, 2000a). Accordingly this dose was used in initial experiments.
After an initial PES study in the absence of the drug, the procedure was repeated following the addition of 100 nM isoprenaline to the perfusate in both WT and KCNE1-/- mouse hearts. Figure 6 shows traces from WT (Fig. 6A and C) and KCNE1-/- hearts (Fig. 6B and D) undergoing PES before (Fig. 6A and B) and after (Fig. 6C and D) the addition of isoprenaline. All experimental traces show the same point in the PES sequence (pacing at 8 Hz). In the absence of isoprenaline, the S2 extrastimuli induced VT in only the KCNE1-/- hearts (Fig. 6B; n = 10). In contrast, a concentration of 100 nM isoprenaline was sufficient to induce sustained VT in both WT (Fig. 6C; n = 7) (mean cycle length 45.7 ms) and KCNE1-/- hearts (Fig. 6D; n = 6) (mean cycle length 46.8 ms) following extrastimuli (S2) at S1S2 intervals that approached those resulting in refractoriness (VERP). Once again, the arrhythmia persisted for 30 s despite ongoing stimulation, by which time the pacing sequence had ended and the arrhythmia was then pace terminated.
Figure 6. Ventricular arrhythmias induced by extrastimuli following addition of isoprenaline in both WT and KCNE1-/- mouse hearts.

Recordings from a WT (A and C) and a KCNE1-/- heart (B and D), before (A and B) and after (C and D) the addition of 100 nM isoprenaline. Both WT and KCNE1-/- hearts show arrhythmia induction following isoprenaline addition. All traces are taken from the same point in the pacing sequence. Pacing is at 8 Hz.
Nifedipine suppresses isoprenaline-induced arrhythmias in WT and KCNE1-/- hearts
The final experiments explored whether nifedipine could prevent VT in the presence of 100 nM isoprenaline during PES. In the absence of nifedipine pretreatment, both WT and KCNE1-/- hearts treated with 100 nM isoprenaline showed VT following the imposition of S2 extrastimuli as described above (Fig. 6C and D). In contrast, pretreatment with 1 μM nifedipine prevented VT in both WT (Fig. 7A and C) and KCNE1-/- hearts (Fig. 7B and D) during PES, before (Fig. 7A (n = 11) and B (n = 12)) and after the addition of isoprenaline (Fig. 7C (n = 7)and D (n = 6)). All experimental traces show the same point in the PES sequence (pacing at 8 Hz).
Figure 7. Suppression of isoprenaline-induced arrhythmias in WT and KCNE1-/- hearts by nifedipine.

Recordings from a WT heart pretreated with 1 μM nifedipine (A) and following the addition of 100 nM isoprenaline (C). Recordings from a KCNE1-/- heart pretreated with 1 μM nifedipine (B) and following the addition of 100 nM isoprenaline (D). Nifedipine pretreatment suppresses arrhythmias in both WT and KCNE1-/- hearts even in the presence of isoprenaline. All traces are taken from the same point in the pacing sequence. Pacing is at 8 Hz.
Taken together the above findings support the proarrhythmic KCNE1-/- mouse as a model of LQT5 and are consistent with a potential prophylactic effect of L-type calcium channel blockade in arrhythmia suppression in LQTS.
DISCUSSION
Conditions associated with arrhythmogenic SCD such as heart failure and LQTS are associated with abnormalities in ion channel function with significant consequences for action potential duration and repolarisation (Tomaselli et al. 1994; Marban, 2002). These relationships have been explored in detail experimentally (Shimizu & Antzelevitch, 2000a,b; Papadatos et al. 2002), with our studies of Langendorff-perfused whole mouse heart preparations now leading to several new conclusions.
