

Abstract
Phosphatidylinositol phosphates (PIPs, e.g. PIP2) and long-chain acyl-CoA esters (e.g. oleoyl-CoA) are potent activators of Katp channels that are thought to link Katp channel activity to the cellular metabolism of PIPs and fatty acids. Here we show that the two types of lipid act by the same mechanism: oleoyl-CoA potently reduced the ATP sensitivity of cardiac (Kir6.2/SUR2A) and pancreatic (Kir6.2/SUR1) Katp channels in a way very similar to PIP2. Mutations (R54Q, R176A) in the C- and N-terminus of Kir6.2 that greatly reduced the PIP2 modulation of ATP sensitivity likewise reduced the modulation by oleoyl-CoA, indicating that the two lipids interact with the same site. Polyvalent cations reduced the effect of oleoyl-CoA and PIP2 on the ATP sensitivity with similar potency suggesting that electrostatic interactions are of similar importance. However, experiments with differently charged inhibitory adenosine phosphates (ATP4-, ADP3- and 2′(3′)-O-(2,4,6-trinitrophenyl)adenosine 5′-monophosphate (TNP-AMP2-)) and diadenosine tetraphosphate (Ap4A5-) ruled out a mechanism where oleoyl-CoA or PIP2 attenuate ATP inhibition by reducing ATP binding through electrostatic repulsion. Surprisingly, CoA (the head group of oleoyl-CoA) did not activate but inhibited Katp channels (IC50 = 265 ± 33 μM). We provide evidence that CoA and diadenosine polyphosphates (e.g. Ap4A) are ligands of the inhibitory ATP-binding site on Kir6.2.
KATP channels are hetero-octameric protein complexes, formed by the association of four inwardly rectifying potassium channel subunits (Kir6.2) and four regulatory sulphonylurea receptor subunits (SUR1 or SUR2). They serve as metabolic sensors in many tissues by linking cellular metabolism to membrane excitability. This function arises from their ability to respond to changes in the intracellular concentrations of ATP and ADP. Intracellular ATP binds to a site on Kir6.2 and inhibits KATP channel activity. MgADP interacts with the SUR and activates KATP channels by antagonizing the inhibitory effect of ATP (Nichols & Lopatin, 1997). In addition, recent work has uncovered two distinct classes of lipids as potent activators of KATP channels: long-chain acyl-coenzyme A (LC-CoA) esters (Branstrom et al. 1998; Gribble et al. 1998; Liu et al. 2001) and phosphatidylinositol phosphates (PIPs) (Fan & Makielski, 1997; Baukrowitz et al. 1998; Shyng & Nichols, 1998). Both types of lipid have been shown to increase the open probability and to reduce the ATP sensitivity of KATP channels. The effect on ATP sensitivity is of particular physiological importance since the amount of ATP inhibition determines the activity of KATP channels in cells. LC-CoA esters represent the metabolizable form of LC-fatty acids, which fuel β-oxidation in the mitochondria. Elevated levels of LC-CoA esters have been reported for different pathophysiological situations (ischaemic heart, diabetes mellitus, obesity) in tissues that strongly express KATP channels, pointing to a physiological or pathophysiological relevance for the regulation of KATP channels by LC-CoA esters (van der Vusse et al. 1992; Larsson et al. 1996; Gribble et al. 1998; Deeney et al. 2000; Liu et al. 2001). For instance, it has been suggested that activation of KATP channels by LC-CoA esters may contribute to the development of glucose insensitivity in pancreatic β cells (Larsson et al. 1996). PIPs represent a mechanism to regulate KATP channels by various receptors linked to the metabolism of PIPs (e.g. PLC, PI-kinases) (Baukrowitz & Fakler, 2000). In addition, not only are PIPs essential for the functioning of KATP channels but also all members of the Kir channel family are thought to interact with PIPs (especially PIP2) (Rohacs et al. 2003). Furthermore, mutations in Kir channels that disrupt these interactions with PIPs can lead to channelopathies such as Andersen's and Bartter's syndromes (Lopes et al. 2002). In contrast, LC-CoA esters appear to be specific modulators of KATP channels since none of the other Kir channels (Kir1.1, Kir2.1, Kir3.4, Kir4.1, Kir7.1) tested so far was activated by LC-CoA esters (Rohacs et al. 2003).
Several basic residues in the N-terminus (e.g. R54) (Cukras et al. 2002; Schulze et al. 2003) and C-terminus (e.g. R176, R177, R192, R206, R301, R314) (Fan & Makielski, 1997; Shyng et al. 2000) of Kir6.2 have been implicated in the binding of PIP2. Mutations at these positions resulted in KATP channels with a low open probability that could be restored to normal upon application of PIP2, consistent with a reduced PIP2 affinity. In addition, mutations of R176 and, in particular, R54 have been shown to reduce the modulation of ATP inhibition by PIP2 (Baukrowitz et al. 1998; Schulze et al. 2003). The interactions of PIP2 with the implicated residues are thought to be mainly electrostatic, e.g. substitutions at position 54 reduce the effect of PIP2 on ATP inhibition in the order R54E > R54Q >R54K = wild-type (WT; Schulze et al. 2003). Further, polyvalent cations (e.g. polylysine, Mg2+, Ca2+), which are thought to bind to the negatively charged PIPs, abolish the effects of PIPs on KATP channels (Fan & Makielski, 1997; Shyng & Nichols, 1998; Krauter et al. 2001). LC-CoA esters are also thought to interact with the Kir6.2 subunits (Branstrom et al. 1998; Gribble et al. 1998), but the involvement of specific residues in the modulation by LC-CoA esters has not been demonstrated.
