

Abstract
Almost all aspects of cardiac function are sensitive to modest changes of temperature. We have examined the thermal sensitivity of intracellular pH regulation in the heart. To do this we determined the temperature sensitivity of pHi, intracellular buffering capacity, and the activity of sarcolemmal acid-extrusion proteins, Na+-H+ exchange (NHE) and Na+-HCO3− co-transport (NBC) in guinea-pig isolated ventricular myocytes. pHi was recorded fluorimetrically with acetoxymethyl (AM)-loaded carboxy-SNARF-1 at either 27 or 37°C. At 27°C, intrinsic (non-CO2-dependent) buffering power (βi) was ˜60 % of that at 37°C. Acid-extrusion (Je) through NHE was ˜50 % slower than at 37°C, consistent with a Q10 of ˜2. In 5 % CO2/HCO3−-buffered conditions, in the presence of 30 μm cariporide to inhibit NHE, acid extrusion via NBC was also slowed at 27°C, suggestive of a comparable Q10. Resting pHi at 27°C was similar in Hepes- or 5 % CO2/HCO3−-buffered superfusates but, in both cases, was ˜0.1 pH units lower at 37°C. The higher the starting pHi, the larger was the thermally induced fall of pHi, consistent with a mathematical model where intrinsic buffers with a low principal pKa (e.g. close to 6.0) are less temperature-sensitive than those with a higher pKa. The high temperature sensitivity of pHi regulation in mammalian cardiac cells has implications for experimental work conducted at room temperature. It also has implications for the ability of intracellular acidosis to generate intracellular Na+ and Ca2+ overload, cardiac injury and arrhythmia in the heart.
Temperature is an important modulator of intracellular protein activity. One category of protein is the group of membrane ion transporters that tightly regulates intracellular pH (pHi). In mammalian cardiac cells, for example, this group includes the sarcolemmal, acid-equivalent extrusion proteins, Na+-H+ exchange (NHE) and Na+-HCO3− co-transport (NBC) (Leem et al. 1999). Intracellular pH is also an important modulator of cell function, affecting contractile (Bountra & Vaughan-Jones, 1989) and electrical activity in the heart (Orchard & Cingolani, 1994; Ch'en et al. 1998). Some effects of temperature on cellular activity may therefore be mediated indirectly through an influence on intracellular pH regulation.
Hypothermia (4–10°C) is commonly used during cardiac surgical procedures (Hearse et al. 1981). In addition, basic experimental work on cardiac tissue is frequently carried out at room temperature. Many of the intracellular ionic changes associated with reduced temperature have yet to be extensively investigated. This is particularly pertinent in the heart, given the intimate homeostatic link between intracellular pH, Ca2+, contractility and electrical rhythm. Overactivity of sarcolemmal Na+-H+ exchange (Scholz et al. 1995; Xue et al. 1996; Hartmann & Decking, 1999; Xiao & Allen, 2000) and possibly NBC (Schafer et al. 2000) has been implicated in cardiac injury and arrhythmogenesis during intracellular acid-overload, observed typically during myocardial ischaemia and reperfusion (Karmazyn, 1999). It is of interest that, under these conditions, hypothermia confers some cardioprotection (Shipolini et al. 1997). Knowledge of the temperature dependence of NHE and NBC may be valuable in understanding the protective mechanisms involved.
A recent study in rat cardiomyocytes indicates that NHE activity is slowed considerably by moderate hypothermia (Hoshino & Avkiran, 2001). That report utilised extracellular superfusates buffered with Hepes rather than a physiologically relevant CO2/HCO3− system. We have therefore studied effects of temperature on acid extrusion transporters in guinea-pig ventricular myocytes exposed to either Hepes- or CO2/HCO3−-buffered superfusates. This enables us to assess more comprehensively the effects of temperature on both NHE and NBC. The rate at which these transporters mediate a change of pHi depends, in part, on intracellular buffering power. Intrinsic (non-CO2-dependent) buffering has been variously reported to increase (Do et al. 1996; Hoshino & Avkiran, 2000), decrease (Graber et al. 1992; van Dijk et al. 1997) or remain unchanged (Kiang et al. 1990) following a temperature reduction. We therefore investigated the temperature sensitivity of intracellular buffering. Finally, the thermal sensitivity of pHi was examined, given that changes in buffering and sarcolemmal acid transport may ultimately affect its steady state value. The consequences of the temperature dependence of pHi regulation in the heart, particularly in relation to its effects on contractility and arrhythmogenesis, are discussed.
