

Abstract
Duchenne muscular dystrophy is a lethal muscle disease caused by absence of the protein dystrophin which is part of a glycoprotein complex located on the intracellular surface of the surface membrane. The precise function of dystrophin and the reason why its absence causes severe muscle damage are unclear. Stretch-induced muscle damage is well recognised in normal muscle and is more severe in muscles from animals lacking dystrophin (mdx mice). It has been proposed that stretch-induced damage underlies the progression of damage in muscular dystrophy. In the present study we confirm that single fibres from mdx muscle are more susceptible to stretch-induced damage and show that there is an associated rise in intracellular sodium concentration ([Na+]i) which is greater than in wild-type mice. We show that this rise in [Na+]i can be prevented by Gd3+, which is an established blocker of stretch-activated channels. mdx fibres have a higher than normal resting [Na+]i and this is also reduced by Gd3+. If Gd3+ is applied over the period in which [Na+]i rises following stretched contraction, it prevents one component of the reduced force. The other component of reduced force is caused by inhomogeneity of sarcomeres and can be minimised by stretching the muscle to its new optimum length. These experiments show that part of the short-term damage caused by stretch in mdx fibres can be prevented by blocking stretch-activated channels.
Duchenne muscular dystrophy is an X-linked condition that affects approximately 1 in 3500 male births (Emery, 1991). It is a degenerative muscle disease causing death through respiratory muscle failure by the end of the second decade with no effective treatment at present. The discovery that the disease was caused by absence of the protein dystrophin has given new impetus to therapy and to attempts at understanding the muscle degeneration which underlies the disease (Hoffman et al. 1987). While gene replacement therapy is of proven value in transgenic mouse models, the success of such therapy in humans has so far been limited by the available viral vectors, by the difficulties in delivering genes to the large mass of multi-nucleated muscle cells and by the absence of stable expression of dystrophin after delivery (for review see Skuk et al. 2002). Two main theories have emerged as to the functional role of dystrophin and the reason(s) its absence causes disabling muscle damage (Lansman & Franco, 1991; McArdle et al. 1995). (i) Dystrophin may have a structural role in maintaining the integrity of the surface membrane during the stress imposed by normal contractions, particularly those involving stretch (Hutter, 1992). (ii) Dystrophin may be involved in the clustering of ion channels on the membrane and its absence may lead to abnormal channel function (Carlson, 1998). A universal accompaniment to dystrophic damage is an increase in muscle calcium which may arise through either of the above, or other, pathways (Gillis, 1999).
If muscles are stretched during contraction (eccentric contractions), they develop a characteristic form of weakness followed by muscle soreness, tenderness and stiffness developing over a few days and recovering over 1-2 weeks (Morgan & Allen, 1999; Warren et al. 2001). Such damage is associated with disorganised sarcomeres (Fridén et al. 1981), increased membrane permeability (McNeil & Khakee, 1992) and changes in intracellular Ca2+ concentration ([Ca2+]i) which include an elevated resting [Ca2+]i but a reduced [Ca2+]i during tetani (Balnave & Allen, 1995; Ingalls et al. 1998). It has been proposed that the elevated resting [Ca2+]i might activate endogenous proteases which damage one or more proteins involved in the coupling between excitation and SR Ca2+ release and cause the reduced tetanic [Ca2+]i and contribute to the muscle weakness (Lamb et al. 1995; Chin & Allen, 1996; Bruton et al. 1996). Ca2+ activation of proteases may also contribute to the subsequent muscle inflammation and degeneration which can occur after eccentric exercise (Belcastro et al. 1998).
Stretch-induced damage is generally more severe in mdx muscle (Head et al. 1992; Petrof et al. 1993; Moens et al. 1993) and it has been shown that delivery of a dystrophin mini gene to mdx fibres reduces stretch-induced damage (Deconinck et al. 1996). However the mechanism by which the absence of dystrophin exacerbates stretch-induced damage is unclear. One possibility is that wild-type fibres contain a stretch-activated channel (Franco & Lansman, 1990) and that in mdx fibres these channels change their properties and become more readily activated by stretch (Franco-Obregon & Lansman, 2002). Since the stretch-activated channels in muscle are non-specific cation channels they could contribute to increases in [Na+]i or [Ca2+]i after eccentric contractions (Franco & Lansman, 1990). mdx fibres have also been shown to have increased permeability to divalent cations using the Mn2+ quench approach (Tutdibi et al. 1999). It is widely believed that eccentric damage causes tears in the membrane which would allow movement of ions such as Na+ and Ca2+ into the fibre (McNeil & Khakee, 1992). A very recent study has suggested increased Ca2+ entry through a store-operated channel in mdx muscles (Vandebrouck et al. 2002). Thus there are a number of possible routes whereby Ca2+ might gain entry to mdx fibres and initiate Ca2+-induced damage pathways.
We have recently shown that in wild-type muscle fibres there is a rise of [Na+]i following eccentric contractions and that this rise can be prevented by application of gadolinium (Gd3+), an established blocker of stretch-activated channels (Yeung et al. 2003). In the present study we dissected single intact mdx muscle fibres and show they are more susceptible to eccentric damage judged by the magnitude of the force deficit after eccentric contractions. We show that in mdx fibres the resting [Na+]i is elevated and can be reduced by Gd3+. Gd3+ also eliminates the elevation of [Na+]i caused by eccentric contractions and, most interestingly, reduces the force deficit after eccentric contractions. This suggests that agents which block stretch-activated channels might have role in reducing muscle damage in muscular dystrophy.
METHODS
Muscle dissection and mounting
The experiments were approved by the Animal Ethical Committee of the University of New South Wales. Mice were killed by cervical dislocation and single flexor brevis muscle fibres were dissected from age-matched (10-14 weeks old) wild-type (Balb-C) and dystrophic mdx mice. The dissection procedure for mdx fibres was identical to that used for wild-type fibres. The fibre was mounted in an experimental chamber between an Akers AE 801 force transducer (SensorNor, Horten, Norway) and the lever of a motor (Model 300H, Cambridge Technology, Cambridge, MA, USA). Stimulating electrodes parallel to the muscle fibre allowed the force to be measured during stimulation while known length changes were imposed on the muscle.