Two mechanisms have been put forward to account for the reduction of ATP inhibition by PIPs. Firstly, PIPs and ATP control the open probability of KATP channels by an allosteric mechanism, with PIPs stabilizing the open state and ATP stabilizing the closed state of the channel (Enkvetchakul et al. 2000). In addition, it has been suggested that PIPs and ATP bind to overlapping (neighbouring) sites on the channel and compete directly for binding (Fan & Makielski, 1999; MacGregor et al. 2002). Given that PIPs and ATP are highly negatively charged molecules, this competition might involve electrostatic repulsion (Deutsch et al. 1994; Fan & Makielski, 1999).
Previous studies have indicated differences in the activation of KATP channels by LC-CoA esters and PIPs. In contrast to PIP2, LC-CoA appeared to reduce the ATP sensitivity of cardiac KATP channels (Liu et al. 2001) more potently compared with pancreatic KATP channels (Gribble et al. 1998). Further, the effects of LC-CoA esters on KATP channels were reported to be insensitive to Ca2+ indicating that electrostatic interactions are less critical for the effect of LC-CoA esters compared with PIP2 (Liu et al. 2001). These results might indicate mechanistic differences for the effects of PIPs and LC-CoA esters on KATP channels. To resolve this issue we evaluated the modulation of KATP channels by oleoyl-CoA and PIP2 by testing cardiac and pancreatic KATP channels, different inhibitory (di)adenosine phosphates (Ap4A, ATP, ADP, AMP), polyvalent cations (polylysine, Mg2+) and mutations that are thought to reduce PIP2 sensitivity.
METHODS
The experiments were carried out with the approval of the local animal care committee Thueringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz.
Mutagenesis, cRNA synthesis and oocyte injection
Murine Kir6.2, rat SUR2A and murine SUR1 were kindly provided by Dr F. M. Ashcroft. Site-directed mutagenesis was performed as described previously (Baukrowitz et al. 1999) and verified by sequencing. For oocyte expression, constructs were subcloned into the pBF expression vector (Fakler et al. 1995). Capped cRNAs were synthesized in vitro using SP6 polymerase (Promega, Heidelberg, Germany) and stored in stock solutions at -70 °C. Xenopus oocytes were surgically removed from adult females under anaesthesia (0.4% 3-aminobenzoic acid ethyl ester) and manually dissected. Frogs were humanely killed after the final oocyte collection. About 50 nl of a solution containing cRNA specific for SUR2A, SUR1 and Kir6.2 subunits was injected into Dumont stage VI oocytes. Oocytes were treated with collagenase type II (Sigma, 0.5 mg ml−1), defolliculated and incubated at 19 °C for 1-3 days prior to use.
Electrophysiology
Giant patch recordings (Baukrowitz et al. 1999) in the inside-out configuration under voltage-clamp conditions were made at room temperature (approximately 23 °C) 3-7 days after cRNA injection. Polylysine (Mr 30 000-70 000), diadenosine tetraphosphate (Ap4A), ATP, ADP and CoA were purchased from Sigma and TNP-AMP from Molecular Probes.
Pipettes were made from thick-walled borosilicate glass, had resistances of 0.2-0.4 MΩ (tip diameter of 20-30 μm) and were filled with (mM): 120 KCl, 10 Hepes and 1.8 CaCl2 (pH adjusted to 7.2 with KOH). Currents were recorded with an EPC9 amplifier (HEKA Electronics, Lamprecht, Germany) and sampled at 1 kHz with the analog filter set to 3 kHz (-3 dB). Solutions were applied to the cytoplasmic side of excised patches via a multi-barrel pipette and had the following composition (mM): 120 KCl, 10 Hepes and 2 K2EGTA (Kint solution). Computational work was performed on a Macintosh G4 using commercial software (IGOR, WaveMetrics) and Excel 2001 (Microsoft).
Preparation of lipid solutions
L-α-Phosphatidyl-D-myo-inositol-4,5-bisphosphate (PIP2, from bovine brain) and oleoyl-CoA were purchased from Sigma, stored as stocks (1 mM) at -20 °C, diluted in Kint solution to final concentrations, sonicated for 15 min and used within 6 h. Initial experiments with oleoyl-CoA produced variable effects on the ATP sensitivity of KATP channels. This variability was most probably due to the absorption of oleoyl-CoA by the polyethylene tubing of our application system. If an oleoyl-CoA-containing solution remained for more than 15 min in the tubing then only small effects on KATP channels were observed. However, if the oleoyl-CoA solution was continuously flowing then oleoyl-CoA produced reproducible effects on KATP channels. For PIP2-containing solutions this effect was not observed.
RESULTS
Oleoyl-CoA reduced the ATP sensitivity of pancreatic and cardiac KATP channels with similar potency
Figure 1 illustrates the effect of oleoyl-CoA on the ATP sensitivity of pancreatic (Kir6.2/SUR1) and cardiac (Kir6.2/SUR2A) KATP channels in giant inside-out patches excised from Xenopus oocytes. Pancreatic KATP channels had an initial Ki(ATP) for ATP inhibition of 18 ± 3 μM and application of 20 μM oleoyl-CoA for 30 s shifted the Ki(ATP) to 1.6 ± 0.3 mM (Fig. 1A), which corresponds to a 99 (± 25)-fold reduction in the ATP sensitivity (Fig. 1C and D). For cardiac KATP channels, we measured a somewhat higher initial ATP sensitivity with a Ki(ATP) of 8 ± 0.4 μM (Fig. 1B). Application of oleoyl-CoA reduced the Ki(ATP) to 1.4 ± 0.1 mM (Fig. 1B), which represents a 180 ± 12-fold reduction in the ATP sensitivity (Fig. 1C and D). For comparison, we also measured the effect of PIP2 (20 μM for 30 s) on the two types of KATP channel and obtained results comparable with those of oleoyl-CoA (Fig. 1D). Thus, oleoyl-CoA potently reduced the ATP sensitivity of pancreatic and cardiac KATP channels, very similar to PIP2. This contrasts with a study on pancreatic KATP channels where only small effects of oleoyl-CoA on ATP inhibition were reported (Gribble et al. 1998). We speculate that this difference might be related to the application procedure of the LC-CoA esters since oleoyl-CoA appeared to be readily absorbed by the polyethylene tubing of the application system in contrast to PIP2 (see Methods).
