

Abstract
Cardiac troponin I (cTnI) is a phosphoprotein subunit of the troponin-tropomyosin complex that is thought to inhibit cardiac muscle contraction during diastole. To investigate the contributions of cTnI phosphorylation to cardiac regulation, transgenic mice were created with the phosphorylation sites of cTnI mutated to alanine. Activation of protein kinase C (PKC) by perfusion of hearts with phorbol-12-myristate-13-acetate (PMA) or endothelin-1 (ET-1) inhibited the maximum ATPase rate by up to 25 % and increased the Ca2+ sensitivity of ATPase activity and of isometric tension by up to 0.15 pCa units. PKC activation no longer altered cTnI phosphorylation, depressed ATPase rates or enhanced myofilament Ca2+ sensitivity in transgenic mice expressing cTnI that could not be phosphorylated on serines43/45 and threonine144 (PKC sites). Modest changes in myosin regulatory light chain phosphorylation occurred in all mouse lines, but increases in myofilament Ca2+ sensitivity required the presence of phosphorylatable cTnI. For comparison, the β-adrenergic agonist isoproterenol caused a 38 % increase in maximum ATPase rate and a 0.12 pCa unit decrease in myofilament Ca2+ sensitivity. These β-adrenergic effects were absent in transgenic mice expressing cTnI that could not be phosphorylated on serines23/24 (protein kinase A, PKA, sites). Overall, the results indicate that PKC and PKA exert opposing effects on actomyosin function by phosphorylating cTnI on distinct sites. A primary role of PKC phosphorylation of cTnI may be to reduce the requirements of the contractile apparatus for both Ca2+ and ATP, thereby promoting efficient ATP utilisation during contraction.
A variety of neurohumoral factors are generated in response to stress that result in profound changes in cardiac function. For example, catecholamines elevated in emergency situations increase heart rate (positive chronotropy) and contraction strength (positive inotropy) by stimulating β-adrenergic receptors (reviewed in Solaro, 2002). These receptors couple to intracellular cyclic AMP and protein kinase A (PKA), which mediate changes in cell and organ function. During stress, the heart also responds to a host of factors released locally or globally that activate intracellular protein kinase C (PKC; Endoh, 1995; Sugden & Bogoyovitch, 1996). Endothelin-1, adenosine, angiotensin II and opiates are among the stress-related agonists that stimulate PKC in the heart. Generally, these agents regulate cardiac contractility and can be protective against ischaemic damage (Cohen et al. 2000), but the mechanisms underlying these actions are poorly understood.
Cardiac troponin I (cTnI) is a subunit of the troponin-tropomyosin complex thought to be an important substrate for both PKC and PKA. It is well documented that the serine23/24 residues near the N-terminus of cTnI are avidly phosphorylated by PKA both in vitro and in vivo (Kranias & Solaro, 1982; Solaro, 2002). It is also recognised that phosphorylation of cTnI upon β stimulation decreases the Ca2+ sensitivity of myofilament activation (Robertson et al. 1982; Hofmann & Lange, 1994; Strang et al. 1994; de Tombe & Steinen 1995; Noland et al. 1995; Zhang et al. 1995). However, the roles played by cTnI phosphorylation in modulating relaxation rate, twitch duration and crossbridge kinetics remain controversial (Winegrad et al. 1986; Hofmann & Lange, 1994; Strang et al. 1994; de Tombe & Steinen, 1995; Zhang et al. 1995; Johns et al. 1997; Saeki et al. 1997; Li et al. 2000; Herron et al. 2001; Kentish et al. 2001; Patel et al. 2001; Pi et al. 2002a; Turnbull et al. 2002).
Among the preferred sites for PKC on cTnI are serines43/45 and threonine144 (Noland et al. 1995; Jideama et al. 1996), and even less is known about the physiological consequences of phosphorylation on these sites. Reasons for this gap in understanding may include the fact that PKC typically phosphorylates several myofilament proteins in parallel and often on multiple sites. Moreover, changes in the phosphorylation of any one protein can be rather modest, particularly with physiologically relevant stimuli (Huang et al. 1997). To overcome these limitations, we created transgenic mice expressing cTnI with known PKC phosphorylation sites mutated to alanine. Mutant cTnIs were expressed on a cTnI-null background to eliminate contributions from native phosphorylatable cTnI. The presence of even low levels of phosphorylatable cTnI (< 15 %) could confound the results due to the highly cooperative nature of the thin-filament regulatory system.
PKC has been shown to inhibit actomyosin ATPase in cardiac myofibrils and in reconstituted myofilaments (Noland & Kuo, 1991; Noland et al. 1995; Jideama et al. 1996). This pioneering work demonstrated that PKC was inhibitory towards ATPase activity, and suggested a mechanistic link with PKC-mediated inhibition of contraction strength (negative inotropy). However, recent work has challenged the notion that PKC primarily mediates negative inotropy in the heart (Huang et al. 1997, 2001; Pi et al. 1997; Pi & Walker 2000). PKC can induce positive inotropy, but how this reconciles with the inhibitory effects of PKC on actomyosin ATPase rates is unknown. Moreover, the question arises whether PKC and PKA pathways in the heart are redundant if both can mediate positive inotropy.
To address these issues, the functional consequences of PKC phosphorylation were examined in myofibrils from transgenic mice. One mouse line expressed cTnI that could not be phosphorylated on the classical PKA sites, serines23/24. Use of this line permitted PKC sites to be examined with a defined phosphorylation status of PKA sites, and eliminated the possibility of cross-phosphorylation of these sites by PKC (Swiderek et al. 1990; Noland et al. 1995, 1996; Jideama et al. 1996). Three previously identified PKC sites were evaluated with the aid of a second mouse line containing cTnI that could not be phosphorylated on any of five total sites (serines23/24 (PKA sites), and serines43/45 and threonine144 (PKC sites); Pi et al. 2002a). Here we report measurements of MgATPase activity and isometric tension in myofibrils isolated from these transgenic mouse hearts after stimulation of PKC signalling pathways. Parallel measurements after stimulation of PKA signalling pathways were also made. The data provide evidence that phosphorylation of cTnI on either PKC or PKA sites regulates myofilament Ca2+ sensitivity and ATP hydrolysis rate, but in distinct ways. Some of this work has been presented in preliminary form (Pi et al. 2002b; Zhang et al. 2002).
METHODS
Genetically modified mice
Mutated cDNA of cTnI was used as a transgene in which codons for Ser23, Ser24, Ser43, Ser45 and Thr144 were converted into Ala codons (cTnI-Ala5), or only Ser23 and Ser24 were converted into Ala codons (cTnI-Ala2). The cardiac-specific α-myosin heavy chain promoter was used to drive expression of the mutated cTnI cDNA in mouse myocardium. By crossing mice heterozygous for the cTnI-knockout allele (Huang et al. 1999) with cTnI transgenic founders, mice expressing cTnI-Ala2 or cTnI-Ala5 on the null background were generated (Pi et al. 2002a).
Preparation of cardiac myofibrils
All animals were handled in accordance with the guidelines of the University of Wisconsin Research Animal Resource Committee. Two different experimental protocols were employed.
Protocol 1
Hearts were rapidly excised following cervical dislocation and were immediately homogenised in relaxing buffer A containing (mm): 5 ATP, 100 KCl, 10 imidazole pH 7.0, 1 MgCl2, 2 EGTA, 1 phenylmethylsulphonyl fluoride (PMSF), 10 benzamidine, 1 DTT, 20 2,3-butanedione monoxime (BDM) and 75 mg 100 ml−1 protease inhibitor cocktail (Complete, Roche Pharmaceuticals). Phosphatase inhibitor calyculin A (15 nm) was added to maintain the phosphorylation status of the myofibrils. The cardiac myofibrils were then skinned in relaxing solution containing 0.33 % Triton X-100 and 1 mg ml−1 bovine serum albumin (BSA). After 4 min of skinning, myofibrils were pelleted, washed and resuspended in relaxing buffer A. Skinned myofibrils were then split into two populations: one for MgATPase assays and one for mechanical measurements. For MgATPase assays, myofibrils were pelleted and resuspended in relaxing buffer B (relaxing buffer A with 2 mm ATP and no BDM) immediately before beginning the assay to reduce background inorganic phosphate (Pi) levels. The protein concentration of the myofibrils was determined after resuspension.
Protocol 2
Hearts were excised from mice anesthetised by peritoneal injection of pentobarbital (20 mg kg−1 body weight) and subjected quickly to aortic cannulation and retrograde perfusion at 37°C on a Langendorff apparatus with modified Ringer's buffer containing (mm): 125 NaCl, 5 KCl, 25 Hepes pH 7.4, 5 pyruvate, 1.2 MgSO4, 11 glucose, 2 NaH2PO4, 1 CaCl2 and 1 mg ml−1 BSA. The buffer was gassed with 95 % O2 and 5 % CO2. After autorhythmic beating was stabilised for 5 min, the hearts were perfused with the designated concentration of agonist for 15 min. Hearts were then divided into four groups based upon the treatment received: control (modified Ringer's buffer only); isoproterenol (Iso, 10 nm); endothelin-1 (ET-1, 10 nm), and phorbol-12-myristate-13-acetate (PMA, 100 nm). At the end of the process, 15 nm calyculin A was added to the solution to maintain the phosphorylation status of myofilament proteins. Hearts were washed in ice-cold Ca2+-free Ringer's buffer to suppress beating, and homogenised in relaxing buffer A. Skinning and splitting of myofibrils for ATPase assays and mechanical measurements were carried out as described for protocol 1.