The dissection was performed in the following solution (mM): 136.5 NaCl, 5 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.4 NaH2PO4 and 11.9 NaHCO3 (pH 8.0). During the experiment, the fibre was superfused with standard solution of the following composition (mM): 121 NaCl, 5 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.4 NaH2PO4, 24 NaHCO3, 5.5 glucose and 0.2 % fetal calf serum (Gibco). The solution was bubbled with 95 % O2-5 % CO2 (pH 7.4). All experiments were performed at room temperature (≈ 22 °C).
The effects of stretch-sensitive channel blockers, gadolinium (Gd3+) and streptomycin, were investigated. A stock solution of 1 M GdCl3 in water was prepared and diluted to 20 μM in standard solution immediately before use. Streptomycin was prepared in a stock solution of 100 mM in water and diluted to 100 μM in standard solution.
Experimental protocol
Both wild-type and mdx muscle fibres were exposed to the protocol as described previously (Yeung et al. 2002a). Fibres were initially set to the length that produced maximal tetanic force (optimum length, Lo). The fibre width was measured at different points along the fibre and the mean diameter was used to calculate the cross-sectional area and to convert force measurements to force/cross-sectional area.
Each muscle fibre was first subjected to 10 isometric tetani as control, followed by 10 eccentric tetani. The muscle fibre was stimulated at 100 Hz, and tetani were 400 ms in duration with a rest period of 3.6 s between tetani (cycle 4 s). The stretch was applied 200 ms after the start of stimulation and the fibre was stretched from Lo to Lo + 40 % over 100 ms (stretching velocity = 4 × Lo s−1). The fibre was returned to Lo between tetani. The force-frequency relationship was established by tetani at 10, 20, 30, 50, 70 and 100 Hz stimulation. The reduction in force at 100 Hz (force deficit) was measured 10 min after the eccentric tetani.
Confocal imaging of [Na+]i using sodium green
The fluorescent indicator sodium green (Molecular Probes, OR, USA) was used to measure [Na+]i. The membrane permeant version sodium green AM was prepared as a 1 mM stock solution in dimethyl sulphoxide (DMSO). Once dissection was complete, the muscle fibre was incubated in 0.5 ml of dissection solution containing 5 μM of sodium green AM for 20 min at room temperature. After loading, the fibre was transferred to the experimental chamber and perfused with the standard solution for 30-45 min to allow hydrolysis. The full experimental protocol was performed on the stage of an inverted confocal microscope (Leica TCS SL, Heidelberg, Germany) so that the fluorescence intensity of sodium green within the fiber could be studied at any time. The fibre was examined with a × 63, numerical aperture 1.2, water immersion objective using a 488 nm illumination line at 25 % maximum power and recording at wavelengths 510-560 nm. This objective has a working distance of 220 μm above the coverslip which formed the base of the chamber. Image processing and analysis were performed using NIH Image (Scion Corporation, MD, USA).
In the preliminary experiments, we varied the loading conditions to achieve optimal imaging. With heavy loading (30 min of 10 μM sodium green AM) there was some evidence of loading into subcellular compartments. With our standard loading protocol (see above) the distribution of sodium green fluorescence was close to uniform (see images in Fig. 1 and Fig. 3).
Figure 1. Calibration of sodium green fluoresence in mouse single fibres.
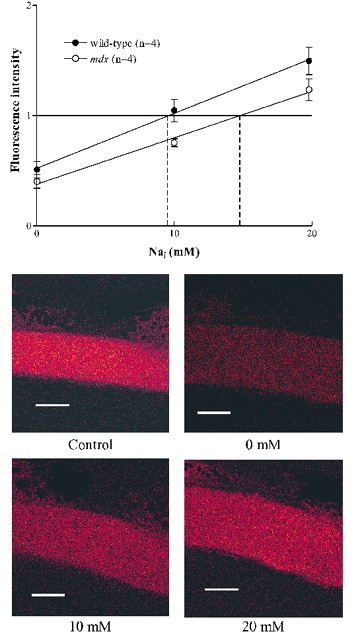
The upper panel shows measurements of fluoresence intensity, all normalized to the intensity in the fibre before calibration. Points are means ± S.E.M. (n = 4) of the fluoresence obtained when the calibrated [Na+]i was 0, 10 or 20 mM. Correction for dye loss during calibration has been applied (see Methods). The estimates of resting [Na+]i were made by linear interpolation of the control fluorescence, indicated by vertical dashed lines. Lower panels show representative images of the sodium green fluoresence from a control fibre and the same fibre during the calibration at 0, 10 and 20 mM [Na+]i. Scale bar 25 μm.
Figure 3. [Na+]i signals from wild-type and mdx muscle fibres following isometric and eccentric contractions.
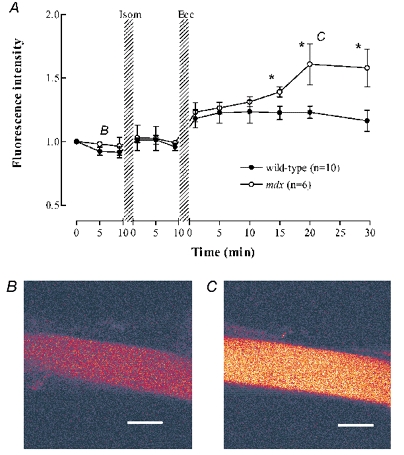
A, sodium green fluorescence normalized to the starting point of each experiment. Wild-type (filled symbols), mdx (open symbols). * Significant difference between wild-type and mdx. The rise in fluorescence after the eccentric contractions was statistically significant for both wild-type and mdx fibres. B and C, representative images of the sodium green fluorescence from one mdx fibre at the times indicated by the B and C labels on panel A. Scale bar 25 μm.