Figure 1. Oleoyl-CoA and PIP2 reduce ATP inhibition of cardiac and pancreatic KATP channels with similar potency.
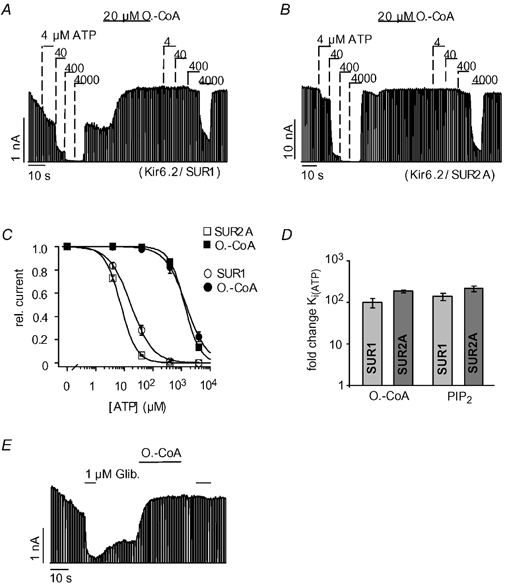
Pancreatic (Kir6.2/SUR1) and cardiac (Kir6.2/SUR2A) KATP currents were measured in giant inside-out patches excised from Xenopus oocytes at -90 mV; inward currents are shown as upward deflections. A, effects of ATP and oleoyl-CoA (O.-CoA) on pancreatic KATP channels at the concentrations indicated. B, effects of ATP and oleoyl-CoA on cardiac KATP channels. C, from experiments such as those in A and B, concentration-response curves were created for ATP inhibition before (□, ○) and after (▪, •) application of 20 μM oleoyl-CoA for 30 s. Continuous lines represent fits to a standard Hill equation: I/Imax= 1/(1 + ([ATP]/Ki(ATP)nH)), error bars represent ±S.E.M. rel. current, relative current. D, the bars represent the fold change ±S.E.M. of the Ki(ATP) for cardiac and pancreatic KATP channels upon application of oleoyl-CoA (as in C) or PIP2 (20 μM for 30 s). E, effects of glibenclamide (Glib) and 10 μM oleoyl-CoA on cardiac KATP channels.
Further, PIP2 is known to reduce the sensitivity of KATP channels to sulphonylureas such as glibenclamide that inhibit KATP channels via the SUR (Koster et al. 1999; Krauter et al. 2001). Figure 1E shows that application of oleoyl-CoA completely abolished the inhibition produced by 1 μM glibenclamide. In summary, the effects of oleoyl-CoA and PIP2 on the inhibition of cardiac and pancreatic KATP channels by ATP and glibenclamide were virtually identical.
R54Q and R176A reduced the modulation of ATP sensitivity by oleoyl-CoA
The residues R54 and R176 in Kir6.2 are likely to interact with PIP2 directly (Huang et al. 1998; Soom et al. 2001; Schulze et al. 2003). Further, the mutations R176A and, especially, R54Q have been shown to markedly reduce the effect of PIP2 on ATP inhibition (Baukrowitz et al. 1998; Shyng & Nichols, 1998; Schulze et al. 2003). We therefore tested whether these mutations also interfered with the effect of oleoyl-CoA on the ATP sensitivity. The activity of R176A (and R54Q) channels declined spontaneously upon patch excision, and application of oleoyl-CoA dramatically increased the channel activity (Fig. 2A) as seen previously with PIP2. The ATP sensitivity of R176A channels was measured subsequent to run down when channel activity was reasonably stable. R176A channels had an initial Ki(ATP) of 6 ± 0.5 μM, which was reduced to 174 ± 23μM upon the application of 20 μM oleoyl-CoA for 30 s (Fig. 2A and B), which represents a 30 (± 4)-fold reduction in the ATP sensitivity. For R54Q channels, application of oleoyl-CoA reduced the Ki(ATP) from 3.2 ± 0.7 to 25 ± 2 μM representing only an 8 (± 2)-fold reduction of the Ki(ATP) (Fig. 2C). In summary, the fold change in ATP sensitivity produced by the oleoyl-CoA application was 180 ± 12 for WT channels (Kir6.2/SUR2A) (Fig. 1D), 30 ± 4 for R176A channels and 8 ± 2 for R54Q channels (Fig. 2D). This outcome closely resembles the effect of R176A and R54Q on the modulation of ATP inhibition by PIP2 (Schulze et al. 2003) and suggests that PIP2 and oleoyl-CoA interact with the same residues to modulate ATP inhibition.
Figure 2. R54Q and R176A reduce oleoyl-CoA modulation of ATP inhibition.
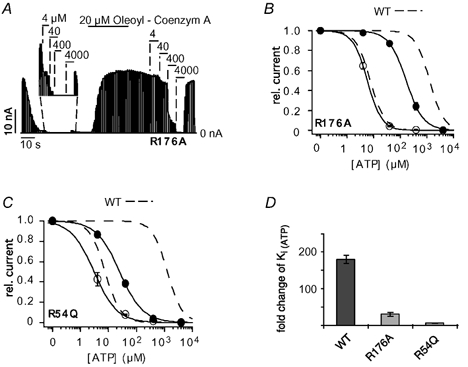
A, ATP sensitivity of R176A (Kir6.2/SUR2A) currents measured subsequent to run down (see magnification) and after application of 20 μM oleoyl-CoA for 30 s. B, concentration-response curves from experiments such as that in A; circles represent data before (○) and after (•) application of oleoyl-CoA; WT concentration-response curves (dashed lines) are shown for comparison. C, concentration-response curves for R54Q channels before (○) and after (•) application of oleoyl-CoA. WT concentration-response curves (dashed lines) are shown for comparison. D, the bars represent the fold change ±S.E.M. for Ki(ATP) upon oleoyl-CoA application for WT, R176A and R54Q channels.