Myofibrillar MgATPase assays
Basal and Ca2+-stimulated myofibrillar MgATPase activities were measured using a modification of the method of Swartz et al. (1999). Incubations were performed in triplicate and Pi measurements in duplicate. For each incubation, a 50 μg sample of myofibrils was added to 1 ml of activation buffer (mm): 130 KCl, 20 Pipes pH 7.0, 4 EGTA, 4 MgCl2, 1 NaN3, 2 ATP, 1 DTT and 1 mg ml−1 BSA (to minimise sticking/clumping of myofibrils), at pCa 9–4. The reaction was incubated at 30°C for 10 min then quenched with 0.5 ml ice-cold 25 % trichloroacetic acid. The ATPase reaction was linear for at least 15 min under these conditions (Fig. 1S, on-line supplement). Precipitated proteins were pelleted at 4°C, and 2 × 200 μl aliquots of the supernatant (containing Pi) were diluted to 1 ml with water and reacted with 44 mm molybdic acid and 1 % polyvinyl alcohol/0.042 % malachite green for 2 min at room temperature. The reaction was then bleached by adding 7.8 % H2SO4, and colour was read at 625 nm with a Beckman DU-620 spectrophotometer. MgATPase activity is expressed as (nmole Pi) min−1 (mg protein)−1 by comparison with a standard curve for phosphate, which was linear to at least 500 nmole Pi in a 1 ml sample volume. Protein was determined by the bicinchoninic acid assay (Pierce, Rockford, IL, USA).
Figure 1. Mouse genotyping.
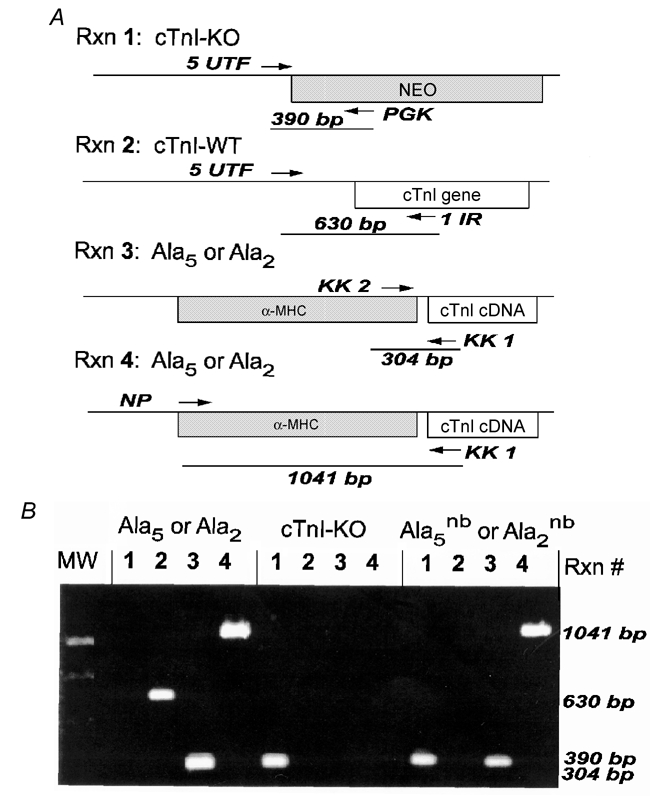
A, cardiac TnI constructs and the PCR primers (horizontal arrows and trivial names UTF, KK1, KK2, NP, PGK and 1IR) used to detect them. Genotyping required four separate PCR reactions on DNA isolated from mouse tails. Reaction 1 (Rxn 1) generated a 390 base pair (bp) fragment for the cTnI-knockout allele (cTnI-KO). Reaction 2 (Rxn 2) generated a 630 bp fragment for the wild-type cTnI gene (WT-cTnI). The Ala2 and Ala5 transgenes containing the α-myosin heavy chain (MHC) promoter were both detected by reaction 3 (Rxn 3; 304 bp fragment) and reaction 4 (Rxn 4; 1041 bp fragment). B, a typical agarose gel showing PCR fragments for the four reactions performed on three different mouse lines. Mouse genotypes (indicated above each set of four lanes) were determined by the pattern of PCR fragments. Ala5nb/Ala2nb mice were positive for the transgene and the cTnI-knockout allele but negative for wild-type cTnI. MW = molecular weight.
Phosphorylation assays
Myofilament protein phosphorylation was monitored by different methods depending upon whether myocytes were skinned (Fig. 2B and Fig. 6C) or intact (Fig. 5C). Skinned myocytes were incubated with a purified kinase and γ-32P-ATP then subjected to SDS-PAGE and autoradiography. Intact myocytes were incubated in 32P-orthophosphate, stimulated with Iso, endothelin or PMA, then subjected to SDS-PAGE and autoradiography. Radioactive bands were quantified by use of a UVP Transilluminator bioimaging system, or by excising bands from the gel and counting in 5 ml of BioSafe scintillation cocktail using a Beckman liquid scintillation counter. With skinned myocytes, untreated controls typically showed little or no radiolabel in the bands of interest, so kinase-treated samples were normalised to the protein content of the phosphorylated band (estimated by a standard curve of BSA loaded onto the gel). With intact myocytes, untreated controls contained basal radiolabel in the bands of interest, so phosphorylation of agonist-treated samples was quantified as the percent increase in 32P content over basal.
Figure 2. Characterisation of TnI expression.
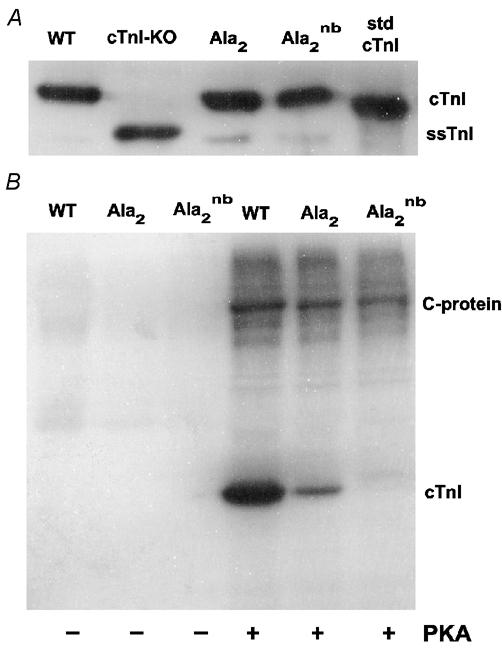
A, Western blot of myofibrils from wild-type (WT), cTnI-Ala2 transgenic founder, and cTnI-Ala2nb mice at 1 month of age. Myofibrils from 15-day-old cTnI-KO mice (lane 2) and purified bovine cTnI (std cTnI, lane 6) are included as controls. ssTnI is slow skeletal TnI. B, autoradiogram of PKA-phosphorylated myofilament proteins. Incorporation of 32P in the absence (left three lanes, labelled −) and presence (right three lanes, labelled +) of a PKA catalytic subunit. 32P-incorporation in cTnI of Ala2 was 14 ± 5 % that of wild-type and in Ala2nb was undetectable. Tissue samples were the same as in A.
Figure 6. Effects of MLCK treatment on tension and myofilament protein phosphorylation.