First, we provide compelling empirical evidence that a mouse in which the entire KCNE1 coding sequence responsible for the β subunit of IKs was deleted exhibits marked cardiac arrhythmogenicity following PES. It is thought that murine cardiac IKs channel expression peaks during the first week of life and, even 7 days postnatally, less than 10 % of ventricular myocytes exhibit channel activity (Wang et al. 1996; Drici et al. 1998; Kupershmidt et al. 1999). GM mice in which KCNE1 is replaced with the LacZ gene, which encodes beta-galactosidase, show that KCNE1 expression is highly restricted to the cardiac conduction system (Kupershmidt et al. 1999; Warth & Barhanin, 2002) strongly implying the involvement of the Purkinje network in the arrhythmias we have observed. It has already been shown that KCNE1-/- mice (Drici et al. 1998; Warth & Barhanin, 2002) show significantly exacerbated adaptation of the QT interval to heart rate resulting in prolonged electrocardiographic QT intervals during bradycardia as is described for some LQTS patients (Neyroud et al. 1998) and that mice in which the KCNQ1 gene, which encodes the α subunit of the IKs channel protein, has been disrupted (Casimiro et al. 2001) or suppressed (Demolombe et al. 2001) also show QT prolongation. Taken together with these reports, our findings support a direct role for IKs, a further KCNE1/KCNQ1 mediated current, or an indirect role for IKs or KCNE1/KCNQ1 in ventricular electrophysiology and arrhythmias in mice.
No spontaneous arrhythmias have been seen in previous studies of IKs deficient mice (Drici et al. 1998; Kupershmidt et al. 1999; Casimiro et al. 2001) and at first sight this may seem surprising given our findings. However, although arrhythmias or premature ventricular complexes in affected patients may occur spontaneously, the human LQTS phenotype is variable, and in some cases it may only be unmasked by some other intervention such as through a side effect of drug therapy (Yang et al. 2002) or through programmed stimulation (Saumarez et al. 2003). Hence the observed differences between our experimental observations and the clinical findings against a fixed genetic background need not be functionally significant. Any discrepancy that may exist could be related to quantitative physiological differences between the human and mouse heart and arrhythmias in the latter case may need to be unmasked by an intervention such as decremental pacing as used in our study.
Secondly, we have, for the first time, adapted the clinical PEFA technique and demonstrated a highly significant difference in the extent to which the EGD is increased at shorter S1S2 intervals in KCNE1-/- compared to WT mouse hearts in parallel with their empirically demonstrated arrhythmogenic properties, reflecting a wider spread in conduction velocities in these hearts. This is a property common to reentry, which is thought to be the mechanism underlying the continued propagation of arrhythmias first initiated by early afterdepolarisations (EADs) in LQTS (el-Sherif et al. 1996; Roden & Anderson, 2000; Antzelevitch & Shimizu, 2002).
Thirdly, we successfully separated the proarrhythmic KCNE1-/- from WT mice through a plot of EGD increase against critical S1S2. We thus parallel results from clinical studies conducted in patients with LQTS with a history of ventricular arrhythmias (Saumarez & Grace, 2000; Saumarez et al. 2003). We thus believe that the present study demonstrates significant similarities and further supports the suitability of the KCNE1-/- mouse as a model for the study of arrhythmogenic mechanisms in LQTS, notwithstanding any physiological differences between the mouse and human heart. The present study also complements other recent findings in mice with targeted disruption of the sodium channel gene Scn5a in which many of the clinical features of the arrhythmia conditions due to Scn5a disruption are reproduced (Papadatos et al. 2002). Additionally, our experimental findings lend cautious support to PEFA (albeit through murine data) as a clinical method to stratify prospectively patients with LQTS into those at risk of SCD, situations in which conventional programmed electrical stimulation (Bhandari et al. 1985) and analysis of the surface electrocardiogram have generally failed.
Finally, our subsequent explorations using the calcium channel blocker nifedipine and the sympathomimetic agent isoprenaline gave results consistent with a hypothesis in which it is a shift in the balance between ICa and IK at the end of the action potential plateau that predisposes to arrhythmogenesis in these murine hearts. This is supported clinically by results from the Survival With Oral D-Sotalol (SWORD) trial which show increased mortality following myocardial infarction associated with use of the potassium channel blocker D-sotalol, presumed primarily to be due to arrhythmias (Waldo et al. 1996). Additionally, the reduced proarrhythmic effects of amiodarone, which also reduces repolarising potassium channel currents, may be attributed to its additional calcium channel blocking properties (Hohnloser et al. 1994).