Calibration of sodium green
An in vivo calibration of [Na+]i was performed on four control and four mdx fibres. The Na+ ionophore gramicidin D and the Na+ pump inhibitor strophanthidin were used to eliminate the Na+ gradient across the membrane. The fibre was exposed to the calibration solution containing (mM unless stated): 2 EGTA, 10 glucose, 10 Hepes, 10 μM gramicidin, 10 μM strophanthidin, pH 7.4 and various concentrations of NaCl (0, 10, 20, 140 mM) with the balance to 140 mM provided by KCl. In all cases the first three solutions applied to the muscle fibre were 0, 10 and 20 mM since the resting [Na+]i always lay between these values. Solutions were applied for about 5 min to allow time for equilibration and in different order in different experiments. In all the calibration experiments the first three points were completed; in two experiments 140 mM Na+ was also included in the calibration series with a final return to 20 mM Na+ to allow assessment of dye loss. Such experiments showed that dye loss averaged 6 % per solution change and therefore in constructing Fig. 1 we corrected each intensity by this amount, i.e. 6 % increase for the first point, 12 % increase for the second point, etc. We also performed the calibrations without this correction for dye loss; without the correction the estimated [Na+]i values were 10-15 % higher but the difference between wild-type and mdx fibres was similar. The resting [Na+]i was determined by linear interpolation of the resting fluorescence with the calibration lines shown in Fig. 1 (indicated by vertical dashed lines).
Figure 1 shows images of the sodium green fluorescence in the control fibre and in the 0, 10 and 20 mM calibration solutions. Note that the fibre swells substantially in the calibration solution, presumably because in the high extracellular KCl solution, the membrane potential is depolarized and Cl− (plus osmotically equivalent H2O) enters the fibre to approach electrochemical equilibrium (Hodgkin & Horowicz, 1959). To offset the apparent reduction in sodium green fluorescence caused by swelling we measured the fluoresence in a rectangular box which was greater than the width of the fibre so that all the fluoresence from a length of the fibre was determined.
Statistics
Data are presented as means ± S.E.M. Student's t test or one-way analysis of variance with the Bonferroni correction for multiple comparisons was used to verify statistical significance. The significance was accepted at P < 0.05.
RESULTS
Effect of eccentric contractions on the mechanical properties of mdx muscle fibres
In the present study we dissected single mdx muscle fibres from the flexor brevis muscle and compared them to wild-type fibres from the same muscle. The mdx fibres ranged in length from 0.6 to 1.2 mm and were similar in length to wild-type muscle fibres. The diameters of the wild-type (31 ± 3 μm, n = 8) and mdx (29 ± 2 μm, n = 9) muscle fibres were also similar. The mdx fibres exhibited a significantly lower force production at 70 and 100 Hz compared to control fibres. For instance at 100 Hz stimulation, the force per cross-sectional area for wild-type fibres was 367 ± 32 kPa (n = 7) compared with 262 ± 19 kPa (n = 9) in mdx fibres (P < 0.05). This is in agreement with observations from other studies (Quinlan et al. 1992; Petrof et al. 1993).
We used an established model of eccentric contraction in which single mouse muscle fibres are subjected to a stretch of 40 % of Lo during 10 maximal tetani (Balnave & Allen, 1995; Yeung et al. 2002a). In the mdx fibres, the reduction of force at all frequencies was substantially greater than that of wild-type fibres after the same eccentric protocol. The 100 Hz force following eccentric contractions decreased to 34 ± 4 % of the control in wild-type fibres and to 23 ± 3 % in mdx mice (P < 0.05). A characteristic of eccentric muscle damage is that there is a greater reduction of force at low frequencies of stimulation than at high frequencies (for review see Morgan & Allen, 1999). In Fig. 2A we confirm this finding in wild-type fibres and show that the same phenomenon occurs in mdx fibres. A further characteristic of eccentric muscle damage is that there is an increase in the optimum muscle length associated with overstretched sarcomeres. In wild-type fibres we previously showed that the present protocol caused a shift in Lo of 14 ± 4 % (Yeung et al. 2003). In Fig. 2B we show that in mdx fibres the shift in Lo was 11 ± 1 %; this was not significantly different from that observed in the wild-type fibres.
Figure 2. Changes in properties of wild type and mdx muscle following eccentric protocol.
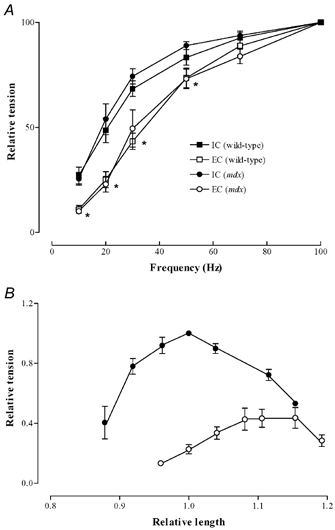
A, comparison of force-frequency relationships following isometric contraction (filled symbols) and following eccentric contractions (open symbols). Data show means ± S.E.M. for 8 wild-type and 9 mdx fibres. Square symbols are wild-type fibres, circular symbols are mdx fibres. Data normalised to 100 Hz tetani. * Significant difference between both sets of isometric and eccentric data. B, force-length relation of mdx fibres before (filled symbols) and after eccentric contractions (open symbols).
Intracellular sodium in wild-type and mdx fibres
Previous studies have shown an elevated [Na+]i in mdx muscles (Dunn et al. 1993). In order to determine whether this was the case for our single fibres, we performed an in vivo calibration of the sodium green fluorescence (see Methods for details). Figure 1 shows representative images of one fibre before the calibration procedure and at 0, 10 and 20 mM [Na+]i during the calibration procedure. The upper panel shows the fluoresence from sodium green normalized to the control fluorescence at 0, 10 and 20 mM imposed [Na+]i. The resting [Na+]i for each fibre was determined by linear interpolation. In wild-type fibres (n = 4) the resting [Na+]i was 9.7 ± 1.1 mM while in mdx fibres (n = 4) the resting [Na+]i was significantly greater at 15.4 ± 1.1 mM (P < 0.05).
Intracellular sodium following isometric and eccentric tetani
We have recently shown that in wild-type fibres, the present protocol of eccentric tetani causes a substantial rise in [Na+]i from about 7 to 16 mM (Yeung et al. 2003). Given that mdx fibre are more susceptible to damage following eccentric contractions, we hypothesised that the changes in [Na+]i following eccentric contractions might be larger. Images of the sodium green fluorescence were recorded regularly before and after isometric and eccentric tetani in both wild-type and mdx fibres. As in our previous study, the distribution of fluorescence was essentially uniform under all conditions so in the present study we simply averaged the fluorescence signal from a large region of the fibre. For the present purpose fluorescence was measured as a fraction of the initial value recognising that the initial fluorescence represents different levels of [Na+]i in wild-type and mdx fibres. Figure 3 shows that there was no significant change to Na+ fluorescence in the wild-type and mdx muscle fibres following isometric tetani. However after eccentric tetani there was a significant rise in sodium green fluorescence in the first measurement after eccentric tetani (about 1 min after the last tetanus) in both normal and mdx fibres. For the first 15 min after eccentric contractions the increase in Na+ fluorescence was not significantly different in wild-type and mdx fibres but thereafter the increase in [Na+]i was larger in mdx compared to wild-type fibres (P < 0.05).
