

Abstract
The reduction of oxygen to water proceeds via one electron at a time. In the mitochondrial respiratory chain, Complex IV (cytochrome oxidase) retains all partially reduced intermediates until full reduction is achieved. Other redox centres in the electron transport chain, however, may leak electrons to oxygen, partially reducing this molecule to superoxide anion (O2−•). Even though O2−• is not a strong oxidant, it is a precursor of most other reactive oxygen species, and it also becomes involved in the propagation of oxidative chain reactions. Despite the presence of various antioxidant defences, the mitochondrion appears to be the main intracellular source of these oxidants. This review describes the main mitochondrial sources of reactive species and the antioxidant defences that evolved to prevent oxidative damage in all the mitochondrial compartments. We also discuss various physiological and pathological scenarios resulting from an increased steady state concentration of mitochondrial oxidants.
The chemistry of oxygen and oxidative stress
Reactive oxygen species (ROS) is a phrase used to describe a variety of molecules and free radicals (chemical species with one unpaired electron) derived from molecular oxygen. Molecular oxygen in the ground state is a bi-radical, containing two unpaired electrons in the outer shell (also known as a triplet state). Since the two single electrons have the same spin, oxygen can only react with one electron at a time and therefore it is not very reactive with the two electrons in a chemical bond. On the other hand, if one of the two unpaired electrons is excited and changes its spin, the resulting species (known as singlet oxygen) becomes a powerful oxidant as the two electrons with opposing spins can quickly react with other pairs of electrons, especially double bonds.
The reduction of oxygen by one electron at a time produces relatively stable intermediates. Superoxide anion (O2−•), the product of a one-electron reduction of oxygen, is the precursor of most ROS and a mediator in oxidative chain reactions. Dismutation of O2−• (either spontaneously or through a reaction catalysed by superoxide dismutases) produces hydrogen peroxide (H2O2), which in turn may be fully reduced to water or partially reduced to hydroxyl radical (OH•), one of the strongest oxidants in nature. The formation of OH• is catalysed by reduced transition metals, which in turn may be re-reduced by O2−•, propagating this process (Liochev & Fridovich, 1999). In addition, O2−• may react with other radicals including nitric oxide (NO•) in a reaction controlled by the rate of diffusion of both radicals. The product, peroxynitrite, is also a very powerful oxidant (Beckman & Koppenol, 1996; Radi et al. 2002b; Fig. 1). The oxidants derived from NO• have been recently called reactive nitrogen species (RNS).
Figure 1. Sites of superoxide formation in the respiratory chain.
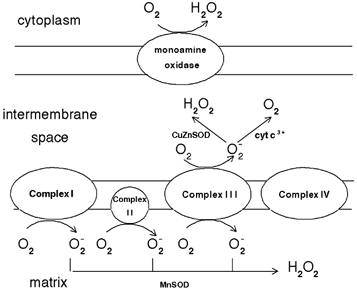
Various respiratory complexes leak electrons to oxygen producing primarily superoxide anion (O2−•). This species may reduce cytochrome c (in the intermembrane space), or may be converted to hydrogen peroxide (H2O2) and oxygen (in both the matrix and the intermembrane space). Increased steady state concentrations of O2−• may reduce transition metals (which in turn react with H2O2 producing hydroxyl radicals (OH•)) or may react with nitric oxide to form peroxynitrite. Both OH• and peroxynitrite are strong oxidants which indiscriminately react with nucleic acids lipids and proteins.
‘Oxidative stress’ is an expression used to describe various deleterious processes resulting from an imbalance between the excessive formation of ROS and/or RNS and limited antioxidant defences (Fig. 1). Whilst small fluctuations in the steady-state concentration of these oxidants may actually play a role in intracellular signalling (Droge, 2002), uncontrolled increases in the steady-state concentrations of these oxidants lead to free radical-mediated chain reactions which indiscriminately target proteins (Stadtman & Levine, 2000), lipids (Rubbo et al. 1994), polysaccharides (Kaur & Halliwell, 1994) and DNA (Richter et al. 1988; LeDoux et al. 1999).
In vivo, O2−• is produced both enzymatically and non-enzymatically. Enzymatic sources include NADPH oxidases located on the cell membrane of polymorphonuclear cells, macrophages and endothelial cells (Babior, 2000; Vignais, 2002; Babior et al. 2002) and cytochrome P450-dependent oxygenases (Coon et al. 1992). The proteolytic conversion of xanthine dehydrogenase to xanthine oxidase provides another enzymatic source of both O2−• and H2O2 (and therefore constitutes a source of OH•) and has been proposed to mediate deleterious processes in vivo (Yokoyama et al. 1990).
The non-enzymatic production of O2−• occurs when a single electron is directly transferred to oxygen by reduced coenzymes or prosthetic groups (for example, flavins or iron sulfur clusters) or by xenobiotics previously reduced by certain enzymes (for example, the anticancer agent adriamycin or the herbicide paraquat). The mitochondrial electron transport chain contains several redox centres that may leak electrons to oxygen, constituting the primary source of O2−• in most tissues.
Detection of ROS and RNS
The formation of ROS and RNS may be monitored using a variety of procedures including fluorometric and spectrophotometric methods, chemiluminescence and electron paramagnetic resonance (Chance et al. 1979; Pou et al. 1989; Tarpey & Fridovich, 2001).
Many of these methods rely on the redox properties of specific ROS or RNS, and therefore are prone to artifacts caused by species of similar reactivity or by reactive intermediates produced by the probe itself (Picker & Fridovich, 1984; Faulkner & Fridovich, 1993; Liochev & Fridovich, 1995, 1998).