Oleoyl-CoA affects KATP channels by an electrostatic mechanism
A defining property for the effects of PIPs on KATP channels is their sensitivity to polyvalent cations such as polylysine and Mg2+, which reverse these effects (Fan & Makielski, 1997; Shyng & Nichols, 1998; Krauter et al. 2001). The effect of polylysine on the modulation of ATP sensitivity by oleoyl-CoA is shown in Fig. 3A: application of 2 μg ml−1 polylysine completely reversed the shift in ATP sensitivity produced by oleoyl-CoA (Fig. 3A and B). Similar results were obtained for the effect of polylysine on the PIP2-mediated shift in ATP sensitivity (data not shown). To estimate the Mg2+ sensitivity of the oleoyl-CoA effect, patches were treated with oleoyl-CoA to reduce ATP inhibition and, subsequently, the effect of Mg2+ on the ATP sensitivity was measured. Mg2+ produced only a little inhibition in the absence of ATP (used as a control), but it greatly increased the inhibition produced by 1 mM ATP (Fig. 3C). Under these conditions, application of 1 mM ATP inhibited about 30 % of the KATP current (no Mg2+), but increasing the Mg2+ concentration successively to 20 mM increased the ATP inhibition up to 98 % (Fig. 3C). We used these values for current inhibition (1 mM ATP) to calculate the Ki(ATP) for the respective Mg2+ concentrations using the Hill equation for ATP inhibition with a Hill coefficient of 1.8. (The Hill coefficient showed little variability (1.76 ± 0.05) under these conditions justifying its use as a fixed parameter.) These approximated Ki(ATP) values were plotted against the Mg2+ concentrations to obtain a concentration-response relationship for the effect of Mg2+ on ATP sensitivity (Fig. 3D). This curve was fitted to the standard Hill equation with an IC50(Mg2+) of 1.3 ± 0.2 mM. The same procedure was used to estimate the Mg2+ sensitivity for the PIP2-induced reduction of ATP sensitivity; this resulted in a similar concentration- response curve with an IC50(Mg2+) of 1.5 ± 0.2 mM (Fig. 3E and F). Thus, the effects of oleoyl-CoA and PIP2 on ATP inhibition displayed similar sensitivities to polylysine and Mg2+, suggesting that electrostatic interactions are of comparable importance for the two lipids.
Figure 3. Oleoyl-CoA reduce the ATP sensitivity by an electrostatic mechanism.
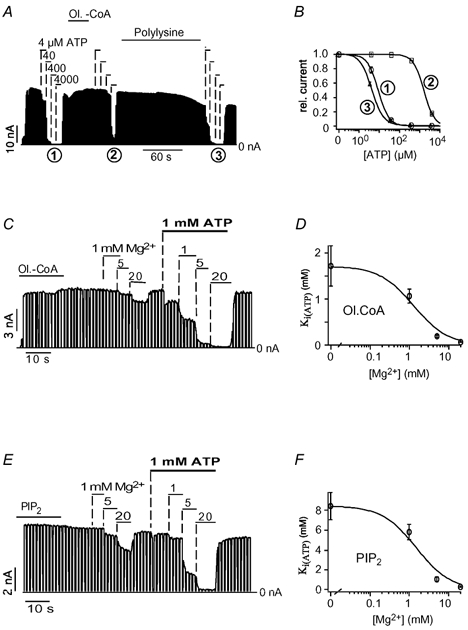
A, effects of 20 μM oleoyl-CoA, 2 μg ml−1 polylysine and ATP on cardiac KATP channels. B, from the experiment in A, concentration-response curves for ATP inhibition were obtained before oleoyl-CoA (1), after oleoyl-CoA (2) and after polylysine (3). C, effects of Mg2+ in the presence and absence of 1 mM ATP on KATP channels subsequent to the application of 20 μM oleoyl-CoA. D, the amount of ATP inhibition was used to estimate the Ki(ATP) using the Hill equation for ATP inhibition (Fig. 1C) with a Hill coefficient of 1.8. A plot of these Ki(ATP) values against the respective Mg2+ concentrations resulted in a concentration-response curve that was fitted to a standard Hill function with an IC50(Mg2+) of 1.3 ± 0.2 mM and Hill coefficient of 1. E, effects of Mg2+ in the presence and absence of 1 mM ATP on KATP channels subsequent to the application of 20 μM PIP2. The amount of ATP inhibition was used to estimate the Ki(ATP) using the Hill equation for ATP inhibition (Fig. 1C) with a Hill coefficient of 1.8. A plot of these Ki(ATP) values against the respective Mg2+ concentrations resulted in a concentration-response curve that was fitted to a standard Hill function with an IC50(Mg2+) of 1.5 ± 0.2 mM and Hill coefficient of 1.