Effects of blockers of stretch-sensitive channels
In our previous study in wild-type fibres we showed that the increase in [Na+]i following eccentric tetani was spatially uniform and blocked by Gd3+ (20 μM), an established blocker of stretch-activated channels (Yeung et al. 2003). In the present experiments we also established that the rise of [Na+]i after eccentric damage in mdx fibres was spatially uniform (compare Fig. 3B and 3C). We therefore tested whether the generalized [Na+]i increase was blocked by Gd3+. In wild-type fibres under resting conditions, 20 μM Gd3+ for 10 min did not produce any significant effect on resting sodium green fluorescence (Fig. 4). In contrast in mdx fibres Gd3+ caused a substantial reduction in sodium green fluorescence (to 59 ± 1 % of control, P < 0.001). Furthermore Gd3+ eliminated the rise of sodium green fluorescence following eccentric contractions and appeared to cause a prolonged reduction in [Na+]i, which did not recover after Gd3+ was removed.
Figure 4. Effects of Gd3+ on [Na+]i signals from wild-type and mdx fibres.
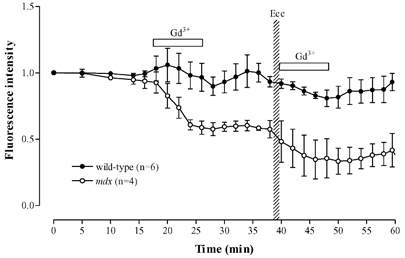
Sodium green fluorescence from wild-type (filled symbols) and mdx (open symbols) muscle fibres. Initial application of Gd3+ was on resting fibres; subsequent application was immediately after eccentric contractions.
We further examined the possible role of stretch-activated channels by the use of streptomycin (100 μM), another non-specific stretch-activated channel inhibitor (Sokabe et al. 1993). In two fibres application of streptomycin to the resting mdx fibres for 10 min reduced sodium green fluorescence to a mean of 80 % (values 75 and 85 %) whereas streptomycin had little effect on the resting fluorescence in wild-type fibres (Yeung et al. 2003). When streptomycin was applied for 10 min after the eccentric protocol, the large rise in sodium green fluoresence seen in Fig. 3 was eliminated. For instance at 6 min after the eccentric protocol the sodium green fluoresence in mdx fibres had risen to 126 ± 5 % while in the streptomycin treated fibres the fluoresence at this time averaged 86 % (values 85 and 87 %). Thus the streptomycin data are similar to the Gd3+ data and suggest there is a class of Na+ permeable channels active in mdx fibres which were not found in wild-type fibres.
Gadolinium and the force deficit after eccentric contractions in mdx muscle
Given that Gd3+ prevents Na+ entry following eccentric contractions, we tested whether Gd3+ could also minimize muscle damage in mdx fibres as measured by the force deficit (Fig. 5). We previously reported that Gd3+ reduced the force deficit in wild-type fibres (Yeung et al. 2003). Gd3+ applied under control conditions to mdx fibres had no effect on the 100 Hz force (first bar in Fig. 5). Following our standard protocol of eccentric contractions force was reduced to 23 ± 3 % of the control value measured 10 min after the contractions. If Gd3+ was added to the solution either before (n = 5) or immediately after (n = 5) the stretched contractions and maintained for 10 min, there was a highly significant improvement of developed force to 46 ± 4 % (P < 0.001). In two experiments we applied streptomycin (100 μM) for 10 min following the eccentric protocol and the developed force in these experiments averaged 40 % (values 39 and 41 %). There was a tendency, which did not reach significance, for Gd3+ to be more effective in preventing damage if it was present during the stretched contraction as well as after. If Gd3+ was removed after 10 min and the recovery measured several minutes after its removal, the improvement was maintained. Thus it seems that the critical time for preventing stretch-induced damage was the period over which [Na+]i was rising.
Figure 5. Recovery of force after eccentric contractions in isolated mdx muscle fibres.
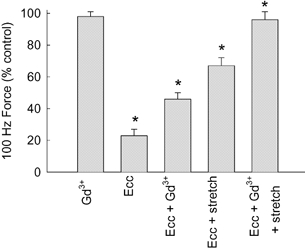
Bars indicate 100 Hz force normalized to control tetani. First bar (Gd3+), force in the presence of 20 μM Gd3+. Second bar (Ecc), force 10 min after eccentric protocol. Third bar (Ecc + Gd3+), 20 μM Gd3+ present for 10 min following the eccentric protocol, force measured at 11 min. Fourth bar (Ecc + stretch), muscle stretched to new optimum length prior to measurement of force at 10 min after eccentric contractions. Fifth bar (Ecc + Gd3++ stretch), 20 μM Gd3+ present for 10 min after eccentric contractions, then stretched to new optimum length prior to force measurement at 11 min. Bars show + 1 S.E.M. compared to control. * Significantly different from preceding bar using one-way ANOVA.
In four experiments the fibre was stretched to the new optimum length after the eccentric contractions and this increased the force to 69 ± 5 % (Fig. 5, P < 0.001). In a further four experiment we both added Gd3+ during the 10 min period prior to the measurement of force and stretched the mdx muscle fibres to the new Lo. As shown in Fig. 5 this combined protocol increased force to 96 ± 5 %, which was not significantly different from either the control or control plus Gd3+. In the two experiments in which streptomycin was added after the eccentric protocol the force after stretching to the new Lo averaged 88 % (values 86 and 90 %). Thus the short-term damage in mdx fibres, at least as monitored by force, seems to be largely reversed by the combination of these two manoeuvres.