Specific inhibitory enzymes may be added to unequivocally identify the species (for example, superoxide dismutase or catalase to eliminate O2−• or H2O2, respectively) but these enzymes do not determine whether these ROS are the primary species or just intermediates formed in the detection reaction. For example, a research group concluded that O2−• was produced by the enzyme glucose oxidase because superoxide dismutase inhibited the reduction of nitroblue tetrazolium in the presence of glucose, when in reality the probe was reacting with the enzyme's prosthetic group and O2−• was formed after this reaction (Al-Bekairi et al. 1994; Liochev & Fridovich, 1995). Similarly, luminol and lucigenin may also produce O2−• during their oxidation, leading to the false conclusion that O2−• was actually formed in the process (Liochev & Fridovich, 1998). When possible, it is recommended to use more than one method to identify specific ROS, particularly if they are based on different properties of a given species. For example, O2−• may oxidize certain probes (adrenaline (epinephrine) and hydroethidine) and also reduce others (cytochrome c or nitroblue tetrazolium; Butler et al. 1975). The choice of methods may also vary depending on the source of ROS under study. For example, if one is monitoring O2−• by the respiratory chain, cytochrome c and nitroblue tetrazolium are usually not good choices because they react with other carriers in the electron transport chain.
Mitochondrial sources of superoxide and nitric oxide
The standard reduction potential for the conversion of molecular oxygen to O2−• is -0.160 V (Wood, 1987). The respiratory chain includes a variety of redox centres with standard reduction potentials between -0.32 V (NAD(P)H) and +0.39 V (cytochrome a3 in Complex IV). Given the highly reducing intramitochondrial environment, various respiratory components, including flavoproteins, iron-sulfur clusters and ubisemiquinone, are thermodynamically capable of transferring one electron to oxygen. Moreover, most steps in the respiratory chain involve single-electron reactions, further favouring the monovalent reduction of oxygen. On the other hand, the mitochondrion possesses various antioxidant defences designed to eliminate both O2−• and H2O2 (see below). As a result, the steady state concentrations of O2−• and H2O2 have been estimated to be around 10−10 M and 5 × 10−9 M, respectively (Cadenas & Davies, 2000).
Over the past 35 years several laboratories have identified a variety of mitochondrial sources of O2−• including several respiratory complexes and individual enzymes. Superoxide formation occurs on the outer mitochondrial membrane, in the matrix and on both sides of the inner mitochondrial membrane (Table 1, Fig. 2). Whilst the O2−• generated in the matrix is eliminated in that compartment, part of the O2−• produced in the intermembrane space may be carried to the cytoplasm via voltage-dependent anion channels (Han et al. 2003).
Table 1.
Compartmental localization of the main mitochondrial source of superoxide anion
| Component | Localization | References |
|---|---|---|
| Complex I | Inner membrane/ | (Turrens & Boveris, 1980; Turrens et al. 1982; |
| (NADH dehydrogenase) | inner side | Genova et al. 2001; Kushnareva et al. 2002) |
| Complex II | Inner membrane/ | (Zhang et al. 1998; Lenaz, 2001) |
| (succinate dehydrogenase) | inner side | |
| Complex III | Inner membrane/ | (Boveris et al. 1976; Cadenas et al. 1977; |
| (ubiquinol-cytochrome c reductase) | inner side | Turrens et al. 1985) |
| Complex III | Inner membrane/ | (Han et al. 2001; Starkov & Fiskum, 2001) |
| (ubiquinol-cytochrome c reductase) | outer side | |
| External NADH dehydrogenase | Inner membrane/ | (Fang & Beattie, 2003) |
| (yeast) | outer side | |
| Glycerolphosphate dehydrogenase | Inner membrane/ | (Drahota et al. 2002) |
| outer side | ||
| Dehydroorotate dehydrogenase | Matrix | (Forman & Kennedy, 1976) |
| Mono amino oxidase | Outer membrane/ | (Hauptmann et al. 1996; Cadenas & Davies, 2000) |
| inner side |
Figure 2. Mechanism of electron flow in the Q-cycle in Complex III.
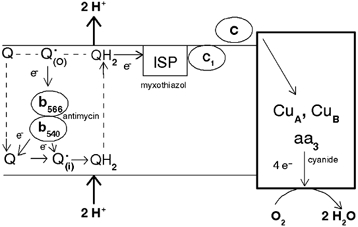
Ubiquinone is reduced either by Complex I or II or by electrons transferred from cytochrome b on the inner side of the membrane (i), producing ubiquinol (QH2). The reduced form transfers one electron to oxygen via the Rieske iron-sulfur protein (ISP), cytochrome c1 and cytochrome c. The terminal oxidase is reduced by four electrons (two by copper atoms and two by cytochromes) which in turn are transferred to oxygen producing water. Cytochrome b is reduced by the ubisemiquinone formed on the outer side (Q•(o)), but this species cannot be formed unless an electron is previously transferred by QH2 to ISP. Superoxide may be produced on both sides of the inner membrane via the autoxidation of Q but the contribution of each pool has not yet been determined.
The relative contribution of every site to the overall O2−• production varies from organ to organ and also depends on whether mitochondria are actively respiring (State 3) or the respiratory chain is highly reduced (State 4) (Barja, 1999). Although Complex III appears to be responsible for most of the O2−• produced in heart and lung mitochondria (Turrens & Boveris, 1980; Turrens et al. 1982), O2−• formation by Complex I appears to be the primary source of O2−• in the brain under normal conditions (Barja & Herrero, 1998; Barja, 1999). Moreover, Complex I is the primary source of ROS in a variety of pathological scenarios ranging from ageing to Parkinson's disease (see below) (Barja & Herrero, 1998; Betarbet et al. 2002; Kushnareva et al. 2002; Nicholls, 2002; Sherer et al. 2003a,b;Trojanowski, 2003).
The rate of O2−• formation by the respiratory chain is controlled primarily by mass action, increasing both when electron flow slows down (increasing the concentration of electron donors, R•) and when the concentration of oxygen increases (eqn (1); Turrens et al. 1982).
| (1) |
The energy released as electrons flow through the respiratory chain is converted into a H+ gradient through the inner mitochondrial membrane (Mitchell, 1977). This gradient, in turn, dissipates through the ATP synthase complex (Complex V) and is responsible for the turning of a rotor-like protein complex required for ATP synthesis (Noji & Yoshida, 2001). In the absence of ADP, the movement of H+ through ATP synthase ceases and the H+ gradient builds up causing electron flow to slow down and the respiratory chain to become more reduced (State IV respiration). As a result, the physiological steady state concentration of O2−• formation increases (Boveris et al. 1972). The formation of O2−• may be further increased in the presence of certain inhibitors (for example rotenone, which inhibits Complex I, or antimycin, an inhibitor of Complex III), which cause those carriers upstream from the site of inhibition to become fully reduced. In Complex I, the primary source of O2−• appears to be one of the iron-sulfur clusters (either N-1 α or N-2; Genova et al. 2001; Kushnareva et al. 2002). In Complex III, most of the O2−• appears to be formed as a result of the autoxidation of ubisemiquinone both on the outer and inner sides of the inner mitochondrial membrane (Table 1).