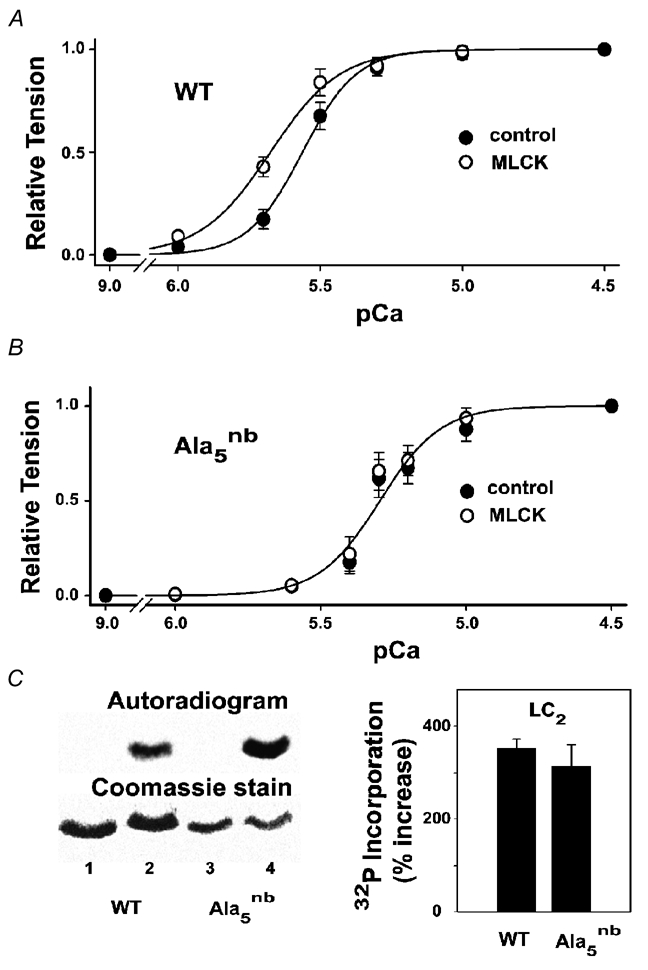
The data points represent the mean ±s.e.m. from six separate experiments, with solid lines (in A and B) illustrating non-linear least-squares fits to the Hill equation. A, pCa-tension curves in wild-type myocytes before and after MLCK (0.5 units ml−1, 10 min, room temperature). The pCa50 values were 5.56 ± 0.03 (control) and 5.67 ± 0.04 (MLCK). Maximum active tension and resting tension in mN mm−2 were 16.2 ± 2 and 1.1 ± 0.2, respectively (control), and 14.7 ± 2 and 1.5 ± 0.2, respectively (MLCK). B, pCa-tension curves in cTnI-Ala5nb before and after the same MLCK treatment. The pCa50 values were 5.24 ± 0.03 (control) and 5.2 ± 0.07 (MLCK). Maximum active tension and resting tension in mN mm−2 were 21.3 ± 3 and 1.0 ± 0.2, respectively (control), and 20.3 ± 3 and 1.4 ± 0.3, respectively (MLCK). C, SDS-PAGE analysis of skinned myocytes treated with MLCK. Lane 1: wild-type, no MLCK; lane 2: wild-type, with MLCK; lane 3: cTnI-Ala5nb, no MLCK; lane 4: cTnI-Ala5nb, with MLCK. The right panel summarises data from four gels obtained by excising LC2, counting and normalising to microgrammes of protein in the LC2 band.
Figure 5. Agonist effects on 32P-phosphate incorporation into myofilament proteins of intact myocytes.
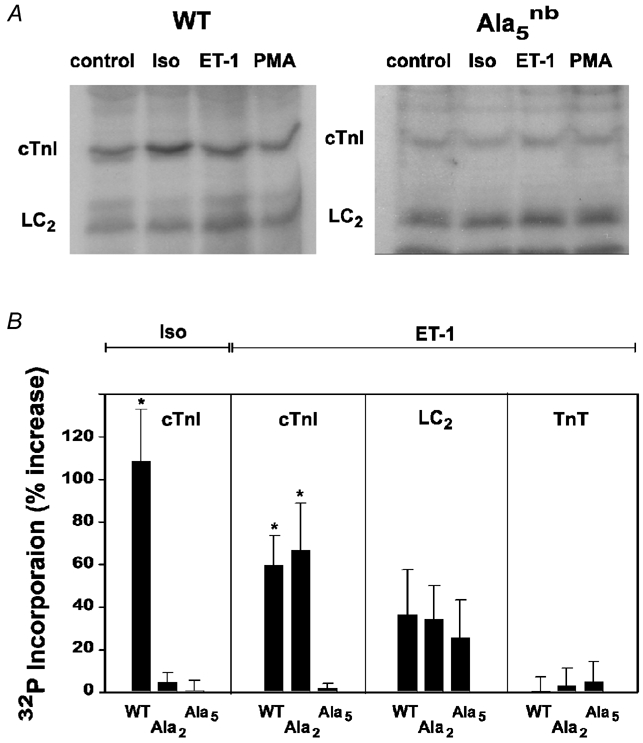
A, autoradiograms of ventricular myocytes from wild-type, cTnI-Ala2nb and cTnI-Ala5nb mice treated with standard doses of the indicated agonists. B, summary of data from a minimum of four autoradiograms. The left panel summarises the effects of 10 nm Iso on cTnI phosphorylation. The other three panels summarise the effects of 10 nm ET-1 on cTnI, LC2 and cardiac troponin T (TnT). The effects of PMA were similar to those of ET-1. *P < 0.05 compared to untreated controls.
Skinned myocytes
A 200 μg sample of Triton X-100-treated myofibrils was incubated for 60 min at 37°C in 200 μl of solution containing 100 μCi (where 1 Ci = 3.7 × 1010 Bq) γ-32P-ATP (Perkin Elmer, Boston, MA, USA) and (mm): 1 ATP, 100 KCl, 10 imidazole pH 7.0, 2 EGTA, 2 MgCl2, 1 PMSF, 10 benzamidine and 75 mg (100 ml of protease inhibitor cocktail)−1 (Complete, Roche Pharmaceuticals). Samples contained either no added kinase, a catalytic subunit of PKA (Sigma, St. Louis, MO, USA), or myosin light chain kinase (MLCK). Each kinase reaction contained 0.05–0.1 units of activity (or 0.25–0.50 units ml−1, where 1 unit = nmoles phosphate transferred min−1). MLCK was purified from chicken gizzard by the method of Adelstein & Klee (1981) and activated by treatment of 50 μl of 0.34 mg ml−1 enzyme with 1 μg trypsin (Sigma) for 20 min at 25°C followed by 1.5 μg soybean trypsin inhibitor. After treatment, MLCK activity was 0.03–0.06 units μg−1 using synthetic smooth muscle myosin regulatory light chain (LC2) peptide as a substrate. Myofibrils were pelleted, washed and solubilised in 200 μl electrophoresis loading buffer with boiling for 5 min. Myofibrillar proteins were separated by SDS-PAGE on 12 % gels, then stained with Coomassie blue, dried and then subjected to autoradiography using X-OMAT film.
Intact myocytes
For 32P incorporation into intact myocytes, freshly isolated mouse myocytes at approximately 106 cells ml−1 were incubated in phosphate-free 1 mm Ca2+ Ringer's buffer containing 1 mCi ml−132P-orthophosphate (Perkin Elmer) for 2 h at 37°C. A 200 μl sample of labelled myocytes was treated with the various agonists for 15 min followed by washing and solubilisation in gel sample buffer containing 50 nm calyculin A to inhibit phosphatases. The method used was essentially as described previously using rat myocytes (Huang et al. 1997), but mouse myocytes are less tolerant to 32P-orthophosphate, so the specific activity of the final labelled myocytes was reduced by fourfold. Samples were prepared for SDS-PAGE by addition of 50 μl of 5 × sample buffer, boiled for 5 min, pelleted to remove insoluble material and electrophoresed as described earlier.
Mechanical measurements in skinned cardiac myofibrils
Isometric force development in skinned myofibrils was measured as described previously (Huang et al. 1999). In brief, the ends of small myofibril bundles (containing 1–2 cardiac myocytes) were glued to the tip of a piezoelectric translator (model 350, Physik Instruments, Cambridge, MA, USA) and a force transducer (Model 403, Cambridge Technology). Active force was recorded at a sarcomere length of 2.2–2.3 μm by transferring the attached myocytes into activating solutions ranging from pCa 9 to pCa 4.5. Tension values were normalised to cross-sectional area by measuring the width of a central segment of the attached myocyte and assuming a rectangular cross-section with a 2:1 width:depth ratio. Values for resting and maximum active tension were in the range 1–2 mN mm−2 and 13–17 mN mm−2, respectively (Table 1). Tension-pCa curves were constructed by subtracting the resting tension (measured at pCa 9) from tension values measured over the range pCa 6–4.5. Maximum active tension was taken as the difference between the tension at pCa 4.5 and that at pCa 9.
Table 1.
Agonist effects on maximum MgATPase activity and maximum active tension in wild-type and mutant myofibrils
| No treatment | 10 nm ET-1 | 100 nm PMA | 10 nm Iso | ||
|---|---|---|---|---|---|
| Wild-type | Maximum ATPase rate | 207 ± 16 | 190 ± 26 | 175 ± 17* | 286 ± 12* |
| Difference | — | n.s. | −16% | +38% | |
| Maximum tension | 14.5 ± 1.1 | 12.8 ± 0.9 | 14.6 ± 1.6 | 12.7 ± 1.1 | |
| Difference | — | n.s. | n.s. | n.s. | |
| Ala2nb | Maximum ATPase rate | 197 ± 10 | 148 ± 15* | 149 ± 15* | 192 ± 18 |
| Difference | — | −25% | −24% | n.s. | |
| Maximum tension | 15.1 ± 1.3 | 14.0 ± 1.3 | 15.1 ± 1.2 | 13.8 ± 1.2 | |
| Difference | — | n.s. | n.s. | n.s. | |
| Ala5nb | Maximum ATPase rate | 176 ± 11 | 168 ± 9 | 165 ± 13 | 172 ± 17 |
| Difference | — | n.s. | n.s. | n.s. | |
| Maximum tension | 12.5 ± 1.2 | 13.3 ± 0.8 | 13.0 ± 1.5 | 13.5 ± 1.3 | |
| Difference | — | n.s. | n.s. | n.s. |
Maximum MgATPase in units of (nmol Pi) min−1 (mg protein)−1 measured at pCa 4. Maximum active tension (total minus resting) in units of μm mm−2. Resting tension values were: 1.5 ± 0.2 (all wild-type), 1.6 ± 0.3 (all Ala2) and 1.8 ± 0.3 (all Ala5) in mm mm−2.