Effects of oleoyl-CoA and PIP2 on the inhibition of KATP channels by various (di)adenosine phosphates
In addition to ATP, KATP channels are also sensitive to inhibition by ADP, AMP (Tucker et al. 1998) and diadenosine polyphosphates (ApnA) such as diadenosine tetraphosphate (Ap4A) (Jovanovic et al. 1996; Martin et al. 1998). ADP and AMP are thought to inhibit KATP channels via the same site and mechanism but with lower affinity than ATP (Tucker et al. 1998; Ribalet et al. 2003). However, inhibition by Ap4A has been suggested to be different from that by ATP (Martin et al. 1998). We compared the effects of oleoyl-CoA and PIP2 on the inhibition of KATP channels by the various (di)adenosine phosphates. Application of 75 μM TNP-AMP, 200 μM ADP, 15 μM ATP and 15 μM Ap4A inhibited between 40 and 60 % of the KATP current (Fig. 4C), thus approximately representing the IC50 concentrations of the respective (di)adenosine phosphates. The trinitrophenol derivative of AMP (TNP-AMP) was used instead of AMP (Ki(AMP) ≈ 1.2 mM) because of its approximately 15-fold higher potency in inhibiting KATP channels (authors’ unpublished data). The various (di)adenosine phosphates were applied at concentrations 10-fold in excess of their estimated IC50 values (Fig. 4C) resulting in virtually complete inhibition of the KATP current. Application of oleoyl-CoA abolished the inhibition for all the tested (di)adenosine phosphates with similar potency (Fig. 4A). Very similar results were obtained for the effect of PIP2 (Fig. 4B), further substantiating the hypothesis that oleyol-CoA and PIP2 act by the same mechanism. Remarkably, oleoyl-CoA and PIP2 reduced the sensitivity to inhibition by the various (di)adenosine phosphates in parallel indicating that the respective IC50 values were reduced by the same factor. Since the (di)adenosine phosphates differ greatly in their molecular charge, these results ruled out a mechanism where oleoyl-CoA or PIP2 attenuates the binding of the (di)adenosine phosphates through direct electrostatic repulsion (see Discussion).
Figure 4. Effects of oleoyl-CoA and PIP2 on inhibition of KATP current by various (di)adenosine phosphates.
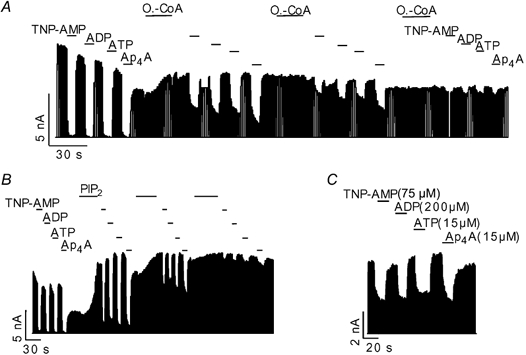
A, effect of application of 750 μM TNP-AMP (AMP), 2000 μM ADP, 15 μM ATP, 15 μM Ap4A and 4 μM oleoyl-CoA on cardiac KATP channels as indicated. B, same concentrations as in A, but with application of 20 μM PIP2 instead of oleoyl-CoA. C, inhibition of KATP channels by (di)adenosine phosphates at the concentrations indicated.
CoA and Ap4A inhibit KATP channels via interaction with the ATP-binding site on Kir6.2
The CoA portion of the LC-CoA molecule is likely to interact with the KATP channel because it contains a highly negatively charged 3′-phosphorylated ADP group. Therefore, we tested whether CoA also activated KATP channels. Surprisingly, CoA caused a fast, reversible and dose-dependent inhibition of KATP channel activity with a Ki(CoA) of 265 ± 33 μM (Fig. 5A). Furthermore, inhibition by CoA was abolished by the application of oleoyl-CoA (Fig. 5A) and PIP2 (Fig. 5B). The inhibitory effect of CoA might arise from a displacement of PIPs from the channel or, alternatively, CoA might interact with the ATP-binding site to cause channel inhibition. These alternatives were tested by employing mutants of Kir6.2 that reduce either the PIP2 sensitivity (R54Q) (Schulze et al. 2003) or the ATP sensitivity (R50E, K185Q) (Tucker et al. 1998). R54Q produced a slight increase in the CoA sensitivity (Fig. 5C) as seen for ATP inhibition (Fig. 2C). In contrast, R50E (Fig. 5D) and K185Q greatly reduced the inhibition by CoA (Fig. 5C). These results suggested that CoA and ATP interact with the same site on Kir6.2. To test directly for competition between CoA and ATP, CoA inhibition was measured in the presence of ATP. Under control conditions (in the absence of ATP), 333 μM CoA inhibited 51 ± 3 % of the KATP current but only 22 ± 4 % in the presence of 50 μM ATP (Fig. 5E and F). This reduction in CoA inhibition is close to the theoretical value (29 %, indicated in Fig. 5F) calculated under the assumption that CoA and ATP compete for the same site to cause inhibition (see figure legend).
Figure 5. CoA causes inhibition by interacting with the ATP-binding site on Kir6.2.
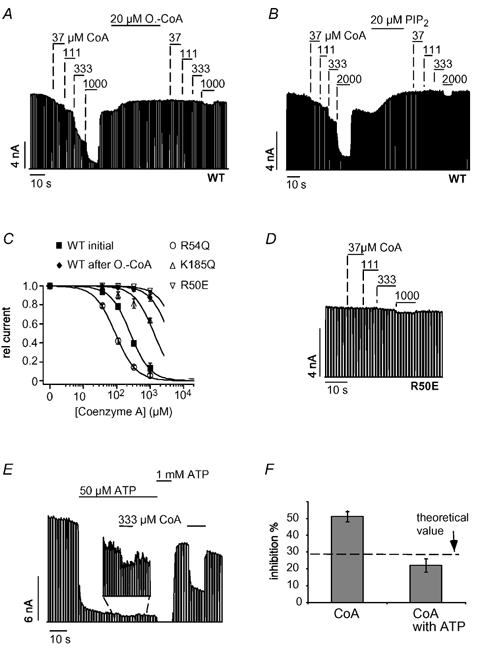
A, CoA inhibited cardiac KATP currents in a concentration-dependent manner and application of 20 μM oleoyl-CoA or 20 μM PIP2 (B) largely reduced CoA inhibition. C, concentration-response curves for CoA inhibition for WT, R50E, R54Q and K185Q channels with fits to standard Hill equations with Ki(ATP) values of 265 ± 33 μM (WT), 6 ± 0.5 mM (R50E), 92 ± 6 μM (R54Q) and 1.9 ± 0.2 mM (K185Q). D, inhibition of R50E channels by different concentrations of CoA. E, inhibition of KATP currents by CoA in the presence and absence of ATP at the concentrations indicated; magnification shows CoA inhibition in the presence of 50 μM ATP. F, from experiments such as that in E, current inhibition was determined for 333 μM oleoyl-CoA in the absence and presence of 50 μM ATP and plotted as means ± S.E.M. The dashed line represents the expected theoretical value for current inhibition by CoA in the presence of 50 μM ATP, assuming direct competition between CoA and ATP. To obtain this value, the amount of CoA inhibition (in the absence of ATP) was used to calculate the ATP concentration (14.2 μM) necessary to produce the same inhibition using a concentration-response curve as in Fig. 1C. Accordingly, the theoretical value represents the expected increase in ATP inhibition produced by a rise in the ATP concentration from 50 μM to (50 + 14.2) μM.