Preliminary reports of some of this work have been published in abstract form (Ch'en et al. 2000, 2001).
METHODS
Isolation of guinea-pig ventricular myocytes
The composition of cell isolation solutions and the detailed procedure have been previously described (Lagadic-Gossmann et al. 1992). In brief, single ventricular myocytes were isolated from 350–450 g albino guinea-pigs (killed by cervical dislocation; procedure conformed with the UK Animals (Scientific Procedures) Act 1986) using enzymatic and mechanical dispersion (0.7 mg ml−1 collagenase, Boehringer Mannheim and 0.04 mg ml−1 protease, Sigma). The cells were suspended in Hepes-buffered Dulbecco's Modified Eagle's Medium and left at room temperature (18–22°C) until use. Only rod-shaped myocytes showing Ca2+ tolerance were used in this work.
Measurement of intracellular pH
SNARF loading and calibration
The intracellular pH of isolated ventricular myocytes was measured using the dual emission fluorophore carboxy-SNARF-1, loaded as the acetoxymethyl (AM) ester. The full procedure for dye loading, measuring pHi, and calibrating dye signals has been described previously (e.g. Buckler & Vaughan-Jones, 1990; Sun et al. 1996). Briefly, isolated myocytes were loaded for 8 min at room temperature with 10 μm carboxy-SNARF-1 AM ester. SNARF fluorescence from individual cells was excited at 540 ± 12 nm and measured simultaneously at 590 ± 5 and 640 ± 5 nm with an inverted microscope converted for epifluorescence (Nikon Diaphot). Signals were digitised at 0.5 kHz (CED 1401) and the emission ratio calculated and converted to a pHi value using the pH ratiometric fluorescence equation (Buckler & Vaughan-Jones, 1990; Sun et al. 1996). The pH calibration data for this equation were obtained in situ from individual cells using the nigericin (10 μm) technique (see below for calibration solutions) and averaged for more than 10 cells from at least three animals. The data were acquired routinely every 2 months for the terms Rmax (maximum emission ratio at pH 5.5), Rmin (minimum ratio at pH 9.5), and F640,max/min (9.5/5.5 pH fluorescence ratio measured at 640 nm), measured at both 27 and 37°C. Typical values at 27°C were 1.648, 0.266 and 1.698, respectively. These predict a −log dissociation constant (pKa) of 7.43 for intracellular SNARF. Typical values at 37°C were 1.623, 0.231 and 2.035, respectively. These predict a pKa of 7.36 for intracellular SNARF. To reduce potential contamination of the cell superfusion system with nigericin (Richmond & Vaughan-Jones, 1997), nigericin calibration was not performed after every experiment; instead, the most recently acquired pH calibration data were used as default values.
Cleaning the superfusion system
The apparatus was thoroughly cleaned following a nigericin calibration. The superfusion lines were replaced and the superfusion chamber and switcher tap (see Richmond & Vaughan-Jones, 1997, for details of superfusion) were dismantled and soaked in ethanol for several hours. This was followed by a soak for at least 12 h in 20 % Decon 75 (Decon Laboratories Ltd, Sussex, UK), and then simmering in deionised water for several hours.
Solutions
Hepes-buffered Tyrode solution
Standard Hepes-buffered Tyrode solution contained (mm): NaCl 140, KCl 4.5, MgCl2 1, CaCl2 2, Hepes 20 and glucose 11. This was adjusted to pH 7.4 at 37 or 27°C with 4 m NaOH.
CO2/HCO3−-buffered Tyrode solution
Standard CO2/HCO3−-buffered Tyrode solution pH 7.4 had the same composition as Hepes-buffered Tyrode solution but with [NaCl] reduced to 125 mm, and 22 mm NaHCO3 substituted for Hepes. All CO2/HCO3−-buffered Tyrode solutions at pH 7.4 were equilibrated with 5 % CO2 and 95 % air for at least 1 h prior to starting experiments.
Calibration solutions
Nigericin calibration solutions contained (mm): KCl 140, MgCl2 1, EGTA 0.5 and nigericin 0.01, buffered with one of the following organic buffers (Sigma, UK): 20 mm Mes (pH 5.5), 20 mm Pipes (pH 6.5), 20 mm Hepes (pH 7.5) or 20 mm Capso (pH 9.5), and were adjusted to the correct pH with 4 m NaOH at 37 or 27°C.