P < 0.05; n.s. = not significant; Data from a minimum of six separate experiments are reported as mean ±s.e.m.
Data analysis
The Ca2+ sensitivity of tension was determined by fitting normalised tension-pCa curves with a Hill equation (P =P0/(1 + K50nH/[Ca2+]nH)), where P0 is maximum active tension, K50 is the [Ca2+] at which 50 % of maximum active tension was produced, and nH is the Hill coefficient. K50 values are reported as pCa50, where pCa50=−log K50. Data were analysed in two ways: (1) by fitting the data obtained for each cell individually and then averaging the derived Hill parameters and (2) by fitting averaged data from many cells to obtain composite Hill parameters. The final numbers derived by these two methods were comparable (see on-line data supplement). MgATPase rates were measured at each pCa value in triplicate and analysed by comparison with an appropriate control treatment (typically wild-type, no agonist) determined on the same experimental day. Basal MgATPase rate measured at pCa 9 was subtracted from all values before analysis. For both tension and MgATPase measurements, the data are expressed as mean ±s.e.m. and were analysed using a one-way ANOVA or a two-tailed unpaired t test after confirming that variances in the two groups were similar. Values of P < 0.05 were considered to be statistically significant.
RESULTS
Two transgenic mouse lines were used in the present study. cTnI-Ala2 expressed cTnI, in which serines23/24 were replaced with alanine, whereas cTnI-Ala5 expressed cTnI, in which serines23/24, serines43/45 and threonine144 of cTnI were all replaced with alanine. In general, the original transgenic founders and their offspring expressed a mixture of both mutant cTnI protein and native (wild-type) cTnI (Pi et al. 2002a and see below). By crossing transgenic mice with cTnI-knockout mice using the breeding strategy described by Pi et al. (2002a), it was possible to obtain mice expressing only the mutant cTnI protein on a cTnI-null background. In the process, the lethal cTnI-null phenotype (Huang et al. 1999) was rescued by the presence of transgenic cTnI. Mice were genotyped by amplifying three different alleles (cTnI-knock out, cTnI-wild-type, and cTnI transgene) using four separate PCR reactions (Fig. 1). Mice harbouring two targeted native cTnI alleles (replaced with a neomycin-resistance cassette) and rescued by a non-phosphorylatable cTnI were designated cTnI-Ala2nb or TnI-Ala5nb mice (nb indicating the null background).
Treatment of cardiac myofibrils with PKA in the presence of 32P-γ-ATP was used to determine whether or not native cTnI was present in transgenic mouse hearts. For example, in the case of cTnI-Ala2 mice, SDS-PAGE and autoradiography revealed significant differences between wild-type, cTnI-Ala2 and cTnI-Ala2nb hearts (Fig. 2). Myofibrils from each mouse line expressed similar levels of PKA phosphorylatable C-protein, whereas phosphorylatable cTnI was reduced in cTnI-Ala2 and undetectable in cTnI-Ala2nb. Our interpretation is that cTnI-Ala2 mice express a mixture of endogenous phosphorylatable cTnI (14 %) and transgenic alanine23/24 cTnI (86 %), whereas cTnI-Ala2nb mice express only the latter. A similar heterogeneity of cTnI expression was observed for cTnI-Ala5 mice, and was abrogated by crossing onto the cTnI null background (Pi et al. 2002a). Because of this homogeneity and because cTnI-Ala2nb and cTnI-Ala5nb mice were generally healthy, had normal litter sizes and presented little or no cardiac pathology, these lines were used for all of the experiments described here.
Myofibrils isolated from cTnI-Ala2nb hearts hydrolysed MgATP and generated tension in a Ca2+-dependent manner similar to wild-type myofibrils (summarised in Table 1). In both cTnI-Ala2nb and wild-type myofibrils, minimum MgATPase rates measured at pCa 9 were approximately 70 (nmole Pi) min−1 (mg protein)−1 (data not shown), whereas maximum rates measured at pCa 4 were approximately 200 (nmole Pi) min−1 (mg protein)−1. Maximum isometric tension was also indistinguishable between cTnI-Ala2nb and wild-type myofibrils. The pCa50 values for half-maximal MgATPase activity and isometric tension were also similar in the pCa range 5.74–5.78 (summarised in Table 2). On this basis, we conclude that the serine to alanine mutations at residues 23 and 24 of cTnI were relatively benign. Moreover, unlike many other mutations in myofilament proteins (Seidman & Seidman, 2001), these changes in serines23/24 did not result in detectable cardiac hypertrophy or cardiomyopathy. The cTnI-Ala2nb mice were used here to examine cTnI phosphorylation by PKC without confounding variations in the phosphorylation state of PKA sites.
Table 2.
Agonist effects on myofilament Ca2+ sensitivity in wild-type and mutant myofibrils
| Treatment | Wild-type | Ala2nb | Ala5nb | ||
|---|---|---|---|---|---|
| ET-1 | ATPase pCa50 | None | 5.69 ± 0.03 | 5.78 ± 0.02 | 5.47 ± 0.04 |
| 10 nm ET-1 | 5.83 ± 0.05* | 5.85 ± 0.02* | 5.39 ± 0.09 | ||
| Δ, Direction | 0.14, Left | 0.07, Left | n.s. | ||
| Tension pCa50 | None | 5.76 ± 0.02 | 5.74 ± 0.02 | 5.53 ± 0.02 | |
| 10 nm ET-1 | 5.81 ± 0.03 | 5.83 ± 0.02* | 5.52 ± 0.05 | ||
| Δ, Direction | n.s. | 0.09, Left | n.s. | ||
| PMA | ATPase pCa50 | None | 5.69 ± 0.03 | 5.78 ± 0.02 | 5.47 ± 0.04 |
| 100 nm PMA | 5.84 ± 0.03* | 5.91 ± 0.03* | 5.43 ± 0.03 | ||
| Δ, Direction | 0.15, Left | 0.13, Left | n.s. | ||
| Tension pCa50 | None | 5.76 ± 0.02 | 5.74 ± 0.02 | 5.53 ± 0.02 | |
| 100 nm PMA | 5.83 ± 0.02* | 5.85 ± 0.03* | 5.51 ± 0.03 | ||
| Δ, Direction | 0.07, Left | 0.11, Left | n.s. | ||
| Iso | ATPase pCa50 | None | 5.69 ± 0.03 | 5.78 ± 0.02 | 5.47 ± 0.04 |
| 10 nm Iso | 5.57 ± 0.04* | 5.76 ± 0.02 | 5.48 ± 0.03 | ||
| Δ, Direction | 0.12, Right | n.s. | n.s. | ||
| Tension pCa50 | None | 5.76 ± 0.02 | 5.74 ± 0.02 | 5.53 ± 0.02 | |
| 10 nm Iso | 5.64 ± 0.04* | 5.73 ± 0.04 | 5.52 ± 0.03 | ||
| Δ, Direction | 0.12, Right | n.s. | n.s. |
pCa50 values were derived from fitting raw data to the Hill equation then averaging data from a minimum of six separate experiments. Data are presented as mean ±s.e.m.
P < 0.05. Δ is the difference.
The available evidence indicates that phosphorylation by PKC decreases the MgATPase rate in cardiac myofibrils (Noland & Kuo, 1991; Jideama et al. 1996; Noland et al. 1996; Pyle et al. 2001). To explore the involvement of cTnI, we treated hearts with 10 nm ET-1 or 100 nm PMA, both of which activate intracellular PKC pathways (Endoh, 1995). The maximal MgATPase rate in wild-type myofibrils was reduced by 8–16 % compared to untreated hearts (Table 1), but only PMA-treated myofibrils achieved statistical significance when data from 12 separate experiments were pooled. A more substantial 25 % reduction in MgATPase rate was observed in cTnI-Ala2nb myofibrils after treatment with ET-1 or PMA (Table 1). This observed difference between wild-type and cTnI-Ala2nb suggests that changes in the phosphorylation status of serines23/24 influences the myofibrillar response to PKC. Regulation of MgATPase activity by PKC was then tested in cTnI-Ala5nb hearts that expressed cTnI with the additional phosphorylation sites serines43/45 and threonine144 mutated to alanine. PKC activators did not inhibit MgATPase activity in cTnI-Ala5nb mice (Table 1), indicating that phosphorylation of these serine and/or threonine residues plays a key role in the inhibition of MgATPase activity.