Although O2−• production increases as the respiratory chain becomes more reduced, not all mitochondrial inhibitors have this effect. Most of the production of O2−• by Complex III is actually inhibited if electron flow between the Rieske Fe-S protein and oxygen is blocked (for example, by myxothiazol, cyanide or cytochrome c depletion; Fig. 2; Turrens et al. 1985). This inhibitory effect indicates that O2−• must be produced as a result of the autoxidation of ubisemiquinone (Q•), an intermediate produced in Complex III during the Q-cycle (Trumpower, 1990; Fig. 2). The first experimental evidence for the Q-cycle came in 1974, when Chance and co-workers reported that addition of oxygen to anaerobic mitochondria caused a transient reduction of cytochrome b, instead of the expected oxidation. This puzzling result became known as the ‘oxidant-induced reduction of cytochrome b‘. According to this model, coenzyme Q is fully reduced in the inner side of the mitochondrial membrane (ubiquinol, QH2) and then migrates to the outer side of the inner membrane carrying 2 H+ that become part of the pool needed to sustain ADP phosphorylation. Once on the outer side of the membrane, one electron is transferred to cytochrome c1 (via the Rieske Fe-S protein), resulting in the formation of Q•. The second electron is needed to reduce cytochrome b, but eventually some electrons leak to oxygen, producing O2−•. Under normal conditions, since cytochrome b cannot be reduced unless an electron is transferred to oxygen, an oxidant is needed for the reduction of this cytochrome. As a result of this process, antimycin, by binding to cytochrome b, increases the steady state concentration of Q•, also increasing O2−• formation (Fig. 2). Myxothiazol (by inhibiting the Rieske Fe-S protein) and cyanide or cytochrome c depletion (by preventing the electrons from reaching molecular oxygen), inhibit the increased O2−• production observed in the presence of antimycin (Turrens et al. 1985). Yet, the inhibition by myxothiazol is not complete, suggesting that there are additional sites in Complex III responsible for O2−• formation (Starkov & Fiskum, 2001;Young et al. 2002; Fig. 2).
The simultaneous formation of O2−• and nitric oxide produces peroxynitrite, a very strong oxidant and nitrating agent. Nitric oxide is a vasodilator resulting from the breakdown of arginine to citrulline, in a reaction catalysed by a family of NADPH-dependent enzymes called nitric oxide synthases. Two separate laboratories have recently discovered that the mitochondrial matrix contains a unique form of nitric oxide synthase (Ghafourifar & Richter, 1997; Giulivi et al. 1998; Alvarez et al. 2003). Although its physiological role is still unclear, the formation of nitric oxide in mitochondria may have important consequences because this compound binds to haem groups from cytochromes (in particular cytochrome oxidase) and inhibits respiration (Poderoso et al. 1996) This may, in turn, stimulate O2−• formation (for example from Complex I; Poderoso et al. 1996), which in turn may react with more nitric oxide forming peroxynitrite, an oxidant capable of inhibiting important enzymes and affecting mitochondrial integrity (Cassina & Radi, 1996; Radi et al. 2002a).
Since NO• formation requires oxygen, the rate at which it is produced varies with the intramitochondrial oxygen concentration (Alvarez et al. 2003). Assuming an intramitochondrial oxygen concentration of 20 μM, Alvarez et al. recently estimated that the intramitochondrial steady-state concentrations of NO• and peroxynitrite in the liver are around 36 nM and 2.2 nM, respectively (Alvarez et al. 2003). Other investigators (Wittenberg & Wittenberg, 1989) have estimated that the intramitochondrial oxygen concentration is considerably lower (3 μM), which would imply lower NO• and peroxynitrite steady-state concentrations.
Mitochondrial antioxidant defences
The deleterious effects resulting from the formation of ROS in the mitochondrion are, to a large extent, prevented by various antioxidant systems. Superoxide is enzymatically converted to H2O2 by a family of metalloenzymes called superoxide dismutases (SOD; Fridovich, 1995). Since O2−• may either reduce transition metals, which in turn can react with H2O2 producing OH• or spontaneously react with NO• to produce peroxynitrite, it is important to maintain the steady-state concentration of O2−• at the lowest possible level. Thus, although the dismutation of O2−• to H2O2 and O2 can also occur spontaneously, the role of SODs is to increase the rate of the reaction to that of a diffusion-controlled process.
The mitochondrial matrix contains a specific form of SOD, with manganese in the active site (Fridovich, 1995), which eliminates the O2−• formed in the matrix or on the inner side of the inner membrane. The expression of MnSOD is further induced by agents that cause oxidative stress, including radiation and hyperoxia, in a process mediated by the oxidative activation of the nuclear transcription factor NFκB (Oberley et al. 1987; Das et al. 1995; Warner et al. 1996; Tsan et al. 2001; Murley et al. 2001).
The steady-state concentration of O2−• in the intermembrane space is controlled by three different mechanisms. Firstly, this compartment contains a different SOD isozyme which contains copper and zinc instead of manganese (CuZnSOD (Okado-Matsumoto & Fridovich, 2001)) and is also found in the cytoplasm of eukaryotic cells. Secondly, the intermembrane space contains cytochrome c which can be reduced by O2−• (k = 107 M−1 s−1, Butler et al. 1975) regenerating oxygen in the process (Fig. 2). The reduced cytochrome c can then transfer electrons to the terminal oxidase. Thus, some of the electrons that escaped the respiratory chain producing O2−• may re-reduce cytochrome c and still contribute to energy production by providing the energy needed to pump H+ through Complex IV. Finally, the spontaneous dismutation of O2−• in the intermembrane space is facilitated by the lower pH in this compartment, resulting from the extrusion of H+ coupled to respiration (Guidot et al. 1995).