Ventricular arrhythmias in LQTS are widely thought to result from triggered activity (Roden & Anderson, 2000; Keating & Sanguinetti, 2001). This has been attributed to a cellular influx (or reduced efflux) of positive ions during repolarisation in late phase 3 or early phase 4 of the action potential, a period dependent on the balance between outward and inward current contributions (Noble, 1992). The resulting imbalance producing transient early (phase 3) or delayed (phase 4) afterdepolarisations might be expected to activate sodium channels, thus generating a further action potential and initiating premature beats. In such a situation, potassium channel inhibition could slow or even reverse normal cell repolarisation thus inducing EADs. Most LQTS subtypes result from loss-of-function mutations in potassium channel genes and this may relate to the EADs demonstrated during adrenaline and isoprenaline infusions in such patients (Shimizu et al. 1991, 1998; Keating & Sanguinetti, 2001). Conversely, increases in inward currents such as L-type calcium currents, could have similar effects. The action of β-adrenergic stimulation that leads to an increase in L-type calcium currents is thought to underlie the mechanism by which some patients with LQTS are prone to ventricular arrhythmias such as torsade de pointes during exercise (Schwartz et al. 2001). The reason that β-adrenergic stimulation does not normally lead to torsade de pointes is due to an observed cAMP-dependent increase in IKs, which results under normal conditions (Yazawa & Kameyama, 1990). Hence β-adrenergic stimulation normally leads to action potential shortening. In LQT1 and LQT5 however, mutations in KCNQ1 and KCNE1, respectively, lead to a functional disruption of IKs and hence inward currents predominate. These findings taken together provide a physiological rationale for the widespread use of β-blockade in LQTS patients (Moss et al. 2000) but also suggest that L-type calcium channel blockade may be of benefit.
In our experiments using nifedipine, we found that pretreatment suppressed arrhythmogenesis in KCNE1-/- hearts during PES, consistent with suppression of EADs through L-type calcium channel blockade. Furthermore, these arrhythmias were suppressed in both KCNE1-/- and WT hearts in the presence of β-adrenergic stimulation with isoprenaline, which was used at a dose sufficiently high to empirically induce arrhythmias following PES even in the WT heart, consistent with an increased predominance of inward calcium current contributions at such elevated isoprenaline concentrations. In addition, using PEFA, the EGD was not decreased following nifedipine pretreatment in these hearts. This is perhaps unsurprising as it is likely that the initiation of arrhythmias as a result of triggered activity and EADs was suppressed with the use of calcium channel blockade without causing a change in the spread of conduction velocities and hence EGD. Our findings suggest that calcium channel blockade may offer a potentially important therapeutic alternative to β-blockade, particularly in patients intolerant of such treatment, or may have a potentially useful synergistic role. In common with β-blockade, calcium channel inhibition would correct imbalances in ion channel currents during repolarisation that may occur in conditions associated with delayed repolarisation such as LQTS (Marban, 2002) and even in heart failure (Beuckelmann et al. 1993; Tomaselli et al. 1994; Tomaselli & Marban, 1999).
Evidence for a possible benefit of calcium channel blockade in patients with torsades de pointes or LQTS is limited and tends to come from isolated case reports (Cosio et al. 1991; Shimizu et al. 1995). There have been conflicting reports for the use of calcium channel blockade in heart failure. The Prospective Randomized Amlodipine Survival Evaluation (PRAISE) trial (Packer et al. 1996; O'Connor et al. 1998) showed a significant reduction in pump failure and sudden death following the use of amlodipine in non-ischaemic heart failure but this was not confirmed in PRAISE-2 (Thackray et al. 2000). Nevertheless, what is clear is that there was no increase in mortality seen in these trials in all forms of heart failure as had been feared following earlier reports with calcium channel blockers (Packer, 1990). Our results thus provide a clear indication for further trials to investigate specifically the potential role that calcium channel blockade may have in the management of arrhythmic risk in LQTS and cardiac failure.
Acknowledgments
We thank Dr Sangeeta Chawla for help in the construction of figures. This work was supported by the British Heart Foundation, the Medical Research Council, the Wellcome Trust, the Leverhulme Trust, the Helen Kirkland Fund for Cardiac Research and the Raymond and Beverly Sackler Medical Research Centre.