The responsiveness of the thin-filament regulatory system to Ca2+ is another parameter that is thought to be regulated by α- and β-adrenergic agonists (Endoh & Blinks, 1988). Treatment of hearts with the PKC activators ET-1 or PMA caused changes in pCa50 values for both MgATPase activity (Fig. 3) and isometric tension (Fig. 4). In this case, the Ca2+ sensitivity of the regulatory system increased (i.e. the curves shifted to the left to lower values of [Ca2+]). Generally, the direction of the change was the same for PMA and ET-1, but the response to PMA was more pronounced. In addition, changes in myofibrillar Ca2+ sensitivity following PKC agonists were typically more readily detected in cTnI-Ala2nb than in wild-type hearts (Table 2). This finding is consistent with the suggestion made above that the effects of PKC are modified by changes in the phosphorylation state of PKA sites. Importantly, PKC activators modulated pCa50 values for both MgATPase and isometric tension in the same leftward direction, and these effects were eliminated by mutation of the three primary PKC-directed serine/threonine residues in cTnI (Fig. 3 and Fig. 4; Table 2).
Figure 3. Agonist effects on pCa versus MgATPase activity.
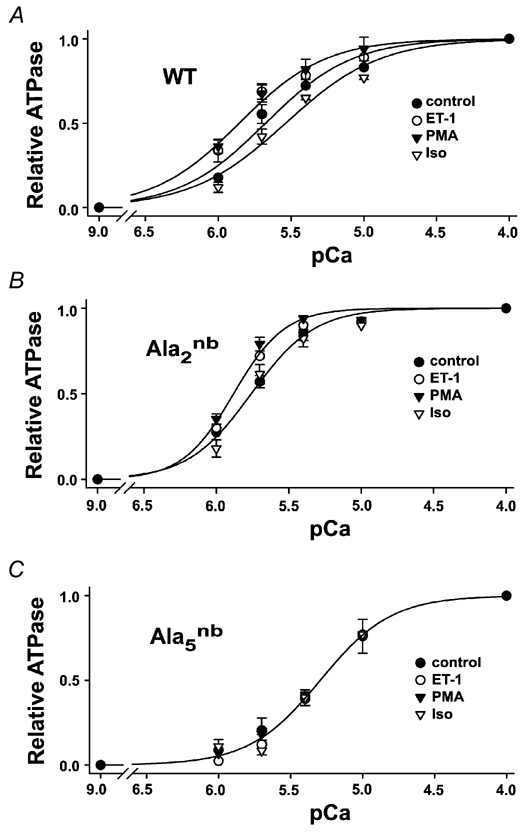
A, wild-type, B, cTnI-Ala2nb and C, cTnI-Ala5nb. Data points represent the mean of six separate experiments in which the ATPase activity at each pCa value was performed in triplicate. The basal ATPase activity measured at pCa 9 was subtracted and the activity at each pCa value was normalised to the maximum at pCa 4 for that experiment. Typical basal and maximum ATPase activities are illustrated and summarised in the on-line data supplement. Solid lines represent non-linear least-squares fits to the Hill equation. pCa50 values are given in Table 2.
Figure 4. Agonist effects on the pCa versus isometric tension relationship.
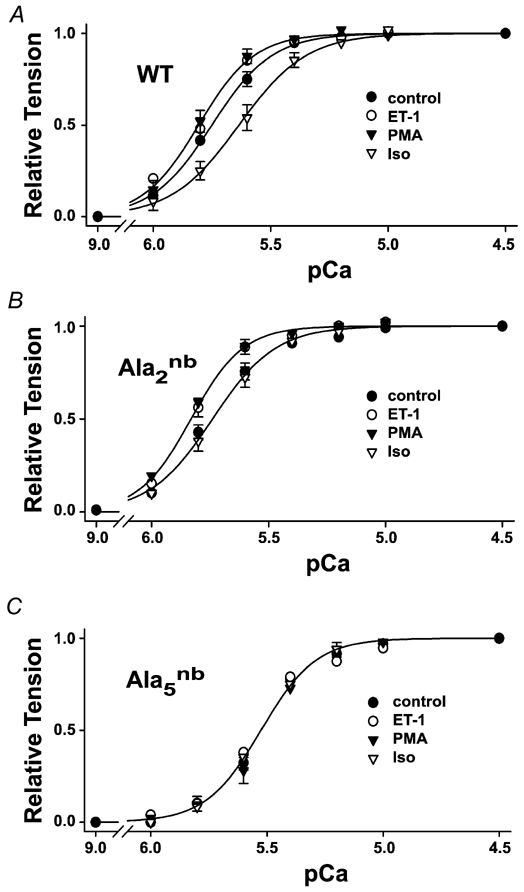
A, wild-type, B, cTnI-Ala2nb and C, cTnI-Ala5nb. Data points represent the mean from six separate experiments in which active tension was determined by subtracting the resting tension at pCa 9 from the measured tension at each pCa value. Active tension at each pCa value was then normalised to maximum tension at pCa 4.5 for that experiment. Solid lines represent non-linear least-squares fits to the Hill equation. pCa50 values are given in Table 2, and maximum active tensions and resting tension are given in Table 1.
Treatment with 100 nm PMA or 10 nm ET-1 promoted statistically significant changes in the phosphorylation of cTnI, as determined by SDS-PAGE and autoradiography of 32P-labelled myocytes (Fig. 5). These changes were observed in wild-type and cTnI-Ala2nb myocytes (Fig. 5), both of which showed similar changes in myofibril function (i.e. increased myofilament Ca2+ sensitivity and decreased MgATPase rate). In cTnI-Ala5nb myocytes, there was no change in the phosphorylation of cTnI (Fig. 5), demonstrating that the functionally relevant PKC sites were no longer present. Interestingly, another myofilament protein, LC2, showed increases in phosphorylation in all three types of myocyte, but these changes were more modest and did not achieve statistical significance (Fig. 5).
To explore the potential impact of LC2 phosphorylation further, we exploited the substrate specificity of MLCK to selectively phosphorylate ventricular LC2. Figure 6 shows that wild-type myofibrils responded to MLCK treatment with a 0.11 pCa unit increase in Ca2+ sensitivity, as determined by the tension-pCa relationship. LC2 in wild-type myocytes was strongly phosphorylated under these conditions (Fig. 6C). In cTnI-Ala5nb myocytes, the same MLCK treatment had little or no effect on the tension-pCa relationship (Fig. 6B), despite similar levels of LC2 phosphorylation in these myocytes (Fig. 6C). Therefore, the influence of LC2 phosphorylation on myofilament Ca2+ sensitivity appears to depend markedly upon the phosphorylation status of PKC sites on cTnI.
For comparison and to validate the mouse models, the effects of β-stimulation on myofilament properties were examined in parallel on each experimental day. The β-adrenergic agonist Iso is known to increase the phosphorylation of serines23/24 and cause a rightward shift of the myofilament Ca2+ sensitivity (Solaro, 2002). In measurements of either MgATPase activity (Fig. 3) or isometric tension (Fig. 4), 10 nm Iso shifted the pCa50 to higher values of [Ca2+] by 0.12 pCa units (Table 2). Moreover, this pronounced rightward shift of Ca2+ sensitivity was not observed in myofibrils that could not be phosphorylated on serines23/24, including cTnI-Ala2nb and cTnI-Ala5nb (Fig. 3 and Fig. 4; Table 2). These observations are consistent with the results of reconstitution and subunit exchange experiments that previously demonstrated the importance of PKA phosphorylation of serines23/24 (Noland et al. 1995; Zhang et al. 1995, Solaro, 2002). We also found no evidence that PKA could phosphorylate other sites on cTnI, such as the PKC sites on cTnI-Ala2nb (Fig. 2C and Fig. 5B). These observations also help to validate the use of these transgenic mouse models for studies of contractile regulation by cTnI.
The same 10 nm Iso treatment that reduced myofilament Ca2+ sensitivity also increased MgATPase rates in wild-type mouse myofibrils. The enhancement of the maximal MgATPase rate was 38 % and statistically significant (Table 1). Moreover, this effect of Iso was no longer observed in myofibrils that could not be phosphorylated on serines23/24, including cTnI-Ala2nb and cTnI-Ala5nb. Thus, under the conditions employed here, phosphorylation of cTnI by PKA resulted in a substantial increase in the rate of MgATP hydrolysis at maximal Ca2+, and mutation of the PKA sites to alanine eliminated this effect. Overall, the results indicate that not only do PKA and PKC have opposing effects on the Ca2+ sensitivity of the thin-filament regulatory system, they also have opposing effects on maximal MgATPase activity.
One of the rationales for creating cTnI-Ala2nb mice was to eliminate possible cross-phosphorylation of PKA sites by PKC, a phenomenon that has been documented for cTnI (Swiderek et al. 1990; Noland et al. 1995, 1996; Jideama et al. 1996). In order to further test whether cross-phosphorylation could influence myofibrillar Ca2+ sensitivity in our system, we treated wild-type hearts simultaneously with ET-1 and Iso (10 nm each). The pCa50 value after this co-stimulation was 5.74 ± 0.03 (n = 5), which is not statistically different from that measured in untreated controls (5.76 ± 0.04, n = 5). This suggests that cTnI phosphorylation on both PKA and PKC sites can have offsetting effects. It also indicates that cross-phosphorylation of PKA sites during intense PKC stimulation can mask the effects of PKC on myofilament Ca2+ sensitivity. Similar offsetting effects were observed with MgATPase activity measurements, as co-stimulation with ET-1 and Iso prevented the increase in MgATPase rate observed with Iso alone (in (nmole Pi) min−1 (mg protein)−1: unstimulated: 206 ± 13; 10 nm Iso: 286 ± 12; 10 nm Iso + 10 nm ET-1: 202 ± 10). These observations suggest that the functional consequences of cTnI phosphorylation on PKC sites depend critically upon the phosphorylation status of PKA sites, and vice versa. Therefore, cross-phosphorylation of PKA sites on cTnI by PKC is likely to be a confounding factor in studies of the kinase regulation of cardiac myofilaments.