Hydrogen peroxide, the product of O2−• dismutation and the main precursor of OH• in the presence of reduced transition metals, is mostly decomposed by the enzyme glutathione peroxidase. In the liver, mitochondria account for about one third of the total glutathione peroxidase activity (Chance et al. 1979). A second glutathione peroxidase associated with the mitochondrial membrane, known as phospholipid-hydroperoxide glutathione peroxidase, is specifically involved in reducing lipid peroxides associated with the membrane(Ursini et al. 1999; Nomura et al. 2000).
Catalase, a major H2O2 detoxifying enzyme found in peroxisomes, is also present in heart mitochondria (Radi et al. 1991). However, this enzyme has not been found in mitochondria from other tissues, including skeletal muscle (Phung et al. 1994).
In addition to cytochrome c, other electron carriers appear to have a detoxifying role against ROS. Ubiquinol (QH2) has been shown to act as a reducing agent in the elimination of various peroxides in the presence of succinate (Bindoli et al. 1982; Eto et al. 1992). Thus, coenzyme Q is a source of O2−• when partially reduced (semiquinone form) and an antioxidant when fully reduced (Beyer, 1990). The inner mitochondrial membrane also contains vitamin E, a powerful antioxidant that interferes with the propagation of free radical-mediated chain reactions (Ham & Liebler, 1995).
Finally, cytochrome c oxidase (Complex IV) may also act as a peroxidase although, given the high Km for H2O2 (0.18 mM), the relevance of this reaction may be negligible (Orii, 1982).
In addition to the antioxidant defences mentioned above, the mitochondrion has a variety of DNA-repairing enzymes to correct errors resulting from oxidative damage. This is very important because, although 95 % of the mitochondrial proteins are encoded by the nuclear DNA, the mitochondrial chromosome (mtDNA) contains genes for several important proteins including subunits of NADH dehydrogenase and cytochrome oxidase and cytochrome b.
Although under normal conditions there is a balance between ROS formation and antioxidants, in several pathological scenarios the antioxidant defences become insufficient resulting in oxidative stress leading often to apoptosis and cell death. Apoptosis (or programmed cell death) is the mechanism used by mammals, plants and other organisms to eliminate redundant or damaged cells (Kroemer et al. 1998; Hoeberichts & Woltering, 2003). Apoptosis may be triggered by extracellular signals (extrinsic pathway) or by intracellular processes (intrinsic pathway). An increased mitochondrial formation of ROS triggers the intrinsic pathway by increasing the permeability of the outer mitochondrial membrane through the opening of transition pores. The opening of the permeability transition pore is favoured by oxidative stress through oxidation of intracellular glutathione and other critical sulfhydryl groups (Chernyak, 1997). As a result of this process, cytochrome c moves from the intermembrane space into the cell's cytoplasm (Liu et al. 1996) where it joins another factor (Apaf-1). In the presence of dATP this complex polymerizes into an oligomer known as ‘apoptosome’. The apoptosome activates a protease (caspase-9), which in turn activates caspase-3. The cascade of proteolytic reactions also activates DNAses and in the end the process results in cell death (Li et al. 1997). Under normal conditions, various anti-apoptotic factors (including Bcl-xL) prevent the mitochondrial permeability transition as long as they remain bound to the outer membrane. This factor is eliminated when another factor, Bax, is translocated to mitochondria, starting apoptosis (Finucane et al. 1999).
The gradual loss of cytochrome c from the intermembrane space during apoptosis favours the mitochondrial formation of O2−• in two ways: (1) cytochrome c is a scavenger of O2−• and (2) as cytochrome c is released, the respiratory chain becomes more reduced because electron flow between Complex III and Complex IV slows down (Cai & Jones, 1998).
Mitochondrial ROS formation during hyperoxia and during hypoxia
As predicted from eqn (1), the mitochondrial production of O2−• must increase with oxygen concentration. The proportion of oxygen converted into O2−• in vitro ([O2] = 220 μM) accounts for about 1-2 % of the overall oxygen consumption. In vivo, particularly in tissues not exposed to atmospheric oxygen, the proportion of oxygen converted O2−• is likely to be smaller since the intramitochondrial oxygen concentration is between 3 and 30 μM (Wittenberg & Wittenberg, 1989; Genova et al. 2001; Alvarez et al. 2003).
As oxygen concentration increases, the rate of mitochondrial O2−• production increases linearly (Turrens et al. 1982). However, the release of H2O2 from mitochondria is biphasic, increasing at a faster rate above 60 % O2 (Turrens et al. 1985). The slower release of H2O2 at lower PO2 suggests that the mitochondrial antioxidant defences can compensate for sudden increases in the concentration of this peroxide. Apparently these defences become overwhelmed at higher PO2, which explains the mitochondrial alterations observed in the lungs of animals exposed to oxygen concentrations around 60 % or higher (Crapo et al. 1983).
Under normobaric hyperoxic conditions, the only organs affected by ROS formation are the lungs, since they the only ones in direct contact with atmospheric oxygen. However, under hyperbaric conditions, more oxygen is dissolved in the plasma, and therefore other tissues become exposed to a hyperoxic environment. Under these conditions, the brain is the first organ to show the effects of an increased ROS formation, resulting in convulsions. Interestingly, this phenomenon was proposed to be associated with oxygen free radicals 15 years before the discovery of O2−• as a normal intracellular metabolite (Gerschman et al. 1954).
The formation of ROS should decrease with hypoxia, since this activity is proportional to ROS (eqn (1)). Yet, various groups have reported a paradoxical increase in oxidative stress under moderately hypoxic conditions (1.5 % O2, equivalent to an oxygen concentration of around 16 μM; Waypa & Schumacker, 2002; Schumacker, 2002). These studies show that when cells are incubated with dichlorofluorescein, a fluorescent probe for ROS, hypoxia increases fluorescence in cells with functioning mitochondria (Chandel et al. 2001). Mutants without a functioning respiratory chain do not show this increase in fluorescence (Chandel et al. 2001; Schroedl et al. 2003). This response is eliminated when cells are made severely hypoxic (Schumacker, 2002).