REFERENCES
- Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- Bhandari AK, Shapiro WA, Morady F, Shen EN, Mason J, Scheinman MM. Electrophysiologic testing in patients with the long QT syndrome. Circulation. 1985;71:63–71. doi: 10.1161/01.cir.71.1.63. [DOI] [PubMed] [Google Scholar]
- Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci U S A. 2001;98:2526–2531. doi: 10.1073/pnas.041398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C, Drici MD, Romey G, Barhanin J, Lazdunski M. HERG and KvLQT1/IsK, the cardiac K+ channels involved in long QT syndromes, are targets for calcium channel blockers. Mol Pharmacol. 1998;54:695–703. [PubMed] [Google Scholar]
- Cosio FG, Goicolea A, Lopez Gil M, Kallmeyer C, Barroso JL. Suppression of Torsades de Pointes with verapamil in patients with atrio-ventricular block. Eur Heart J. 1991;12:635–638. doi: 10.1093/oxfordjournals.eurheartj.a059952. [DOI] [PubMed] [Google Scholar]
- Demolombe S, Lande G, Charpentier F, van Roon MA, van den Hoff MJ, Toumaniantz G, Baro I, Guihard G, Le Berre N, Corbier A, De Bakker J, Opthof T, Wilde A, Moorman AF, Escande D. Transgenic mice overexpressing human KvLQT1 dominant-negative isoform. Part I: phenotypic characterisation. Cardiovasc Res. 2001;50:314–327. doi: 10.1016/s0008-6363(01)00231-0. [DOI] [PubMed] [Google Scholar]
- Drici MD, Arrighi I, Chouabe C, Mann JR, Lazdunski M, Romey G, Barhanin J. Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ Res. 1998;83:95–102. doi: 10.1161/01.res.83.1.95. [DOI] [PubMed] [Google Scholar]
- Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97:142–146. doi: 10.1161/01.cir.97.2.142. [DOI] [PubMed] [Google Scholar]
- el-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome. Tridimensional mapping of activation and recovery patterns. Circ Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- Hohnloser SH, Klingenheben T, Singh BN. Amiodarone-associated proarrhythmic effects. A review with special reference to torsade de pointes tachycardia. Ann Intern Med. 1994;121:529–535. doi: 10.7326/0003-4819-121-7-199410010-00009. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD, Magnuson MA, Roden DM. Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circ Res. 1999;84:146–152. doi: 10.1161/01.res.84.2.146. [DOI] [PubMed] [Google Scholar]
- Langendorff O. Untersuchungen am überlebenden Säugethierherzen. Pflugers Arch ges Physiol. 1895;61:291–332. [Google Scholar]
- Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest. 2000;106:1447–1455. doi: 10.1172/JCI10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Spear JF, Guarnieri T, Weisfeldt ML, De Langen CD, Becker LC, Moore EN. Cesium chloride-induced long QT syndrome: demonstration of afterdepolarizations and triggered activity in vivo. Circulation. 1985;72:1092–1103. doi: 10.1161/01.cir.72.5.1092. [DOI] [PubMed] [Google Scholar]
- Marban E. Cardiac channelopathies. Nature. 2002;415:213–218. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, Vincent GM, Locati EH, Priori SG, Napolitano C, Medina A, Zhang L, Robinson JL, Timothy K, Towbin JA, Andrews ML. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- Neyroud N, Maison-Blanche P, Denjoy I, Chevret S, Donger C, Dausse E, Fayn J, Badilini F, Menhabi N, Schwartz K, Guicheney P, Coumel P. Diagnostic performance of QT interval variables from 24-h electrocardiography in the long QT syndrome. Eur Heart J. 1998;19:158–165. doi: 10.1053/euhj.1997.0730. [DOI] [PubMed] [Google Scholar]
- Noble D. Ionic mechanisms determining the timing of ventricular repolarization: significance for cardiac arrhythmias. Ann N Y Acad Sci. 1992;644:1–22. doi: 10.1111/j.1749-6632.1992.tb30998.x. [DOI] [PubMed] [Google Scholar]
- O'Connor CM, Carson PE, Miller AB, Pressler ML, Belkin RN, Neuberg GW, Frid DJ, Cropp AB, Anderson S, Wertheimer JH, Demets DL. Effect of amlodipine on mode of death among patients with advanced heart failure in the PRAISE trial. Prospective Randomized Amlodipine Survival Evaluation. Am J Cardiol. 1998;82:881–887. doi: 10.1016/s0002-9149(98)00496-2. [DOI] [PubMed] [Google Scholar]
- Packer M. Calcium channel blockers in chronic heart failure. The risks of ‘physiologically rational’ therapy. Circulation. 1990;82:2254–2257. doi: 10.1161/01.cir.82.6.2254. [DOI] [PubMed] [Google Scholar]