Chemicals
When 15 mm ammonium chloride was used, it was added directly to solutions without osmotic compensation. Hoe 642 (cariporide) was kindly provided by Aventis (formerly Hoechst AG, Germany) and added as a solid to perfusates shortly before use. All other chemicals were from Sigma (UK) and Merck (UK).
Calculation of acid-equivalent fluxes
Acid-equivalent fluxes (Je) were estimated as the product of the rate of pHi recovery (dpHi/dt) following an intracellular acid load (typically pre-pulsing with 15 mm NH4Cl), and the total intracellular buffering power (βtot).
The value of dpHi/dt was determined by computer as the least squares regression line fitted to the initial change of calibrated pHi sampled at 0.5 s intervals over a 0.5–1 min time period. The pHi associated with the calculated dpHi/dt was taken to be the mid-point pHi of linear best fit. Note that comparisons of acid-equivalent fluxes in the present work were made at a common mid-point pHi.
The term βtot is the sum of intrinsic buffering power (βi) plus buffering power due to CO2/HCO3− (βCO2). βi in guinea-pig ventricular myocytes at 37°C has been estimated as:
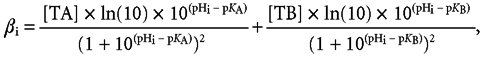 |
(1) |
where [TA] and [TB] are the total concentrations of buffer A and buffer B, respectively. The values for the parameters were calculated as: [TA]= 84.22 mm, [TB]= 29.38 mm, pKA= 6.03 and pKB= 7.57 (Leem et al. 1999), and have been used in the present work. The value for βCO2 was estimated using the Henderson-Hasselbach equation as: βCO2= 2.303[HCO3−]i (Leem et al. 1999). This assumes the same pKa for CO2/HCO3− and the same solubility for CO2 on either side of the sarcolemma.
Experimental determination of intracellular buffering power
In experiments where βi (mm) was determined experimentally, intracellular acid loads of differing magnitude were induced by ammonium removal and the consequent change in pHi was used to compute intrinsic buffering power, βi (Roos & Boron, 1981):
Removing extracellular NH4+ results in the depletion of intracellular NH4+ that exits as NH3, leaving behind H+i. Since [H+]i can thus be equated with [NH4+]i at the moment of removal, the expression for βi becomes:
This can then be evaluated with [NH4+]i expressed as a rearrangement of the Henderson-Hasselbalch equation:
where C is the total extracellular concentration of the weak base and assuming the dissociation constant of NH4+ to be the same intracellularly and extracellularly at both 37 and 27°C. Note that the method assumes a rapid equilibration of NH3 across the sarcolemma and that during the acid loading phase, significant transmembrane transport of acid-equivalents does not occur.
By a similar method, prepulsing a cell with sodium acetate (salt of a weak acid) induces an intracellular alkalosis that may be used to estimate βi. The concentration of intracellular base added is assumed to equal [Acetate]i at the end of the prepulse period:
where C is the total extracellular concentration of sodium acetate, assuming the pKa of acetic acid remains unchanged within the cytoplasmic compartment.
Determination of bath temperature changes
To ensure that pHi measurements were made at the appropriate temperature, the time course of change in bath temperature on changing heater settings for the superfusate was measured. In standard Hepes-buffered solution, pHo 7.4, the heater temperature was step-changed from 27 to 37°C and the bath temperature was monitored. Approximately 60 s was needed to raise the bath temperature to 37°C (data not shown). All temperature-induced effects on pHi in the present work were therefore measured ≈1 min after changing the heater temperature.
RESULTS
Thermal sensitivity of carboxy-SNARF-1 ratio
Given that we assess pHi regulation by monitoring intracellular SNARF fluorescence, we initially investigated the dye's temperature sensitivity. Figure 1A shows results from a typical series of calibration experiments of carboxy-SNARF-1 using the nigericin technique (Sun et al. 1996), performed at 27 and 37°C (filled triangles and circles, respectively). The fluorescence ratios obtained were different at the two temperatures and, for a given pHi, fell with an increase of temperature.
Figure 1. Temperature dependence of SNARF ratio.