DISCUSSION
The PKC family of kinases has received considerable attention in the cardiovascular system in recent years because of its potential role in contractile regulation, cardioprotection, heart development and disease progression (Endoh, 1995; Sugden & Bogoyevitch, 1995; Cohen et al. 2000; Solaro, 2002). One of the well-characterised PKC substrates in the heart is cTnI. Kuo and co-workers (Venema & Kuo, 1993; Noland et al. 1995, 1996; Jideama et al. 1996) performed a systematic analysis of cTnI phosphorylation in reconstituted myofibrils and identified three PKC sites: serine43, serine45 and threonine144. The results of the present study provide evidence that phosphorylation of one or more of these sites on cTnI plays a central role in depressing myofilament ATPase activity and in enhancing myofilament Ca2+ sensitivity. The reduction of ATP hydrolysis rates with PKC activation observed here confirm previous measurements in other species (Winegrad et al. 1986; Noland & Kuo, 1991; McClellan et al. 1996; Sakei et al. 1998; Pyle et al. 2001) and help to validate our experimental system. Also supporting the validity of the system was the observed decrease in myofibrillar Ca2+ sensitivity associated with the phosphorylation of serines23/24, as reported in other species (Solaro, 2002).
Whether or not myofilament Ca2+ sensitivity can be altered by PKC treatment has been controversial. It has been reported that PKC activation causes an increase (Puceat et al. 1990; Clement et al. 1992; Terzic et al. 1992; Noland & Kuo, 1993; Pyle et al. 2001), a decrease (Gwathmey & Hajjar, 1990; Takeishi et al. 1998; Burkart et al. 2003) or no change (Venema et al. 1993; Noland et al. 1995; 1996; Strang & Moss, 1995; Jideama et al. 1996) in the Ca2+ responsiveness of the myofilaments in different experimental contexts. Notably, it has been shown by several investigators that certain PKC isoforms can mimic PKA by phosphorylating serines23/24 (Swiderek et al. 1990; Jideama et al. 1996; Noland et al. 1996), but this possibility of cross-phosphorylation of serines23/24 by PKC was not considered in most of the studies cited above. To address this issue, we eliminated the possibility of cross-phosphorylation of serines23/24 by mutating both to alanine. We employed an agonist stimulation protocol involving the perfusion of intact beating hearts to mimic physiological conditions as much as possible. Using this strategy, we were able to demonstrate reproducible changes in both the MgATPase rate and Ca2+ sensitivity upon stimulation of PKC pathways. Furthermore, these changes were more consistently detected (and more robust) when the possibility of cross-phosphorylation was eliminated. These changes were then abolished by mutation of three PKC sites on cTnI to non-phosphorylatable alanine.
Our observations with PKC stimulation are probably not unique to mice or to our experimental protocol because qualitatively similar effects have been reported in rats using phorbol ester treatment of isolated ventricular myocytes. A PKC-mediated increase in the Ca2+ sensitivity of force was reported by Terzic et al. (1992) and Puceat et al. (1990), but these observations were not readily reproduced (Solaro, 2002). By monitoring myofibrillar ATPase activity, Pyle et al. (2001) observed a phorbol-ester-mediated decrease in maximum ATPase rate and an increase in Ca2+ sensitivity similar to that reported here. Intriguingly, a recent report has described a similar increase in myofilament Ca2+ sensitivity and a similar decrease in crossbridge cycling rate due to troponin modification in failing human hearts (Knott et al. 2002). Moreover, these effects were reversed by treatment of troponin with a phosphatase. Another study also showed enhanced myofilament Ca2+ sensitivity in failing human hearts that was reversed by phosphatase treatment (van der Velden et al. 2003). This study found a poor correlation between the level of LC2 phosphorylation and myofilament Ca2+ sensitivity, leading to the conclusion that other myofilament phosphoproteins must play a role. To our knowledge, the results presented here provide the first direct link between the enhanced Ca2+ sensitivity of myofilaments and phosphorylation of cTnI on serines43/45 and/or threonine144.
PKC activation in the myocardium may lead to increased phosphorylation of several myofilament proteins, notably cTnI, cTnT and LC2 (Clement et al. 1992; Noland & Kuo, 1993; Huang et al. 1997; Montgomery et al. 2001). In our hands, cTnI phosphorylation always showed the largest changes in phosphorylation, cTnT the smallest and LC2 somewhere in between (Fig. 5; Huang et al. 1997). Evidence has been presented from several laboratories demonstrating parallel phosphorylation of cTnI and LC2 with PKC activation (Venema et al. 1993; Damron et al. 1995; Huang et al. 1997). In addition, LC2 phosphorylation has been linked to a leftward shift of the Ca2+ dependence of MgATPase or tension (Morano et al. 1985; Sweeney & Stull, 1986; Clement et al. 1992; Noland & Kuo, 1993), an effect that is potentiated in the presence of PKC (Clement et al. 1992). LC2 phosphorylation on the order of a 15 % increase has recently been shown to play a role in the positive inotropic response to α-adrenergic stimulation (Andersen et al. 2002).
The data presented here suggest the intriguing possibility that cTnI and LC2 phosphorylation synergistically sensitise the myofilaments to Ca2+. Evidence for this stems from the observation that eliminating PKC sites on cTnI had two rather dramatic effects. It prevented PKC activators from sensitising the myofilaments to Ca2+, and it greatly blunted the effects of LC2 phosphorylation on Ca2+ sensitivity. Such a synergistic interaction between cTnI phosphorylation and LC2 phosphorylation may also help to explain certain characteristics of PKC phosphorylation sites on cardiac myofilament proteins. First, many myofilament proteins, including cTnI and LC2, appear to exhibit substantial pre-existing phosphorylation of PKC sites under basal conditions. Second, it has been difficult to correlate functional changes with PKC phosphorylation of myofilament proteins, in large part because following agonist, kinase or phosphatase treatment, multiple proteins change their phosphorylation state and these changes are usually modest (substoichiometric; Damron et al. 1995; Huang et al. 1997; van der Velden et al. 2003). The expression of non-phosphorylatable cTnI proteins on a null background may afford significant advantages in studies of the functional consequences of cTnI phosphorylation, by eliminating both basal and stimulated phosphorylation of known sites and by eliminating the uncertainty of low but possibly influential changes in endogenous cTnI phosphorylation.
Considerable light has recently been shed on the relative contributions of cTnI and TnT in mediating the effects of PKC on altered contractile function (Montgomery et al. 2001). In this study, TnT phosphorylation could be completely prevented by transgenic expression of fast skeletal TnT in the mouse heart. This manipulation abolished the decrease of maximum tension caused by phorbol ester, suggesting that TnT phosphorylation mediated this tension decline. Interestingly in these non-phosphorylatable TnT transgenic mice, phosphorylation of TnI increased with phorbol ester treatment in parallel with an increase in Ca2+ sensitivity of tension (although the data were not interpreted in this way). These authors emphasised that the functional consequences of cTnI phosphorylation were quite different in the presence and absence of TnT phosphorylation, suggesting interdependencies between these phosphoproteins (Montgomery et al. 2001).
In our experiments, agonist treatment had no effect on maximum isometric tension in any mouse lines investigated in response to ET-1 or PMA. We cannot completely rule out that such changes in maximum tension occurred, because a change of <15 % could have gone undetected in our experiments. However, we also did not observe significant changes in TnT phosphorylation, the parameter that Montgomery et al. (2001) correlated with reduced tension. Reasons for differences in TnT phosphorylation in the present study and the study of Montgomery et al. (2001) are unknown. We did observe a statistically significant 25 % decrease in maximum MgATPase rate following PKC activation, and a statistically significant 38 % increase following PKA activation, but no change in maximum tension (< 15 %). Other investigators have interpreted changes in MgATPase rate that are uncoupled from changes in isometric tension as reflecting modulation of the tension cost of contraction (de Tombe & Steinen, 1995; Saeki et al. 1997). It is premature to interpret our results in this way, because MgATPase rates and isometric tension were not measured under the same conditions (e.g. sarcomere length, crossbridge strain, ATP/ADP and Pi ratios). These differences may also account for subtle differences in the effects of agonist stimulation when measured by MgATPase activity versus isometric tension.