- Packer M, O'Connor CM, Ghali JK, Pressler ML, Carson PE, Belkin RN, Miller AB, Neuberg GW, Frid D, Wertheimer JH, Cropp AB, Demets DL. Effect of amlodipine on morbidity and mortality in severe chronic heart failure. Prospective Randomized Amlodipine Survival Evaluation Study Group. N Engl J Med. 1996;335:1107–1114. doi: 10.1056/NEJM199610103351504. [DOI] [PubMed] [Google Scholar]
- Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Anderson ME. The pause that refreshes, or does it? Mechanisms in torsades de pointes. Heart. 2000;84:235–237. doi: 10.1136/heart.84.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Balser JR, George AL, Jr, Anderson ME. Cardiac ion channels. Annu Rev Physiol. 2002;64:431–475. doi: 10.1146/annurev.physiol.64.083101.145105. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Saumarez RC, Chojnowska L, Derksen R, Pytkowski M, Sterlinski M, Huang CL, Sadoul N, Hauer RN, Ruzyllo W, Grace AA. Sudden death in non-coronary heart disease is associated with delayed paced ventricular activation. Circulation. 2003;107:2595–2600. doi: 10.1161/01.CIR.0000068342.96569.A1. [DOI] [PubMed] [Google Scholar]
- Saumarez RC, Grace AA. Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res. 2000;47:11–22. doi: 10.1016/s0008-6363(00)00096-1. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267–268. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000a;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Effects of a K(+) channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation. 2000b;102:706–712. doi: 10.1161/01.cir.102.6.706. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Kurita T, Matsuo K, Suyama K, Aihara N, Kamakura S, Towbin JA, Shimomura K. Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long-QT syndrome. Circulation. 1998;97:1581–1588. doi: 10.1161/01.cir.97.16.1581. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Ohe T, Kurita T, Kawade M, Arakaki Y, Aihara N, Kamakura S, Kamiya T, Shimomura K. Effects of verapamil and propranolol on early afterdepolarizations and ventricular arrhythmias induced by epinephrine in congenital long QT syndrome. J Am Coll Cardiol. 1995;26:1299–1309. doi: 10.1016/0735-1097(95)00313-4. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Ohe T, Kurita T, Takaki H, Aihara N, Kamakura S, Matsuhisa M, Shimomura K. Early afterdepolarizations induced by isoproterenol in patients with congenital long QT syndrome. Circulation. 1991;84:1915–1923. doi: 10.1161/01.cir.84.5.1915. [DOI] [PubMed] [Google Scholar]
- Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- Thackray S, Witte K, Clark AL, Cleland JG. Clinical trials update: OPTIME-CHF, PRAISE-2, ALL-HAT. Eur J Heart Fail. 2000;2:209–212. doi: 10.1016/s1388-9842(00)00080-5. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–2539. doi: 10.1161/01.cir.90.5.2534. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron. 1996;17:1251–1264. doi: 10.1016/s0896-6273(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Waldo AL, Camm AJ, Deruyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP. Effect of D-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral D-Sotalol. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- Wang L, Feng ZP, Kondo CS, Sheldon RS, Duff HJ. Developmental changes in the delayed rectifier K+ channels in mouse heart. Circ Res. 1996;79:79–85. doi: 10.1161/01.res.79.1.79. [DOI] [PubMed] [Google Scholar]
- Warth R, Barhanin J. The multifaceted phenotype of the knockout mouse for the KCNE1 potassium channel gene. Am J Physiol Regul Integr Comp Physiol. 2002;282:R639–648. doi: 10.1152/ajpregu.00649.2001. [DOI] [PubMed] [Google Scholar]
- Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, Hohnloser SH, Shimizu W, Schwartz PJ, Stanton M, Murray KT, Norris K, George AL, Jr, Roden DM. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- Yazawa K, Kameyama M. Mechanism of receptor-mediated modulation of the delayed outward potassium current in guinea-pig ventricular myocytes. J Physiol. 1990;421:135–150. doi: 10.1113/jphysiol.1990.sp017937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–2163. doi: 10.1161/hc4301.098254. [DOI] [PubMed] [Google Scholar]
- Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]