DISCUSSION
In these experiments single mdx fibres were dissected with intact tendons and subjected to length changes while [Na+]i and force were measured. In older mdx mice regenerating muscle fibres can take up bizzare shapes (Head et al. 1992) which would preclude dissection of intact single fibres but in the 10- to 14-week-old mice which we used this did not present a problem. The mdx fibres selected during dissection were roughly the same size as wild-type fibres but showed a moderate reduction in force/unit area confirming earlier studies (Quinlan et al. 1992; Petrof et al. 1993). We also confirm that in this single fibre model of muscular dystrophy that stretch-induced damage is greater than in wild-type fibres as previously shown in whole muscles (Head et al. 1992; Petrof et al. 1993; Moens et al. 1993).
Resting [Na+]i in normal and mdx fibres
Estimates of resting [Na+]i in frog skeletal muscle varied between 5 and 22 mM when reviewed by Godt & Maughan (1988). In mammalian fibres there is also variability with estimated values of 13 and 7 mM (Dunn et al. 1993; Yeung et al. 2003). Dunn et al. (1993) also measured the [Na+]i in mdx muscles and reported it to be 23.5 ± 0.7 mM in mouse diaphragm and 24 ± 2 mM in the mouse gastrocnemius. Another study, however, found no significant increase in [Na+]i in either muscles from Duchenne dystrophy or mdx muscle fibres (Turner et al. 1991). Thus our resting [Na+]i of 9.7 ± 1.1 mM in wild-type fibres and 15.4 ± 1.8 mM in mdx fibres are both within the published range.
The increase in resting [Na+]i in mdx fibres could arise either by increased influx or decreased efflux of Na+. There is evidence that both Na+ pump activity and content are increased in mdx mice (Anderson, 1991; Dunn et al. 1995). We showed that Gd3+ and streptomycin both reduced the elevated [Na+]i in mdx fibres while neither had any such effect on wild-type fibres. Thus these results suggest that the cause of the elevated [Na+]i is increased Na+ influx through a Gd3+/streptomycin-sensitive pathway. The reduction of resting fluoresence caused by Gd3+ is quite large (41 % reduction) while that for streptomycin is smaller (20 % reduction). From Fig. 1 it appears that Gd3+ caused the resting [Na+]i to decrease to ≈5 mM whereas the decrease in streptomycin was to ≈10 mM. One possible explanation for the large decrease caused by Gd3+ would be if Gd3+ can enter mdx fibres and quench the sodium green signal. However we excluded this possibility by measuring sodium green fluoresence in response to 10 mM Na+in vitro and showing that even 20 μM Gd3+ did not cause quenching. Another possible reason for the large fall of [Na+]i in Gd3+ -treated mdx fibres is that the increased Na+ pump activity noted above would tend to lower [Na+]i to below the wild-type level when the increased influx was blocked.
Based on the results with stretch-activated channel blockers, we postulate that in mdx fibres a period of eccentric contractions switches some stretch-activated channels into a high permeability mode and causes the increased Na+ flux and [Na+]i which we observed. In the next section we discuss the evidence for such channels and possible reasons why these channels are activated in mdx fibres but not in wild-type fibres. The fact that we see an elevated [Na+]i even in resting fibres which have not been exposed to eccentric contractions suggests that some of these channels may stay switched open for very long periods, perhaps because dystrophin is necessary for the normal regulation of these channels.
[Na+]i in normal and mdx fibres following eccentric contractions
The [Na+]i measurements indicate a substantially greater increase in [Na+]i after eccentric contractions in mdx compared to wild-type fibres. We have previously shown that this increase in [Na+]i in wild-type fibres is abolished by removing Na+ from the extracellular space and that the Na+ pump appears to function normally (Yeung et al. 2003). Thus the increase in [Na+]i appears to arise from an increase in Na+ permeability. Our key observation is that the stretch-induced rise in [Na+]i is abolished by Gd3+. Gd3+ blocks stretch-sensitive channels at 5-10 μM concentrations (Yang & Sachs, 1989) but is a non-specific agent which also blocks L-type Ca2+ channels (Lacampagne et al. 1994), store-operated Ca2+ channels (Flemming et al. 2002) and some Na+ and K+ channels (Elinder & Arhem, 1994). We established that the concentration we used (20 μM Gd3+) has no obvious effect on tetanic contraction, which shows that it does not have drastic effects on normal excitability or contraction. The chemical species of Gd3+ which produces channel block is uncertain because Gd3+ binds phosphate, carbonate and proteins very tightly, and all of these are present in our solutions (Caldwell et al. 1998; Yeung et al. 2003). We also showed that another chemically dissimilar blocker of stretch-activated channels (streptomycin) had similar effects. Thus it seems probable that the route of entry of Na+ following stretch is a stretch-activated channel. This conclusion is supported by McBride et al. (2000) who showed that following eccentric contractions there was a depolarization of the resting membrane potential which also arose through increased permeability to Na+.
However there are several aspects of our results which do not fit simply with this hypothesis. First, eccentric contractions over 1 min caused an increase in [Na+]i that persisted at least 20-30 min. Most stretch-sensitive channels which have been described generally open and close with application and removal of stretch reasonably fast (1-2 min) (Sachs, 1991). This suggests that either the channels are locked into an open state by a preceding period of stretch or that channels are associated with some component of the membrane, such as the T-tubules or vacuoles (Takekura et al. 2001; Yeung et al. 2002a), which remains stretched long after the eccentric contractions. Interestingly the membrane depolarization observed by McBride et al. (2000) and attributed to stretch-activated channels persisted for more than 24 h. Second, the rise of [Na+]i was greater in mdx than in wild-type muscles. This suggests that the absence of dystrophin influences stretch-activated channels. In fact there is strong evidence that the properties of stretch-activated channels differ in wild-type and mdx muscles (Franco-Obregon & Lansman, 2002). Stretch-sensitive channels in mdx fibres show a tendency to be either near fully open or fully closed and stretch could induce them to change in either direction (Franco-Obregon & Lansman, 2002). Thus we postulate that in mdx fibres a period of eccentric contractions switches a net excess of such channels into the open position and generates the increased Na+ permeability which we observed. Some of these channels may stay semi-permanently open explaining the increased [Na+]i in resting mdx fibres discussed above. A third interesting observation is that after Gd3+ had lowered the resting [Na+]i, the [Na+]i failed to recover when Gd3+ was removed from resting fibres. This result could be explained if Gd3+ is also capable of switching channels from their permanently open to their permanently closed state (Franco-Obregon & Lansman, 2002). Alternatively it may be that strongly charged ions such as Gd3+ are only slowly removed after washout from some binding sites on the membrane.