The biological significance of the effects of PKC on myofilament function is not yet completely clear. We propose that PKC signalling reduces the expenditure of ATP both by slowing crossbridge turnover (Venema & Kuo, 1993; Strang & Moss, 1995; McClellan et al. 1996; Lester & Hofmann, 2000; Pyle et al. 2001; Knott et al. 2002) and by reducing the requirement for Ca2+ pumping. The enhanced Ca2+ sensitivity may also be associated with a slowing of Ca2+ dissociation from troponin C, and prolongation of attached crossbridges during the force-generating or work-performing cycle. All of these changes could contribute to a more efficient working heart (McClellan et al. 1996; Saeki et al. 1997), and possibly represent a beneficial fine-tuning of contractile performance under conditions of chronic stress. The two transgenic mouse lines used here, cTnI-Ala2nb and cTnI-Ala5nb, will be instrumental in testing this hypothesis further with direct measurements of contractile efficiency and economy.
The rate of ATP hydrolysis by actomyosin is related to myosin crossbridge turnover dynamics, and the effects of PKA on both MgATPase rate and crossbridge dynamics are controversial (Winegrad et al. 1986; Hofmann & Lange, 1994; Strang et al. 1994; de Tombe & Steinen, 1995; Saeki et al. 1997; Herron et al. 2001; Kentish et al. 2001; Patel et al. 2001; Turnbull et al. 2002). Some investigators report an increase in maximum ATPase rate (Winegrad et al. 1986), tension cost (Saeki et al. 1997) and crossbridge cycling rate (Strang et al. 1994; Herron et al. 2001; Kentish et al. 2001; Patel et al. 2001; Turnbull et al. 2002), whereas others report no change in these parameters following PKA activation (Hofmann & Lange, 1994; de Tombe & Steinen, 1995; Noland et al. 1995). It may be that the species or age of experimental animals and other experimental conditions are critical factors. Nevertheless, we observed a substantial (38 %) increase in maximal MgATPase rate in wild-type mouse hearts that was absent in both cTnI-Ala2nb and cTnI-Ala5nb hearts under the same conditions. This indicates that in mice, at least under some experimental conditions, β-adrenergic signalling stimulates ATP turnover by the cardiac myofibrillar apparatus as a result of phosphorylation of cTnI on serines23/24.
In summary, expression of non-phosphorylatable cTnI mutants in cTnI-null mouse hearts revealed that PKC enhances myofilament Ca2+ sensitivity and depresses MgATPase rates, at least in part by phosphorylation of cTnI. These regulatory effects on cardiac myofibrils were the opposite of the effects of PKA activation, which resulted in a reduced myofilament Ca2+ sensitivity and enhanced MgATPase rates. Thus, even though PKA and PKC signalling may both promote positive inotropy under some conditions, in part by stimulating L-type Ca2+ channels (He et al. 2000), their effects on myofilament function are quite distinct. Phosphorylation of cTnI on separate sites appears to be capable of upregulating or downregulating ATP turnover rates as well as the amount of Ca2+ required for activation of cardiac myofilaments. The significance of these findings may be related to the involvement of neurohumoral factors in the response of the heart to acute versus chronic stress. Activation of PKA pathways in the heart during the fight-or-flight response would increase crossbridge turnover (Strang et al. 1994; Kentish et al. 2001), decrease myofilament Ca2+ sensitivity and increase relaxation rate (Robertson et al. 1982; Zhang et al. 1995; Li et al. 2000; Pi et al. 2002a). Each of these changes would support the heart's need for maximising power output (Herron et al. 2001), which is energetically costly but necessary in acutely stressful situations. In contrast, we propose that under conditions of chronic stress, PKC signalling reduces the consumption of ATP, both by slowing crossbridge turnover (Venema & Kuo, 1993; Strang & Moss, 1995; McClellan et al. 1996; Lester & Hofmann, 2000; Pyle et al. 2001; Knott et al. 2002) and by reducing the requirements for pumping Ca2+. Enhanced myofilament Ca2+ sensitivity may have the added advantage of slowing Ca2+ dissociation from troponin C (Robertson et al. 1982), thereby prolonging attached crossbridge lifetimes when the rapid shortening of heart myofilaments is not necessary. The new data support the concept that phosphorylation of cTnI on distinct sites is instrumental in switching the heart from one type of stress response to another. Finally, the mouse models used here will provide an avenue for further investigation into the role of cTnI phosphorylation in normal and pathological processes at levels of organisation from molecules to cells to organs.
Acknowledgments
This work was supported by NIH grant P01 HL47053. The authors thank Dr R. J. Solaro for providing the wild-type cardiac TnI clone, Dr Xupei Huang for purification of MLCK in the laboratory of Dr Primal de Lanerolle at the University of Illinois Chicago, and Dr John Neumann at the University of Cincinnati for generating the transgenic founder mice.
REFERENCES
- Adelstein RS, Klee CB. Purification and characterization of smooth muscle myosin light chain kinase. J Biol Chem. 1981;256:7501–7509. [PubMed] [Google Scholar]
- Andersen GG, Qvigstad E, Schiander I, Aass H, Osnes JB, Skomedal T. Alpha(1)AR-induced positive inotropic response in heart is dependent on myosin light chain phosphorylation. Am J Physiol. 2002;283:H1471–1480. doi: 10.1152/ajpheart.00232.2002. [DOI] [PubMed] [Google Scholar]
- Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- Clement O, Puceat M, Walsh MP, Vassort G. Protein kinase C enhances myosin light-chain kinase effects on force development and ATPase activity in rat single skinned cardiac cells. Biochem J. 1992;285:311–317. doi: 10.1042/bj2850311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: from adenosine receptor of KATP channel. Annu Rev Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- Damron DS, Darvish A, Murphy L, Sweet W, Moravec CS, Bond M. Arachidonic acid-dependent phosphorylation of troponin I and myosin light chain 2 in cardiac myocytes. Circ Res. 1995;76:1011–1019. doi: 10.1161/01.res.76.6.1011. [DOI] [PubMed] [Google Scholar]
- De Tombe PP, Stienen GJ. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ Res. 1995;76:734–741. doi: 10.1161/01.res.76.5.734. [DOI] [PubMed] [Google Scholar]
- Endoh M. The effects of various drugs on the myocardial inotropic response. Gen Pharmacol. 1995;26:1–31. doi: 10.1016/0306-3623(94)00144-c. [DOI] [PubMed] [Google Scholar]
- Endoh M, Blinks JR. Actions of sympathomimetic amines on the Ca2+ transients and contractions of rabbit myocardium: reciprocal changes in myofibrillar responsiveness to Ca2+ mediated through alpha- and beta-adrenoceptors. Circ Res. 1988;62:247–265. doi: 10.1161/01.res.62.2.247. [DOI] [PubMed] [Google Scholar]
- Gwathmey JK, Hajjar RJ. Effect of protein kinase C activation on sarcoplasmic reticulum function and apparent myofibrillar Ca2+ sensitivity in intact and skinned muscles from normal and diseased human myocardium. Circ Res. 1990;67:744–752. doi: 10.1161/01.res.67.3.744. [DOI] [PubMed] [Google Scholar]
- He JQ, Pi YQ, Walker JW, Kamp TJ. Endothelin-1 and photoreleased diacylglycerol increase L-type Ca current by activation of protein kinase C in rat ventricular myocytes. J Physiol. 2000;524:807–820. doi: 10.1111/j.1469-7793.2000.00807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron TJ, Korte FS, McDonald KS. Power output is increased after phosphorylation of myofibrillar proteins in rat skinned cardiac myocytes. Circ Res. 2001;89:1184–1190. doi: 10.1161/hh2401.101908. [DOI] [PubMed] [Google Scholar]
- Hofmann PA, Lange JH., III Effects of phosphorylation of troponin I and C protein on isometric tension and velocity of unloaded shortening in skinned single cardiac myocytes from rats. Circ Res. 1994;74:718–726. doi: 10.1161/01.res.74.4.718. [DOI] [PubMed] [Google Scholar]
- Huang L, Wolska BM, Montgomery DE, Burkart EM, Buttrick PM, Solaro RJ. Increased contractility and altered Ca(2+) transients of mouse heart myocytes conditionally expressing PKCbeta. Am J Physiol. 2001;280:C1114–1120. doi: 10.1152/ajpcell.2001.280.5.C1114. [DOI] [PubMed] [Google Scholar]
- Huang X, Pi Y, Lee KJ, Henkel AS, Gregg RG, Powers PA, Walker JW. Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Circ Res. 1999;84:1–8. doi: 10.1161/01.res.84.1.1. [DOI] [PubMed] [Google Scholar]
- Huang XP, Pi Y, Lokuta AJ, Greaser ML, Walker JW. Arachidonic acid stimulates protein kinase C-epsilon redistribution in heart cells. J Cell Sci. 1997;110:1625–1634. doi: 10.1242/jcs.110.14.1625. [DOI] [PubMed] [Google Scholar]