It has been proposed that the increase in ROS formation during hypoxia may modulate the activation of one or more hypoxia-inducible factors (HIF; Chandel et al. 2001), a group of proteins that regulate the expression of genes involved in the adaptation to hypoxic conditions (Semenza, 2002). In the presence of oxygen, proline residues 402 and 564 of HIF-1 α become hydroxylated, which eventually leads to ubiquitination and degradation of this factor (Semenza, 2002), thus preventing the induction of a hypoxic stress response. Schroedl et al. suggested that the stabilization of HIF under hypoxic conditions requires mitochondrial ROS formation, as this process is lost in cells without a functioning respiratory chain (Schroedl et al. 2003). They proposed that the mitochondrial formation of ROS is required to stabilize HIF-1 α under hypoxic conditions while the proline hydroxylase activity would be involved in an ‘on or off’ type of response under anoxic conditions (Schroedl et al. 2003).
The proposed increase in ROS formation during hypoxia is difficult to explain. Given the high affinity of cytochrome oxidase for oxygen, at low PO2 any remaining oxygen should be reduced to water by the terminal oxidase. Two factors may contribute to an increase mitochondrial O2−• formation. Firstly, under hypoxic conditions, low concentrations of NO• may still be produced (5-10 % of the normal steady state) since the Km for oxygen of the mitochondrial nitric oxide synthase is around 30-40 μM (Alvarez et al. 2003). Secondly, nitric oxide may bind and inhibit cytochrome oxidase, resulting in an increase in its Km for oxygen and an increased reduction of electron carriers located upstream from the terminal oxidase (Cooper & Davies, 2000), favouring O2−• formation at low oxygen concentrations. Still more research is needed to clarify the extent of ROS formation in the response of tissues to hypoxia.
Mitochondrial ROS formation resulting from exposure to xenobiotics
Several xenobiotics interact with the mitochondrial electron transport chain, increasing the rate of O2−• production through two different mechanisms. Some of these compounds stimulate oxidative stress because they block electron transport, increasing the reduction level of carriers located upstream of the inhibition site. Other xenobiotics may accept an electron from a respiratory carrier and transfer it to molecular oxygen (redox cycling), stimulating O2−• formation without inhibiting the respiratory chain.
It has been proposed that Parkinson's disease may result from exposure to sublethal concentrations of inhibitors of Complex I. The current hypothesis is that these inhibitors stimulate O2−• formation by Complex I, causing the cells to undergo apoptosis (Sherer et al. 2003a,b). Compounds such as MPP+ (1-methyl-4-pyridinium), a metabolite of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a toxic ‘designer drug’ from the 1980s which causes a Parkinson's-like syndrome, block electron flow through Complex I (Trojanowski, 2003). Moreover, exposure of animals to rotenone is accompanied by formation of deposits of α-synuclein similar to those observed in various related brain amyloidoses, suggesting that inhibition of Complex I may have an important role in other neuropathies (Trojanowski, 2003).
The antitumour agent adriamycin (doxorubicin) and other anthracyclines constitute examples of redox cycling xenobiotics. These compounds accept electrons directly from Complex I, causing myocardial toxicity through an increase in the steady-state concentration of O2−• (Jung & Reszka, 2001).
Changes in mitochondrial antioxidant defences: apoptosis and ageing
Oxidative stress may also result from deficiencies in antioxidant defences. Both genetic factors and ageing may cause an increased mitochondrial steady-state concentration of ROS.
The first example involves the mitochondrial form of SOD. The gene for MnSOD (also known as SOD2) is located in the nuclear DNA and the enzyme must be transported into the matrix after translation, where it forms the active homotetramer (Fridovich, 1995; Sutton et al. 2003). The efficiency of MnSOD transport depends upon the mitochondrial targeting sequence (MTS), a signal polypeptide that causes the protein to be imported into the matrix. The sequence for this MTS presents a genetic dimorphism, in which the amino acid alanine in the ninth position is replaced by valine. In this case the tertiary structure of the MTS is altered and the change in tertiary structure interferes with MnSOD translocation into the matrix, resulting in a 40 % lower SOD activity in this compartment (Sutton et al. 2003). At least two reports have correlated this mutation with an increased incidence of Parkinson's disease, stressing the correlation between oxidative stress and this disease (Shimoda-Matsubayashi et al. 1996; Grasbon-Frodl et al. 1999).
Amyotrophic lateral sclerosis (ALS) is another neurological disease that has been associated with oxidative stress. About 10 % of the cases of familial ALS have been linked to a mutation in the gene coding for CuZnSOD (Valentine & Hart, 2003). This mutation causes misfolding, and also prevents the effective import of this enzyme to the inter-membrane space, thus increasing the steady-state concentration of O2−• in this compartment, a process that may lead to apoptosis (Okado-Matsumoto & Fridovich, 2002).
Another genetic mutation indirectly associated with increased formation of ROS is the mutation in one of the subunits of Complex I responsible for Leber hereditary optic neuropathy that causes neuronal apoptosis. In a recent article, the investigators manipulated the expression of MnSOD in order to increase the intramitochondrial steady state of O2−• in normal cells, resulting in the same histopathological changes observed in Leber's disease (Qi et al. 2003).
Although mitochondria express a variety of protective defences (antioxidant and repair enzymes as well as low molecular weight antioxidants), it has been proposed that the oxidation of proteins and the slow accumulation of DNA lesions resulting from the continuous formation of ROS may contribute to the ageing process (Sohal et al. 1993; Ames et al. 1995). Some of these lesions affect the rate of electron flow and lead to an increased formation of ROS, which supports the observed correlation between the rate of mitochondrial O2−• and H2O2 formation and lifespan among several species (Lopez-Torres et al. 1993; Ku et al. 1993).