What causes the force deficit after eccentric contractions in mdx fibres?
Early studies on muscles subjected to eccentric contractions showed that the sarcomere pattern was disrupted and the muscles exhibited localised regions with over- and under-stretched sarcomeres (Fridén et al. 1981). The maximum force can only be produced in a muscle in which all sarcomeres are isometric and at their optimum length. A muscle with a mixture of long and short sarcomeres is unstable and the strongest sarcomeres will shorten at the expense of the weaker sarcomeres. The force-velocity relation dictates that shortening sarcomeres produced less force so that the total force exerted by such a muscle is reduced. Stretching the muscle to the new optimum length minimizes the additional compliance of the overstretched sarcomeres and some of the force reduction associated with the inhomogeneity is eliminated (Wood et al. 1993; Talbot & Morgan, 1996). Interestingly the shift in optimum length was not greater in wild-type compared to mdx fibres suggesting that another component of muscle damage must be greater in the mdx muscles. This conclusion is strengthened by the observation that in permeabilized fibres, in which excitation-contraction coupling is eliminated, eccentric damage is similar in wild-type and mdx fibres (Lynch et al. 2000).
What causes the component of muscle damage which is not corrected by stretching the muscles to the new optimum length? In wild-type mouse muscles the tetanic [Ca2+]i is reduced after eccentric damage and this component of damage can be reversed by application of caffeine, a drug which increases opening of SR Ca2+ channels and can therefore correct the reduction of tetanic [Ca2+]i (Warren et al. 1993; Balnave & Allen, 1995; Ingalls et al. 1998). In the present study we show that there is an increase in Na+ permeability probably caused by increased opening of stretch-activated channels. Since the stretch-activated channels described in muscle are generally non-specific cation channels (Franco & Lansman, 1990; Sigurdson et al. 1992), it seems possible that the previously described increase in resting [Ca2+]i following eccentric contractions also arises by Ca2+ entry through the same stretch-activated channel (Balnave & Allen, 1995; Ingalls et al. 1998). Another possibility is that the raised [Na+]i in mdx fibres could lead to an increased Ca2+ influx on the Na+-Ca2+ exchanger (Gilbert & Meissner, 1982; Balnave & Allen, 1998). As noted in the Introduction, a possible mechanism to explain the reduced force is that the elevated resting [Ca2+]i may activate proteases. There is good evidence that elevated [Ca2+]i can cause a reduction of SR Ca2+ release though it is not clear whether protease damage to the ryanodine receptor is necessarily involved (Lamb et al. 1995; Chin & Allen, 1996; Bruton et al. 1996). Thus we propose that the stretch-activated channels opened by eccentric contractions also allow Ca2+ entry which contributes to the force deficit by reducing SR Ca2+ release. Gd3+ exerts protection by blocking this entry pathway.
Another possible contributor to reduced force after eccentric damage is the persisting acidosis of about 0.17 pH units (Yeung et al. 2002b). It is possible that this acidosis is bigger in mdx muscles because Dunn et al. (1992) showed that extrusion of protons was slower in mdx muscles. On the other hand, the fact that Gd3+ reversed much of this component of the force deficit suggests that any contribution of acidosis is quite small. One concern is that this acidosis might artefactually increase the sodium green signal but this seems unlikely because calibrations show that the effects of pH on sodium green fluorescence are negligible (Amorino & Fox, 1995).
A further possible contributor to the force deficit is the depolarisation of ≈15 mV following eccentric contractions (McBride et al. 2000). Our results suggest that this depolarization might be larger in mdx mice because the Na+ influx is increased. This might cause partial inactivation of the Na+ channels and reduce the amplitude of the action potential and hence of force. For instance, Cairns et al. (1997) showed that a depolarization of ≈15 mV produced by increased extracellular K+ reduced tetanic force by about 5 %. Thus elimination of the depolarization by Gd3+ might produce a recovery of this size.
Therapeutic possibilities of stretch-activated channel blockers
In terms of possible therapy our most interesting finding is that Gd3+ appeared capable of preventing the one component of the short-term stretch-induced damage. Would Gd3+ also protect against the longer-term damage resulting in fibre degeneration observed after severe stretch-induced damage and/or in muscles lacking dystrophin? Clearly direct experiments are needed but the prospects seem at least encouraging. First, Gd3+ has been used in intact animals without obvious detrimental effects at least in the short term (Takagi et al. 1999) but the fact that it binds tightly to phosphate, carbonate and protein probably make it unsuitable for long term use (Caldwell et al. 1998). The recently described spider venom peptide GsMTx-4 is a more potent and specific stretch-activated channel inhibitor but as a peptide probably would not make an optimal therapeutic agent (Suchyna et al. 2000). Aminoglycoside antibiotics, including streptomycin, gentamicin and neomycin, are another class of stretch-activated channel blockers (Sokabe et al. 1993) which have been widely used in intact animals for their antibacterial properties. For instance, McBride et al. (2000) have shown that addition of streptomycin to the drinking water of rats prevents the depolarization following eccentric contractions which is another consequence of the opening of stretch-activated channels. Thus there are existing agents which could be used to test this principle in intact animals. Second, it is widely believed that the elevated [Ca2+]i after stretch-induced damage has a role in the longer term fibre degeneration, probably by activating proteases (Turner et al. 1988; Belcastro et al. 1998). Thus if stretch-activated channel blockers can prevent the rise of [Ca2+]i in the same manner that they prevent the rise of [Na+]i then they might prove valuable in reducing the damage to intact muscles.
Acknowledgments
This work was supported by the Australian Research Council and the National Health and Medical Research Council of Australia. E.W.Y. was supported by grant A-PE65 from the Hong Kong Polytechnic University.