- Jideama NM, Noland TA, Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun YA, Kuo JF. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem. 1996;271:23277–23283. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- Johns EC, Simnett SJ, Mulligan IP, Ashley CC. Troponin I phosphorylation does not increase the rate of relaxation following laser flash photolysis of diazo-2 in guinea-pig skinned trabeculae. Pflügers Arch. 1997;433:842–844. doi: 10.1007/s004240050353. [DOI] [PubMed] [Google Scholar]
- Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- Knott A, Purcell I, Marston S. In vitro motility analysis of thin filaments from failing and non-failing human heart: troponin from failing human hearts induces slower filament sliding and higher Ca(2+) sensitivity. J Mol Cell Cardiol. 2002;34:469–482. doi: 10.1006/jmcc.2002.1528. [DOI] [PubMed] [Google Scholar]
- Kranias EG, Solaro RJ. Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature. 1982;298:182–184. doi: 10.1038/298182a0. [DOI] [PubMed] [Google Scholar]
- Lester JW, Hofmann PA. Role for PKC in the adenosine-induced decrease in shortening velocity of rat ventricular myocytes. Am J Physiol Heart. 2000;279:H2685–2693. doi: 10.1152/ajpheart.2000.279.6.H2685. [DOI] [PubMed] [Google Scholar]
- Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol. 2000;278:H769–779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- McClellan G, Weisberg A, Winegrad S. Effect of endothelin-1 on actomyosin ATPase activity. Implications for the efficiency of contraction. Circ Res. 1996;78:1044–1050. doi: 10.1161/01.res.78.6.1044. [DOI] [PubMed] [Google Scholar]
- Montgomery DE, Chandra M, Huang Q, Jin J, Solaro RJ. Transgenic incorporation of skeletal TnT into cardiac myofilaments blunts PKC-mediated depression of force. Am J Physiol. 2001;280:H1011–1018. doi: 10.1152/ajpheart.2001.280.3.H1011. [DOI] [PubMed] [Google Scholar]
- Morano I, Hofmann F, Zimmer M, Ruegg JC. The influence of P-light chain phosphorylation by myosin light chain kinase on the calcium sensitivity of chemically skinned heart fibres. FEBS Lett. 1985;189:221–224. doi: 10.1016/0014-5793(85)81027-9. [DOI] [PubMed] [Google Scholar]
- Noland TA, Jr, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V, Solaro RJ, Kuo JF. Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca(2+)-stimulated MgATPase of reconstituted actomyosin S-1. J Biol Chem. 1995;270:25445–25454. doi: 10.1074/jbc.270.43.25445. [DOI] [PubMed] [Google Scholar]
- Noland TA, Jr, Kuo JF. Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca2(+)-stimulated actomyosin MgATPase activity. J Biol Chem. 1991;266:4974–4978. [PubMed] [Google Scholar]
- Noland TA, Jr, Kuo JF. Phosphorylation of cardiac myosin light chain 2 by protein kinase C and myosin light chain kinase increases Ca(2+)-stimulated actomyosin MgATPase activity. Biochem Biophys Res Commun. 1993;193:254–260. doi: 10.1006/bbrc.1993.1617. [DOI] [PubMed] [Google Scholar]
- Noland TA, Jr, Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, Solaro RJ, Kuo JF. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996;35:14923–14931. doi: 10.1021/bi9616357. [DOI] [PubMed] [Google Scholar]
- Patel JR, Fitzsimons DP, Buck SH, Muthuchamy M, Wieczorek DF, Moss RL. PKA accelerates rate of force development in murine skinned myocardium expressing alpha- or beta-tropomyosin. Am J Physiol. 2001;280:H2732–2739. doi: 10.1152/ajpheart.2001.280.6.H2732. [DOI] [PubMed] [Google Scholar]
- Pi Y, Kemnitz KR, Zhang D, Kranias EG, Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ Res. 2002a;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- Pi Y, Sreekumar R, Huang X, Walker JW. Positive inotropy mediated by diacylglycerol in rat ventricular myocytes. Circ Res. 1997;81:92–100. doi: 10.1161/01.res.81.1.92. [DOI] [PubMed] [Google Scholar]
- Pi Y, Walker JW. Diacylglycerol and fatty acids synergistically increase cardiomyocyte contraction via activation of PKC. Am J Physiol. 2000;279:H26–34. doi: 10.1152/ajpheart.2000.279.1.H26. [DOI] [PubMed] [Google Scholar]
- Pi YQ, Kemnitz K, Walker JW. Control of myocardial MgATPase by troponin I phosphorylation. Biophysical Journal. 2002b;82:596a. [Google Scholar]
- Puceat M, Clement O, Lechene P, Pelosin JM, Ventura-Clapier R, Vassort G. Neurohormonal control of calcium sensitivity of myofilaments in rat single heart cells. Circ Res. 1990;67:517–524. doi: 10.1161/01.res.67.2.517. [DOI] [PubMed] [Google Scholar]
- Pyle WG, Lester JW, Hofmann PA. Effects of kappa-opioid receptor activation on myocardium. Am J Physiol. 2001;281:H669–678. doi: 10.1152/ajpheart.2001.281.2.H669. [DOI] [PubMed] [Google Scholar]
- Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–263. [PubMed] [Google Scholar]
- Saeki Y, Kobayashi T, Minamisawa S, Sugi H. Protein kinase A increases the tension cost and unloaded shortening velocity in skinned rat cardiac muscle. J Mol Cell Cardiol. 1997;29:1655–1663. doi: 10.1006/jmcc.1997.0401. [DOI] [PubMed] [Google Scholar]
- Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104:557–567. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- Solaro R. Modulation of Cardiac Myofilament Activity by Protein Phosphorylation. Vol. 1. New York: Blackwell Science Inc; 2002. [Google Scholar]
- Strang KT, Moss RL. Alpha 1-adrenergic receptor stimulation decreases maximum shortening velocity of skinned single ventricular myocytes from rats. Circ Res. 1995;77:114–120. doi: 10.1161/01.res.77.1.114. [DOI] [PubMed] [Google Scholar]
- Strang KT, Sweitzer NK, Greaser ML, Moss RL. Beta-adrenergic receptor stimulation increases unloaded shortening velocity of skinned single ventricular myocytes from rats. Circ Res. 1994;74:542–549. doi: 10.1161/01.res.74.3.542. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Bogoyevitch MA. Intracellular signalling through protein kinases in the heart. Cardiovasc Res. 1995;30:478–492. [PubMed] [Google Scholar]
- Swartz DR, Zhang D, Yancey KW. Cross bridge-dependent activation of contraction in cardiac myofibrils at low pH. Am J Physiol. 1999;276:H1460–1467. doi: 10.1152/ajpheart.1999.276.5.H1460. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Stull JT. Phosphorylation of myosin in permeabilized mammalian cardiac and skeletal muscle cells. Am J Physiol. 1986;250:C657–660. doi: 10.1152/ajpcell.1986.250.4.C657. [DOI] [PubMed] [Google Scholar]
- Swiderek K, Jaquet K, Meyer HE, Schachtele C, Hofmann F, Heilmeyer LM., Jr Sites phosphorylated in bovine cardiac troponin T and I. Characterization by 31P-NMR spectroscopy and phosphorylation by protein kinases. Eur J Biochem. 1990;190:575–582. doi: 10.1111/j.1432-1033.1990.tb15612.x. [DOI] [PubMed] [Google Scholar]
- Takeishi Y, Chu G, Kirkpatrick DM, Li Z, Wakasaki H, Kranias EG, King GL, Walsh RA. In vivo phosphorylation of cardiac troponin I by protein kinase Cbeta2 decreases cardiomyocyte calcium responsiveness and contractility in transgenic mouse hearts. J Clin Invest. 1998;102:72–78. doi: 10.1172/JCI2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzic A, Puceat M, Clement O, Scamps F, Vassort G. Alpha 1-adrenergic effects on intracellular pH and calcium and on myofilaments in single rat cardiac cells. J Physiol. 1992;447:275–292. doi: 10.1113/jphysiol.1992.sp019002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull L, Hoh JF, Ludowyke RI, Rossmanith GH. Troponin I phosphorylation enhances crossbridge kinetics during beta-adrenergic stimulation in rat cardiac tissue. J Physiol. 2002;542:911–920. doi: 10.1113/jphysiol.2002.022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Velden J, Papp Z, Boontje NM, de Zaremba Jong JW, Janssen PML, Hasenfuss G, Steinen GJM. Cardiovasc Res. 2003;57:505–514. doi: 10.1016/s0008-6363(02)00662-4. [DOI] [PubMed] [Google Scholar]
- Venema RC, Kuo JF. Protein kinase C-mediated phosphorylation of troponin I and C-protein in isolated myocardial cells is associated with inhibition of myofibrillar actomyosin MgATPase. J Biol Chem. 1993;268:2705–2711. [PubMed] [Google Scholar]
- Venema RC, Raynor RL, Noland TA, Jr, Kuo JF. Role of protein kinase C in the phosphorylation of cardiac myosin light chain 2. Biochem J. 1993;294:401–406. doi: 10.1042/bj2940401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winegrad S, Weisberg A, Lin LE, McClellan G. Adrenergic regulation of myosin adenosine triphosphatase activity. Circ Res. 1986;58:83–95. doi: 10.1161/01.res.58.1.83. [DOI] [PubMed] [Google Scholar]
- Zhang D, Pi YQ, Kemnitz KR, Walker JW. Effects of troponin I phosphorylation on myocardial contraction. Biophys J. 2002;82:397a. [Google Scholar]
- Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]