The ageing process has also been associated with deficiencies in the mitochondrial DNA repair system. The mitochondrial DNA does not contain histones, and therefore is less protected against oxidative stress than the nuclear DNA. As a result the mitochondrial DNA shows a 10- to 20-fold increase in the content of 8-hydroxyguanine, the product of guanine oxidation (McCord & Fridovich, 1988). Since the mitochondrial chromosome codes for some electron carriers, mtDNA damage may indirectly inhibit respiration and stimulate ROS formation. Cockayne syndrome, a human condition that causes premature ageing, has been associated with a deficiency in the mitochondrial enzyme required for DNA repair that catalyses the removal of 8-hydroxyguanine (Bohr et al. 1998).
Other studies have shown a more direct correlation between oxidative stress and ageing. First, there is a correlation between accumulation of oxidized proteins and lifespan, further linking oxidative stress with ageing (Sohal et al. 1993). In a more direct approach, another study showed that overexpression of catalase and SOD results in a 25 % increase in the lifespan of Drosophila melanogaster (Orr & Sohal, 1994).
The sex-related differences in lifespan across species, with females usually living longer than males, also correlate with differences in antioxidant defences. This difference appears to be associated with the production of oestrogens, since it is not observed in ovariectomized animals (Asdell et al. 1967). Although the mechanism behind this effect has not yet been elucidated, part of the oestrogen-dependent protection is associated with an increased mitochondrial concentration of glutathione and glutathione peroxidase activity (Borras et al. 2003).
Conclusion
The mitochondrial respiratory chain constitutes the main intracellular source of ROS in most tissues. The steady-state concentration of these oxidants is maintained at non-toxic levels by a variety of antioxidant defences and repair enzymes. The delicate balance between antioxidant defences and ROS production may be disrupted by either deficient antioxidant defences, inhibition of electron flow or exposure to xenobiotics. This imbalance appears as a common denominator in various pathological processes in which the resulting oxidative insult causes tissue damage and, eventually, cell death.
REFERENCES
- Al Bekairi AM, Nagi MN, Shoeb HA, Al Sawaf HA. Evidence for superoxide radical production by a simple flavoprotein: glucose oxidase. Biochem Mol Biol Int. 1994;34:233–238. [PubMed] [Google Scholar]
- Alvarez S, Valdez LB, Zaobornyj T, Boveris A. Oxygen dependence of mitochondrial nitric oxide synthase activity. Biochem Biophys Res Commun. 2003;305:771–775. doi: 10.1016/s0006-291x(03)00818-0. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Mitochondrial decay in aging. Biochim Biophys Acta. 1995;1271:165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- Asdell SA, Doornenbal H, Joshi SR, Sperling GA. The effects of sex steroid hormones upon longevity in rats. J Reprod Fertil. 1967;14:113–120. doi: 10.1530/jrf.0.0140113. [DOI] [PubMed] [Google Scholar]
- Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life. 2000;50:267–269. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- Barja G, Herrero AJ. Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J Bioenerg Biomembr. 1998;30:235–243. doi: 10.1023/a:1020592719405. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide superoxide and peroxynitrite: the good the bad and the ugly. Am J Physiol. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson's disease. Bioessays. 2002;24:308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- Beyer RE. The participation of coenzyme Q in free radical production and antioxidation. Free Radic Biol Med. 1990;8:545–565. doi: 10.1016/0891-5849(90)90154-b. [DOI] [PubMed] [Google Scholar]
- Bindoli A, Cavallini L, Jocelyn P. Mitochondrial lipid peroxidation by cumene hydroperoxide and its prevention by succinate. Biochim Biophys Acta. 1982;681:496–503. doi: 10.1016/0005-2728(82)90192-x. [DOI] [PubMed] [Google Scholar]
- Bohr V, Anson RM, Mazur S, Dianov G. Oxidative DNA damage processing and changes with aging. Toxicol Lett. 1998:102–103. 47–52. doi: 10.1016/s0378-4274(98)00280-x. [DOI] [PubMed] [Google Scholar]
- Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med. 2003;34:546–552. doi: 10.1016/s0891-5849(02)01356-4. [DOI] [PubMed] [Google Scholar]
- Boveris A, Cadenas E, Stoppani AOM. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J. 1976;156:435–444. doi: 10.1042/bj1560435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J, Jayson GG, Swallow AJ. The reaction between the superoxide anion radical and cytochrome c. Biochim Biophys Acta. 1975;408:215–222. doi: 10.1016/0005-2728(75)90124-3. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Boveris A, Ragan CI, Stoppani AOM. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef heart mitochondria. Arch Biochem Biophys. 1977;180:248–257. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation oxidative stress and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Cai JY, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Mathieu M, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 2001;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyak BV. Redox regulation of the mitochondrial permeability transition pore. Biosci Rep. 1997;17:293–302. doi: 10.1023/a:1027384628678. [DOI] [PubMed] [Google Scholar]
- Coon MJ, Ding X, Pernecky SJ, Vaz ADN. Cytochrome P450: Progress and predictions. FASEB J. 1992;6:669–673. doi: 10.1096/fasebj.6.2.1537454. [DOI] [PubMed] [Google Scholar]
- Cooper CE, Davies NA. Effects of nitric oxide and peroxynitrite on the cytochrome oxidase K(m) for oxygen: implications for mitochondrial pathology. Biochim Biophys Acta. 2000;1459:390–396. doi: 10.1016/s0005-2728(00)00176-6. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Freeman BA, Barry BE, Turrens JF, Young SL. Mechanisms of hyperoxic injury to the pulmonary microcirculation. Physiologist. 1983;26:170–176. [PubMed] [Google Scholar]