We showed that Ap4A and ATP inhibition were similarly affected by oleoyl-CoA and PIP2 (Fig. 4A and B) and asked, therefore, whether Ap4A also interacts with the ATP site. Figure 6A shows the effect of oleoyl-CoA on the inhibition by Ap4A in more detail. As seen with ATP inhibition, application of oleoyl-CoA shifted the concentration- response curve by several orders of magnitude (Fig. 6B). Further, we tested for the impact of the mutations R50E and K185Q on Ap4A inhibition. R50E and K185Q channels were clearly less sensitive to Ap4A compared with WT channels. Ap4A inhibited WT channels with a Ki(Ap4A) of 10 ± 1 μM (Fig. 6B), whereas the Ki(Ap4A) of R50E channels was 440 ± 77 μM and that of K185Q channels was 200 ± 35 μM (Fig. 6B). Moreover, ATP and Ap4A competed for inhibition of KATP channel activity (as seen for CoA). Application of 15 μM Ap4A inhibited 71 ± 1 % of the KATP current in the absence of ATP but only 32 ± 2 % in the presence of 40 μM ATP (Fig. 6C and D). This value is close to the theoretical value (34 %; indicated in Fig. 6C) calculated under the assumption that Ap4A and ATP bind to the same site (see figure legend). We also tested for competition between CoA and Ap4A and found that CoA inhibition was reduced in the presence of Ap4A (Fig. 6D) as expected if Ap4A and CoA compete for binding (Fig. 6D). We conclude that CoA and Ap4A inhibit KATP channels via interaction with the inhibitory ATP site on Kir6.2.
Figure 6. Ap4A and ATP compete for the same site to inhibit KATP channels.
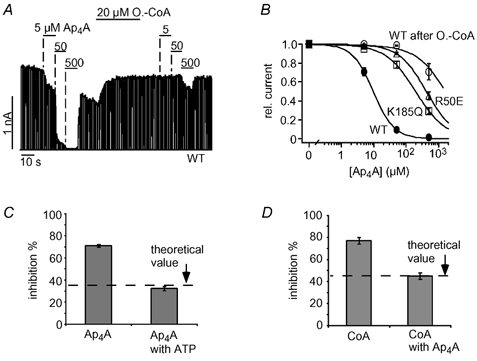
A, Ap4A inhibited cardiac KATP currents in a concentration-dependent manner and application of 20 μM oleoyl-CoA greatly reduced Ap4A inhibition. B, concentration-response curves for Ap4A inhibition of WT, R50E and K185Q channels, and WT channels after oleoyl-CoA (A); fits to standard Hill equations with Ki(Ap4A) values of 10 ± 1 μM (WT), 438 ± 77 μM (R50E) and 200 ± 35 μM (K185Q). C, inhibition of KATP currents by 16 μM Ap4A in the presence and absence of 40 μM ATP is plotted as means ± S.E.M. The dashed line represents the expected theoretical value for current inhibition by 16 μM Ap4A in the presence of 40 μM ATP assuming direct competition between Ap4A and ATP. To obtain this value, the amount of Ap4A inhibition (in the absence of ATP) was used to calculate the ATP concentration (14.5 μM) necessary to produce the same inhibition using a concentration-response curve as in Fig. 1C. Accordingly, the theoretical value represents the expected increase in ATP inhibition produced by a rise in the ATP concentration from 40 μM to (40 + 14.5) μM. D, inhibition of KATP currents by 333 μM CoA in the presence and absence of 40 μM Ap4A is plotted as means ± S.E.M. The dashed line represents the expected theoretical value for current inhibition by 333 μM CoA in the presence of 40 μM Ap4A assuming direct competition between CoA and Ap4A (the theoretical value was calculated as described in C).
DISCUSSION
Here we show that the effects of oleoyl-CoA and PIP2 on KATP channels are virtually indistinguishable in all respects tested: (i) oleoyl-CoA and PIP2 reduced the ATP sensitivity of cardiac and pancreatic KATP channels with similar potency, (ii) both lipids abolished the inhibition by various other (di)adenosine phosphates (Ap4A, ADP, AMP) and the sulphonylurea glibenclamide, (iii) the effects of the two lipids on KATP channels displayed similar sensitivities to polycations (polylysine, Mg2+), and (iv) the mutations R54Q and R176Q reduced the effects of oleoyl-CoA and PIP2 on ATP sensitivity to similar extents. From these results we conclude that LC-CoA esters and PIPs modulate KATP channels via the same mechanism and interaction sites. This outcome fits nicely with a recent study by Rohacs et al. (2003) reporting that the PIP specificity of a Kir channel correlated with the ability to be activated by LC-CoA esters. Kir channels (Kir1.1, Kir2.1, Kir7.1) that discriminated between the different PIPs (e.g. PI(4,5)P2, PI(3,4)P2 and PI(3,4,5)P3) were not activated by LC-CoA esters. In contrast, KATP channels, which are not selective for the different PIPs, are potently activated by oleoyl-CoA. Rohacs et al. (2003) suggested that the low selectivity of the lipid interaction site on KATP channels allows the accommodation of PIPs as well as LC CoA esters.