REFERENCES
- Amorino GP, Fox MH. Intracellular Na+ measurements using sodium green tetraacetate with flow cytometry. Cytometry. 1995;21:248–256. doi: 10.1002/cyto.990210305. [DOI] [PubMed] [Google Scholar]
- Anderson JE. Myotube phospholipid synthesis and sarcolemmal ATPase activity in dystrophic (mdx) mouse muscle. Biochem Cell Biol. 1991;69:835–841. doi: 10.1139/o91-124. [DOI] [PubMed] [Google Scholar]
- Balnave CD, Allen DG. Intracellular calcium and force in single mouse muscle fibres following repeated contractions with stretch. J Physiol. 1995;488:25–36. doi: 10.1113/jphysiol.1995.sp020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balnave CD, Allen DG. Evidence for Na+/Ca2+ exchange in intact single skeletal muscle fibers from the mouse. Am J Physiol. 1998;274:C940–946. doi: 10.1152/ajpcell.1998.274.4.C940. [DOI] [PubMed] [Google Scholar]
- Belcastro AN, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Mol Cell Biochem. 1998;179:135–145. doi: 10.1023/a:1006816123601. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Lannergren J, Westerblad H. Effects of repetitive tetanic stimulation at long intervals on excitation-contraction coupling in frog skeletal muscle. J Physiol. 1996;495:15–22. doi: 10.1113/jphysiol.1996.sp021570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns SP, Hing WA, Slack JR, Mills RG, Loiselle DS. Different effects of raised [K+]o on membrane potential and contraction in mouse fast- and slow-twitch muscle. Am J Physiol. 1997;273:C598–611. doi: 10.1152/ajpcell.1997.273.2.C598. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol. 1998;275:C619–621. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- Carlson CG. The dystrophinopathies: an alternative to the structural hypothesis. Neurobiol Dis. 1998;5:3–15. doi: 10.1006/nbdi.1998.0188. [DOI] [PubMed] [Google Scholar]
- Chin ER, Allen DG. The role of elevations in intracellular Ca2+ concentration in the development of low frequency fatigue in mouse single muscle fibres. J Physiol. 1996;491:813–824. doi: 10.1113/jphysiol.1996.sp021259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck N, Ragot T, Marechal G, Perricaudet M, Gillis JM. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc Natl Acad Sci U S A. 1996;93:3570–3574. doi: 10.1073/pnas.93.8.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JF, Bannister N, Kemp GJ, Publicover SJ. Sodium is elevated in mdx muscles: ionic interactions in dystrophic cells. J Neurol Sci. 1993;114:76–80. doi: 10.1016/0022-510x(93)90052-z. [DOI] [PubMed] [Google Scholar]
- Dunn JF, Burton KA, Dauncey MJ. Ouabain sensitive Na+/K+-ATPase content is elevated in mdx mice: implications for the regulation of ions in dystrophic muscle. J Neurol Sci. 1995;133:11–15. doi: 10.1016/0022-510x(95)00167-z. [DOI] [PubMed] [Google Scholar]
- Dunn JF, Tracey I, Radda GK. A 31P-NMR study of muscle exercise metabolism in mdx mice: evidence for abnormal pH regulation. J Neurol Sci. 1992;113:108–113. doi: 10.1016/0022-510x(92)90272-m. [DOI] [PubMed] [Google Scholar]
- Elinder F, Arhem P. Effects of gadolinium on ion channels in the myelinated axon of Xenopus laevis: four sites of action. Biophys J. 1994;67:71–83. doi: 10.1016/S0006-3495(94)80456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AE. Population frequencies of inherited neuromuscular diseases-A world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- Flemming R, Cheong A, Dedman AM, Beech DJ. Discrete store-operated calcium influx into an intracellular compartment in rabbit arteriolar smooth muscle. J Physiol. 2002;543:455–464. doi: 10.1113/jphysiol.2002.023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A, Lansman JB. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J Physiol. 1990;427:361–380. doi: 10.1113/jphysiol.1990.sp018176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Obregon A, Lansman JB. Changes in mechanosensitive channel gating following mechanical stimulation in skeletal muscle myotubes from the mdx mouse. J Physiol. 2002;539:391–407. doi: 10.1113/jphysiol.2001.013043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridén J, Sjöström M, Ekblom B. A morphological study of delayed muscle soreness. Experientia. 1981;37:506–507. doi: 10.1007/BF01986165. [DOI] [PubMed] [Google Scholar]
- Gilbert JR, Meissner G. Sodium-calcium ion exchange in skeletal muscle sarcolemmal vesicles. J Membr Biol. 1982;69:77–84. doi: 10.1007/BF01871244. [DOI] [PubMed] [Google Scholar]
- Gillis JM. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil. 1999;20:605–625. doi: 10.1023/a:1005545325254. [DOI] [PubMed] [Google Scholar]
- Godt RE, Maughan DW. On the composition of of the cytosol of relaxed skeletal muscle of the frog. Am J Physiol. 1988;254:C591–604. doi: 10.1152/ajpcell.1988.254.5.C591. [DOI] [PubMed] [Google Scholar]
- Head SI, Williams DA, Stephenson DG. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc R Soc Lond B Biol Sci. 1992;248:163–169. doi: 10.1098/rspb.1992.0058. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Horowicz P. The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J Physiol. 1959;148:127–160. doi: 10.1113/jphysiol.1959.sp006278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Hutter OF. The membrane hypothesis of Duchenne muscular dystrophy: quest for functional evidence. J Inherit Metab Dis. 1992;15:565–577. doi: 10.1007/BF01799615. [DOI] [PubMed] [Google Scholar]
- Ingalls CP, Warren GL, Williams JH, Ward CW, Armstrong RB. E-C coupling failure in mouse EDL muscle after in vivo eccentric contractions. J App Physiol. 1998;85:58–67. doi: 10.1152/jappl.1998.85.1.58. [DOI] [PubMed] [Google Scholar]
- Lacampagne A, Gannier F, Argibay J, Garnier D, Le Guennec JY. The stretch-activated ion channel blocker gadolinium also blocks L-type calcium channels in isolated ventricular myocytes of the guinea-pig. Biochim Biophys Acta. 1994;1191:205–208. doi: 10.1016/0005-2736(94)90250-x. [DOI] [PubMed] [Google Scholar]