- Das KC, Lewis-Molock Y, White CW. Thiol modulation of TNFa and Il−1 induced MnSOD gene expression and activation of NF-kappaB. Mol Cell Biochem. 1995;148:45–57. doi: 10.1007/BF00929502. [DOI] [PubMed] [Google Scholar]
- Drahota Z, Chowdhury SK, Floryk D, Mracek T, Wilhelm J, Rauchova H, Lenaz G, Houstek J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J Bioenerg Biomembr. 2002;34:105–113. doi: 10.1023/a:1015123908918. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Eto Y, Kang D, Hasegawa E, Takeshige K, Minakami S. Succinate-dependent lipid peroxidation and its prevention by reduced ubiquinone in beef heart submitochondrial particles. Arch Biochem Biophys. 1992;295:101–106. doi: 10.1016/0003-9861(92)90493-g. [DOI] [PubMed] [Google Scholar]
- Fang J, Beattie DS. External alternative NADH dehydrogenase of Saccharomyces cerevisiae: a potential source of superoxide. Free Radic Biol Med. 2003;34:478–488. doi: 10.1016/s0891-5849(02)01328-x. [DOI] [PubMed] [Google Scholar]
- Faulkner K, Fridovich I. Luminol and lucigenin as detectors for O2−. Free Radic Biol Med. 1993;15:447–451. doi: 10.1016/0891-5849(93)90044-u. [DOI] [PubMed] [Google Scholar]
- Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Kennedy J. Dihydroorotate-dependent superoxide production in rat brain and liver. A function of the primary dehydrogenase. Arch Biochem Biophys. 1976;173:219–224. doi: 10.1016/0003-9861(76)90252-6. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- Genova ML, Ventura B, Giuliano G, Bovina C, Formiggini G, Parenti CG, Lenaz G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001;505:364–368. doi: 10.1016/s0014-5793(01)02850-2. [DOI] [PubMed] [Google Scholar]
- Gerschman R, Gilbert DL, Nye SW, Dwyer P, Fenn WO. Oxygen poisioning and X-irradiation: a mechanism in common. Science. 1954;119:623–626. doi: 10.1126/science.119.3097.623. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J Biol Chem. 1998;273:11038–11043. doi: 10.1074/jbc.273.18.11038. [DOI] [PubMed] [Google Scholar]
- Grasbon-Frodl EM, Kosel S, Riess O, Muller U, Mehraein P, Graeber MB. Analysis of mitochondrial targeting sequence and coding region polymorphisms of the manganese superoxide dismutase gene in German Parkinson disease patients. Biochem Biophys Res Commun. 1999;255:749–752. doi: 10.1006/bbrc.1998.9998. [DOI] [PubMed] [Google Scholar]
- Guidot DM, Repine JE, Kitlowski AD, Flores SC, Nelson SK, Wright RM, McCord JM. Mitochondrial respiration scavenges extramitochondrial superoxide anion via a nonenzymatic mechanism. J Clin Invest. 1995;96:1131–1136. doi: 10.1172/JCI118100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham AJ, Liebler DC. Vitamin E oxidation in rat liver mitochondria. Biochemistry. 1995;34:5754–5761. doi: 10.1021/bi00017a007. [DOI] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann N, Grimsby J, Shih JC, Cadenas E. The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch Biochem Biophys. 1996;335:295–304. doi: 10.1006/abbi.1996.0510. [DOI] [PubMed] [Google Scholar]
- Hoeberichts FA, Woltering EJ. Multiple mediators of plant programmed cell death: interplay of conserved cell death mechanisms and plant-specific regulators. Bioessays. 2003;25:47–57. doi: 10.1002/bies.10175. [DOI] [PubMed] [Google Scholar]
- Jung K, Reszka R. Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev. 2001;49:87–105. doi: 10.1016/s0169-409x(01)00128-4. [DOI] [PubMed] [Google Scholar]
- Kaur H, Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation: nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994;350:9–12. doi: 10.1016/0014-5793(94)00722-5. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Ku H-H, Brunk UT, Sohal RS. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian. Free Radic Biol Med. 1993;15:621–627. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux SP, Driggers WJ, Hollensworth BS, Wilson GL. Repair of alkylation and oxidative damage in mitochondrial DNA. Mutat Res. 1999;434:149–159. doi: 10.1016/s0921-8777(99)00026-9. [DOI] [PubMed] [Google Scholar]
- Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liochev SI, Fridovich I. Superoxide from glucose oxidase or from nitroblue tetrazolium. Arch Biochem Biophys. 1995;318:408–410. doi: 10.1006/abbi.1995.1247. [DOI] [PubMed] [Google Scholar]
- Liochev SI, Fridovich I. Lucigenin as mediator of superoxide production: revisited. Free Radic Biol Med. 1998;25:926–928. doi: 10.1016/s0891-5849(98)00121-x. [DOI] [PubMed] [Google Scholar]
- Liochev SI, Fridovich I. Superoxide and iron: partners in crime. IUBMB Life. 1999;48:157–161. doi: 10.1080/713803492. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lopez-Torres M, Perez-Campo R, Rojas C, Cadenas S, Barja G. Maximum life span in vertebrates: Relationship with liver antioxidant enzymes glutathione system ascorbate urate sensitivity to peroxidation true malondialdehyde in vivo H2O2, and basal and maximum aerobic capacity. Mech Ageing Dev. 1993;70:177–199. doi: 10.1016/0047-6374(93)90047-u. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase: the first twenty years (1968–1988) Free Radic Biol Med. 1988;5:363–369. doi: 10.1016/0891-5849(88)90109-8. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Vectorial chemiosmotic processes. Annu Rev Biochem. 1977;46:996–1005. doi: 10.1146/annurev.bi.46.070177.005024. [DOI] [PubMed] [Google Scholar]
- Murley JS, Kataoka Y, Hallahan DE, Roberts JC, Grdina DJ. Activation of NFkappaB and MnSOD gene expression by free radical scavengers in human microvascular endothelial cells. Free Radic Biol Med. 2001;30:1426–1439. doi: 10.1016/s0891-5849(01)00554-8. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Int J Biochem Cell Biol. 2002;34:1372–1381. doi: 10.1016/s1357-2725(02)00077-8. [DOI] [PubMed] [Google Scholar]
- Noji H, Yoshida M. The rotary machine in the cell ATP synthase. J Biol Chem. 2001;276:1665–1668. doi: 10.1074/jbc.R000021200. [DOI] [PubMed] [Google Scholar]
- Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley LW, St Clair DK, Autor AP, Oberley TD. Increase in manganese superoxide dismutase activity in the mouse heart after X-irradiation. Arch Biochem Biophys. 1987;254:69–80. doi: 10.1016/0003-9861(87)90082-8. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci U S A. 2002;99:9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orii Y. The cytochrome c peroxidase activity of cytochrome oxidase. J Biol Chem. 1982;257:9246–9248. [PubMed] [Google Scholar]
- Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- Phung CD, Ezieme JA, Turrens JF. Hydrogen peroxide metabolism in skeletal muscle mitochondria. Arch Biochem Biophys. 1994;315:479–482. doi: 10.1006/abbi.1994.1528. [DOI] [PubMed] [Google Scholar]
- Picker SD, Fridovich I. On the mechanism of production of superoxide radical by reaction mixtures containing NADH, phenazine methosulfate and nitroblue tetrazolium. Arch Biochem Biophys. 1984;228:155–158. doi: 10.1016/0003-9861(84)90056-0. [DOI] [PubMed] [Google Scholar]
- Poderoso JJ, Carreras MC, Lisdero C, Riobó N, Schöpfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- Pou S, Hassett DJ, Britigan BE, Cohen MS, Rosen GM. Problems associated with spin trapping oxygen-centered free radicals in biological systems. Anal Biochem. 1989;177:1–6. doi: 10.1016/0003-2697(89)90002-x. [DOI] [PubMed] [Google Scholar]
- Qi X, Lewin AS, Hauswirth WW, Guy J. Optic neuropathy induced by reductions in mitochondrial superoxide dismutase. Invest Ophthalmol Vis Sci. 2003;44:1088–1096. doi: 10.1167/iovs.02-0864. [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R. Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem. 2002a;383:401–409. doi: 10.1515/BC.2002.044. [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002b;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–22034. [PubMed] [Google Scholar]
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2003;283:L922–931. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- Schumacker PT. Hypoxia anoxia and O2 sensing: the search. Am J Physiol Cell Mol Physiol. 2002;283:L918–921. doi: 10.1152/ajplung.00205.2002. [DOI] [PubMed] [Google Scholar]
- Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett. 2003a;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003b;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Shimoda-Matsubayashi S, Matsumine H, Kobayashi T, Nakagawa-Hattori Y, Shimizu Y, Mizuno Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. A predictive evidence for conformational change to influence mitochondrial transport and a study of allelic association in Parkinson's disease. Biochem Biophys Res Commun. 1996;226:561–565. doi: 10.1006/bbrc.1996.1394. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Agarwal S, Dubey A, Orr WC. Protein oxidative damage is associated with life expectancy of houseflies. Proc Natl Acad Sci U S A. 1993;90:7255–7259. doi: 10.1073/pnas.90.15.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G. Myxothiazol induces H2O2 production from mitochondrial respiratory chain. Biochem Biophys Res Commun. 2001;281:645–650. doi: 10.1006/bbrc.2001.4409. [DOI] [PubMed] [Google Scholar]
- Sutton A, Khoury H, Prip-Buus C, Cepanec C, Pessayre D, Degoul F. The Ala16Val genetic dimorphism modulates the import of human manganese superoxide dismutase into rat liver mitochondria. Pharmacogenetics. 2003;13:145–157. doi: 10.1097/01.fpc.0000054067.64000.8f. [DOI] [PubMed] [Google Scholar]
- Tarpey MM, Fridovich I. Methods of detection of vascular reactive species. Nitric oxide superoxde hydrogen peroxide and peroxynitrite. Circ Res. 2001;89:224–236. doi: 10.1161/hh1501.094365. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ. Rotenone neurotoxicity: a new window on environmental causes of Parkinson's disease and related brain amyloidoses. Exp Neurol. 2003;179:6–8. doi: 10.1006/exnr.2002.8082. [DOI] [PubMed] [Google Scholar]
- Trumpower BL. The protonmotive Q cycle. J Biol Chem. 1990;265:11409–11412. [PubMed] [Google Scholar]
- Tsan MF, Clark RN, Goyert SM, White JE. Induction of TNF-a and MnSOD by endotoxin: role of membrane CD14 and Toll-like receptor-4. Am J Physiol Cell Physiol. 2001;280:C1422–1430. doi: 10.1152/ajpcell.2001.280.6.C1422. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, Freeman BA, Levitt JG, Crapo JD. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch Biochem Biophys. 1982;217:401–410. doi: 10.1016/0003-9861(82)90518-5. [DOI] [PubMed] [Google Scholar]
- Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, Flohe L. Dual function of the selenoprotein PHGPx during sperm maturation. Science. 1999;285:1393–1396. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner BB, Stuart L, Gebb S, Wispé JR. Redox regulation of manganese superoxide dismutase. Am J Physiol. 1996;271:L150–158. doi: 10.1152/ajplung.1996.271.1.L150. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Schumacker PT. O2 sensing in hypoxic pulmonary vasoconstriction: the mitochondrial door re-opens. Respir Physiol Neurobiol. 2002;132:81–91. doi: 10.1016/s1569-9048(02)00051-4. [DOI] [PubMed] [Google Scholar]
- Wittenberg BA, Wittenberg JB. Transport of oxygen in muscle. Annu Rev Physiol. 1989;51:857–878. doi: 10.1146/annurev.ph.51.030189.004233. [DOI] [PubMed] [Google Scholar]
- Wood PM. The two redox potentials for oxygen reduction to superoxide. Trends Biochem Sci. 1987;12:250–251. [Google Scholar]
- Yokoyama Y, Beckman JS, Beckman TK, Wheat JK, Cash TG, Freeman BA, Parks DA. Circulating xanthine oxidase: potential mediator of ischemic injury. Am J Physiol. 1990;258:G564–570. doi: 10.1152/ajpgi.1990.258.4.G564. [DOI] [PubMed] [Google Scholar]
- Young TA, Cunningham CC, Bailey SM. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: studies using myxothiazol. Arch Biochem Biophys. 2002;405:65–72. doi: 10.1016/s0003-9861(02)00338-7. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu L, Yu CA. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J Biol Chem. 1998;273:33972–33976. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]