On the mechanism of oleoyl-CoA/PIP2 activation of KATP channels
Our measurements with polyvalent cations (polylysine, Mg2+) indicate that oleoyl-CoA acts on KATP channels by an electrostatic mechanism as shown previously for PIP2. Even before the discovery of negative lipids as modulators of KATP channels, Deutsch and coworkers (1994) postulated the existence of a negative charge density on the KATP channel that controls ATP inhibition. In excised patches from cardiac myocytes these authors observed a large increase in ATP sensitivity in the presence of polyvalent cations such as Mg2+, Ca2+ and polylysine. Deutsch and coworkers (1994) suggested that these cations might screen a negative charge density located in the neighbourhood of the ATP-binding site that reduces the binding of ATP via electrostatic repulsion. The half-maximal effect of Mg2+ to increase ATP inhibition was observed at a concentration of about 2 mM (Deutsch et al. 1994). In good agreement, the half-maximal concentration for Mg2+ to abolish the effect of oleoyl-CoA or PIP2 on ATP inhibition was about 1.4 mM (Fig. 3D and F), suggesting that these negatively charged lipids could indeed account for the charge density proposed for native cardiac KATP channels. However, we found that oleoyl-CoA and PIP2 reduced the sensitivity to inhibition produced by various (di)adenosine phosphates with virtually the same potency despite the fact that these molecules differ greatly in their charge (Ap4A5-, ATP4-, ADP3-, TNP-AMP2-). This finding rules out the above-stated electrostatic mechanism (Deutsch et al. 1994), which would predict a much larger effect of the negative charge density (oleoyl-CoA/PIP2) on the inhibition by e.g. ATP4- compared with AMP2-. Therefore, the polyvalent cations most probably act by blocking electrostatic interactions necessary for oleoyl-CoA and PIP2 to bind to the KATP channel but do not interfere directly with the binding of ATP.
The results on the (di)adenosine phosphates argue against electrostatic interactions between ATP and oleoyl-CoA/PIP2 and, thus, are consistent with an allosteric mechanism where oleoyl-CoA/PIP2 reduces the sensitivity to ATP by increasing the open state stability (Enkvetchakul et al. 2000). However, the results are also consistent with a mechanism where oleoyl-CoA/PIP2 and the adenosine phosphates bind in a mutually exclusive manner to an overlapping site on the channel (Fan & Makielski, 1999; MacGregor et al. 2002), because this would also predict no difference for the effect of oleoyl-CoA/PIP2 on the various (di)adenosine phosphates as observed. Therefore, our results cannot distinguish between these two scenarios but rule out a mechanism where oleoyl-CoA/PIP2 and ATP bind to neighbouring sites that are close enough to allow electrostatic cross-talk.
CoA and (di)adenosine polyphosphates are ligands of the ATP-binding site onKir6.2
To our surprise, we found that CoA, in contrast to oleoyl-CoA, inhibited KATP channels. This inhibition was abolished by oleoyl-CoA and PIP2 and reduced by mutations (R50E, K185Q) of residues that are thought to be involved in the binding of the adenosine phosphates (ATP, ADP, AMP) (Tucker et al. 1998; Ribalet et al. 2003). Further, ATP reduced the apparent sensitivity of KATP channels to inhibition by CoA as though the two molecules directly competed for inhibition. These results suggest that ATP and CoA bind to the same site. Intriguingly, CoA contains a 3′-phosphorylated ADP group that is likely to mediate the interaction with the ATP (adenosine phosphate)-binding site. However, because of its negative charge, the 3′-phosphorylated ADP group is also likely to mediate the interaction with the lipid-binding site. As stated previously (Shyng et al. 2000; Cukras et al. 2002; Schulze et al. 2003), the sites are most probably not identical since the sensitivity to oleoyl-CoA/PIP2 and CoA/ATP is affected by different sets of mutations (e.g. Fig. 2 and Fig. 5). In other words, free CoA interacts with the inhibitory ATP-binding site whereas CoA as part of oleoyl-CoA interacts with the activatory lipid-binding site.
While screening for mechanistic differences between oleoyl-CoA and PIP2, we used diadenosine tetraphosphate (Ap4A), which is a potent inhibitor of cardiac (Jovanovic et al. 1996) and pancreatic KATP channels (Martin et al. 1998). It has been proposed that Ap4A and ATP inhibit KATP channels by different mechanisms (Martin et al. 1998). However, our results strongly suggest that Ap4A and ATP interact with the same site on Kir6.2. Similar to ATP, oleoyl-CoA/PIP2 and R50E/K185Q reduced the inhibition of KATP channels by Ap4A. Moreover, we showed competition between Ap4A and ATP for the inhibition of KATP channels. These results question the physiological relevance of ApnA as regulators of KATP channels. ApnA have been proposed to be involved in glucose-dependent insulin secretion in pancreatic β cells because ApnA concentrations rise upon glucose stimulation and inhibition of KATP channel activity is known to trigger insulin secretion. However, the intracellular concentrations of ATP in β cells are in the range 3-5 mM (Niki et al. 1989). Thus, given the observed competition between ATP and Ap4A (Fig. 6C and D), intracellular ATP should completely prevent the inhibition of KATP channels by ApnA. The same logic rules out a physiological relevance for inhibition of KATP channels by CoA. However, these findings might be valuable for structural models of the ATP-binding site since they should allow the binding of Ap4A as well as the bulky CoA.