- Lamb GD, Junankar PR, Stephenson DG. Raised intracellular [Ca2+] abolishes excitation-contraction coupling in skeletal muscle fibres of rat and toad. J Physiol. 1995;489:349–362. doi: 10.1113/jphysiol.1995.sp021056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansman JB, Franco A. What does dystrophin do in normal muscle. J Muscle Res Cell Motil. 1991;12:409–411. doi: 10.1007/BF01738325. [DOI] [PubMed] [Google Scholar]
- McArdle A, Edwards RH, Jackson MJ. How does dystrophin deficiency lead to muscle degeneration?-Evidence from the mdx mouse. Neuromuscul Disord. 1995;5:445–456. doi: 10.1016/0960-8966(95)00001-4. [DOI] [PubMed] [Google Scholar]
- McBride TA, Stockert BW, Gorin FA, Carlsen RC. Stretch-activated ion channels contribute to membrane depolarization after eccentric contractions. J App Physiol. 2000;88:91–101. doi: 10.1152/jappl.2000.88.1.91. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- Moens P, Baatsen PH, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Muscle Res Cell Motil. 1993;14:446–451. doi: 10.1007/BF00121296. [DOI] [PubMed] [Google Scholar]
- Morgan DL, Allen DG. Early events in stretch-induced muscle damage. J App Physiol. 1999;87:2007–2015. doi: 10.1152/jappl.1999.87.6.2007. [DOI] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan JG, Johnson SR, Mckee MK, Lyden SP. Twitch and tetanus in mdx mouse muscle. Muscle Nerve. 1992;15:837–842. doi: 10.1002/mus.880150713. [DOI] [PubMed] [Google Scholar]
- Sachs F. Mechanical transduction by membrane ion channels: a mini review. Mol Cell Biochem. 1991;104:57–60. doi: 10.1007/BF00229804. [DOI] [PubMed] [Google Scholar]
- Sigurdson W, Ruknudin A, Sachs F. Calcium imaging of mechanically induced fluxes in tissue-cultured chick heart: role of stretch-activated ion channels. Am J Physiol. 1992;262:H1110–1115. doi: 10.1152/ajpheart.1992.262.4.H1110. [DOI] [PubMed] [Google Scholar]
- Skuk D, Vilquin JT, Tremblay JP. Experimental and therapeutic approaches to muscular dystrophies. Curr Opin Neurol. 2002;15:563–569. doi: 10.1097/00019052-200210000-00007. [DOI] [PubMed] [Google Scholar]
- Sokabe M, Hasegawa N, Yamamori K. Blockers and activators for stretch-activated ion channels of chick skeletal muscle. Ann N Y Acad Sci. 1993;707:417–420. doi: 10.1111/j.1749-6632.1993.tb38086.x. [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Johnson JH, Hamer K, Leykam JF, Gage DA, Clemo HF, Baumgarten CM, Sachs F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J Gen Physiol. 2000;115:583–598. doi: 10.1085/jgp.115.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi S, Miyazaki T, Moritani K, Miyoshi S, Furukawa Y, Ito S, Ogawa S. Gadolinium suppresses stretch-induced increases in the differences in epicardial and endocardial monophasic action potential durations and ventricular arrhythmias in dogs. Jpn Circ J. 1999;63:296–302. doi: 10.1253/jcj.63.296. [DOI] [PubMed] [Google Scholar]
- Takekura H, Fujinami N, Nishizawa T, Ogasawara H, Kasuga N. Eccentric exercise-induced morphological changes in the membrane systems involved in excitation-contraction coupling in rat skeletal muscle. J Physiol. 2001;533:571–583. doi: 10.1111/j.1469-7793.2001.0571a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JA, Morgan DL. Quantitative analysis of sarcomere non-uniformities in active muscle following a stretch. J Muscle Res Cell Motil. 1996;17:261–268. doi: 10.1007/BF00124247. [DOI] [PubMed] [Google Scholar]
- Turner PR, Fong PY, Denetclaw WF, Steinhardt RA. Increased calcium influx in dystrophic muscle. J Cell Biol. 1991;115:1701–1712. doi: 10.1083/jcb.115.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PR, Westwood T, Regen CM, Steinhardt RA. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- Tutdibi O, Brinkmeier H, Rudel R, Fohr KJ. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J Physiol. 1999;515:859–868. doi: 10.1111/j.1469-7793.1999.859ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandebrouck C, Martin D, Colson-van Schoor M, Debaix H, Gailly P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol. 2002;158:1089–1096. doi: 10.1083/jcb.200203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren GL, Ingalls CP, Lowe DA, Armstrong RB. Excitation-contraction uncoupling: major role in contraction-induced muscle injury. Exercise Sport Sci Rev. 2001;29:82–87. doi: 10.1097/00003677-200104000-00008. [DOI] [PubMed] [Google Scholar]
- Warren GL, Lowe DA, Hayes DA, Karwoski CJ, Prior BM, Armstrong RB. Excitation failure in eccentric contraction-induced injury of mouse soleus muscle. J Physiol. 1993;468:487–499. doi: 10.1113/jphysiol.1993.sp019783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SA, Morgan DL, Proske U. Effects of repeated eccentric contractions on structure and mechanical properties of toad sartorius muscle. Am J Physiol. 1993;265:C792–800. doi: 10.1152/ajpcell.1993.265.3.C792. [DOI] [PubMed] [Google Scholar]
- Yang XC, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science. 1989;243:1068–1071. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]
- Yeung EW, Ballard HJ, Bourreau JP, Allen DG. Intracellular sodium in mammalian muscle fibers following eccentric contractions. J App Physiol. 2003;94:2475–2482. doi: 10.1152/japplphysiol.01128.2002. [DOI] [PubMed] [Google Scholar]
- Yeung EW, Balnave CD, Ballard HJ, Bourreau JP, Allen DG. Development of T-tubular vacuoles in eccentrically damaged mouse muscle fibres. J Physiol. 2002a;540:581–592. doi: 10.1113/jphysiol.2001.013839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung EW, Bourreau JP, Allen DG, Ballard HJ. The effect of eccentric contraction-induced injury on force and intracellular pH in rat skeletal muscles. J App Physiol. 2002b;92:93–99. doi: 10.1152/jappl.2002.92.1.93. [DOI] [PubMed] [Google Scholar]