Acknowledgments
The authors appreciate the excellent technical support by Dr Hariolf Fritzenschaft, and thank Dr Klaus Benndorf and Dr Christoph Biskup for critically reading the manuscript. This work was supported by grant Ba 1793 from the Deutsche Forschungsgemeinschaft (to T.B.).
REFERENCES
- Baukrowitz T, Fakler B. K(ATP) channels: Linker between phospholipid metabolism and excitability. Biochem Pharmacol. 2000;60:735–740. doi: 10.1016/s0006-2952(00)00267-7. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Tucker SJ, Schulte U, Benndorf K, Ruppersberg JP, Fakler B. Inward rectification in KATP channels: a pH switch in the pore. EMBO J. 1999;18:847–885. doi: 10.1093/emboj/18.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branstrom R, Leibiger IB, Leibiger B, Corkey BE, Berggren PO, Larsson O. Long chain coenzyme A esters activate the pore-forming subunit (Kir6. 2) of the ATP-regulated potassium channel. J Biol Chem. 1998;273:31395–31400. doi: 10.1074/jbc.273.47.31395. [DOI] [PubMed] [Google Scholar]
- Cukras CA, Jeliazkova I, Nichols CG. The role of NH2-terminal positive charges in the activity of inward rectifier KATP channels. J Gen Physiol. 2002;120:437–446. doi: 10.1085/jgp.20028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeney JT, Gromada J, Hoy M, Olsen HL, Rhodes CJ, Prentki M, Berggren PO, Corkey BE. Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI beta-cells) J Biol Chem. 2000;275:9363–9368. doi: 10.1074/jbc.275.13.9363. [DOI] [PubMed] [Google Scholar]
- Deutsch N, Matsuoka S, James NW. Surface charge and properties of cardiac ATP-sensitive K+ channels. J Gen Physiol. 1994;104:773–800. doi: 10.1085/jgp.104.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B, Brändle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward-rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 1995;80:149–154. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- Fan Z, Makielski JC. Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem. 1997;272:5388–5395. doi: 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- Fan Z, Makielski JC. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K(+) channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J Gen Physiol. 1999;114:251–270. doi: 10.1085/jgp.114.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Proks P, Corkey BE, Ashcroft FM. Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J Biol Chem. 1998;273:26383–26387. doi: 10.1074/jbc.273.41.26383. [DOI] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Jovanovic A, Zhang S, Alekseev AE, Terzic A. Diadenosine polyphosphate-induced inhibition of cardiac KATP channels: operative state-dependent regulation by a nucleoside diphosphate. Pflugers Arch. 1996;431:800–802. doi: 10.1007/BF02253848. [DOI] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Nichols CG. Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J Gen Physiol. 1999;114:203–213. doi: 10.1085/jgp.114.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauter T, Ruppersberg JP, Baukrowitz T. Phospholipids as modulators of KATP channels: Distinct mechanisms for control of sensitivity to sulphonylureas, K+ channel openers, and ATP. Mol Pharmacol. 2001;59:1086–1093. [PubMed] [Google Scholar]
- Larsson O, Deeney JT, Branstrom R, Berggren PO, Corkey BE. Activation of the ATP-sensitive K+ channel by long chain acyl-CoA. A role in modulation of pancreatic beta-cell glucose sensitivity. J Biol Chem. 1996;271:10623–10626. doi: 10.1074/jbc.271.18.10623. [DOI] [PubMed] [Google Scholar]
- Liu GX, Hanley PJ, Ray J, Daut J. Long-chain acyl-coenzyme A esters and fatty acids directly link metabolism to K(ATP) channels in the heart. Circ Res. 2001;88:918–924. doi: 10.1161/hh0901.089881. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002;34:933–944. doi: 10.1016/s0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- MacGregor GG, Dong K, Vanoye CG, Tang L, Giebisch G, Hebert SC. Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc Natl Acad Sci U S A. 2002;99:2726–2731. doi: 10.1073/pnas.042688899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Pintor J, Rovira JM, Ripoll C, Miras-Portugal MT, Soria B. Intracellular diadenosine polyphosphates: a novel second messenger in stimulus-secretion coupling. FASEB J. 1998;12:1499–1506. doi: 10.1096/fasebj.12.14.1499. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Niki I, Ashcroft FM, Ashcroft SJ. The dependence on intracellular ATP concentration of ATP-sensitive K-channels and of Na,K-ATPase in intact HIT-T15 beta-cells. FEBS Lett. 1989;257:361–364. doi: 10.1016/0014-5793(89)81572-8. [DOI] [PubMed] [Google Scholar]
- Ribalet B, John SA, Weiss JN. Molecular basis for kir6. 2 channel inhibition by adenine nucleotides. Biophys J. 2003;84:266–276. doi: 10.1016/S0006-3495(03)74847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T, Lopes CM, Jin T, Ramdya PP, Molnar Z, Logothetis DE. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A. 2003;100:745–750. doi: 10.1073/pnas.0236364100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze D, Krauter T, Fritzenschaft H, Soom M, Baukrowitz Phosphatidylinositol 4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir channels. A tale of an active and a silent PIP2 site in the N terminus. J Biol Chem. 2003;278:10500–10505. doi: 10.1074/jbc.M208413200. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Cukras CA, Harwood J, Nichols CG. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J Gen Physiol. 2000;116:599–608. doi: 10.1085/jgp.116.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Soom M, Schonherr R, Kubo Y, Kirsch C, Klinger R, Heinemann SH. Multiple PIP2 binding sites in Kir2. 1 inwardly rectifying potassium channels. FEBS Lett. 2001;490:49–53. doi: 10.1016/s0014-5793(01)02136-6. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Vusse GJ, Glatz JF, Stam HC, Reneman RS. Fatty acid homeostasis in the normoxic and ischemic heart. Physiol Rev. 1992;72:881–940. doi: 10.1152/physrev.1992.72.4.881. [DOI] [PubMed] [Google Scholar]