A, intracellular SNARF fluorescence ratio was calibrated by superfusing nigericin (10 μm) solutions of pH 5.5, 6.5, 7.0, 7.5 and 8.5 at 37°C (•) and 27°C (▴). The arrow indicates that at pHi values close to typical pHi, decreasing temperature at a constant pHi causes an increase in SNARF ratio. B, temperature-dependent shifts of intracellular SNARF ratio that are independent of pHi. Extracellular pH was kept constant at 7.0 in the presence of 10 μm nigericin while the temperature was decreased from 37 to 27°C. Note that while pHi will be constant under these conditions, there was nevertheless a large increase in SNARF ratio.
The dynamic range of the dye's fluorescence ratio increased with increasing temperature: Rmax/Rmin was 6.19 at 27°C and 7.03 at 37°C. These results are in good agreement with work on carboxy-SNARF-1 in single mouse muscle fibres (Westerblad et al. 1997). The pKa values of SNARF at the two temperatures were similar: 7.43 at 27°C and 7.36 at 37°C, and are consistent with previous estimates in vitro and intracellularly (Buckler & Vaughan-Jones, 1990; Blank et al. 1992; Westerblad et al. 1997).
Given the above temperature-dependent changes in the spectral properties of SNARF, care must be taken to use calibration data acquired at the relevant temperature. This is emphasised in Fig. 1B, which shows the direct effect of temperature on the intracellular SNARF ratio. The cell's pHi was clamped throughout the experiment at 7.0 by superfusing a high K+o solution containing 10 μm nigericin (see Methods). Lowering the temperature by 10°C reversibly increased the SNARF ratio despite that fact that pHo, and hence pHi, would have been constant. In this case, the observed shift in the intracellular ratio was related directly to the temperature change and not to any change in pHi. The two values of SNARF ratio obtained in Fig. 1B at 37 and 27°C concurred with the calibration plots in Fig. 1A, as denoted by the asterisk. SNARF calibration parameters appropriate for a given temperature have therefore been used throughout the present work.
Thermal sensitivity of intrinsic buffering
To ensure the accurate evaluation of acid-equivalent efflux (Je), the corresponding intrinsic buffering power (βi) needs to be calculated at the same temperature. The value of βi has recently been estimated over a wide pHi range (6.2–7.7) at 37°C (Leem et al. 1999; Zaniboni et al. 2003), but has not been established for 27°C.
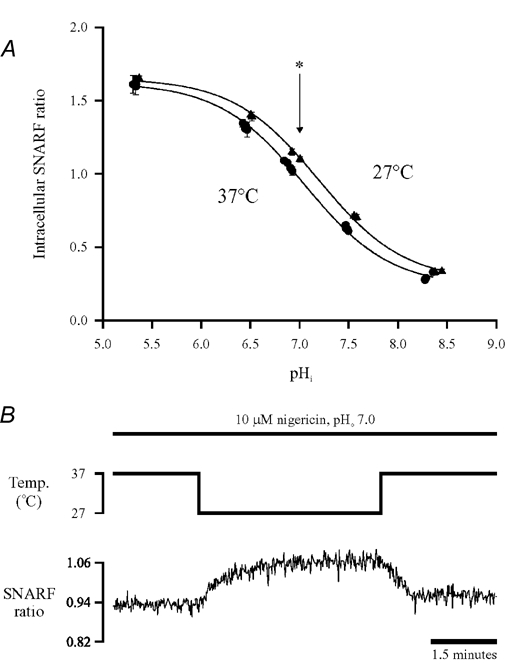
Figure 2 illustrates an experiment where intrinsic buffering power was estimated at both temperatures. A cell was superfused with Hepes-buffered Tyrode solution, i.e. nominally free of CO2/HCO3−, in the presence of 30 μm cariporide (Hoe 642), a high affinity inhibitor of Na+-H+ exchange (Scholz et al. 1995), to prevent sarcolemmal acid efflux. In some experiments, Cl− was also removed from the superfusate (gluconate-substituted) to inhibit acid-equivalent influx on sarcolemmal Cl−-OH− exchange (CHE), although this made no significant difference (data not shown). The cell was acid-loaded by using the ammonium pre-pulse technique (see Methods), and the fall of pHi was recorded at 27 and 37°C. As illustrated in Fig. 2, incremental reduction of extracellular ammonium concentration during the pre-pulse period (40, 20, 10, 5 mm and zero) induced a step-wise fall of pHi that was subsequently used to estimate βi. In other experiments (not shown; see Methods), in order to measure βi at higher values of pHi, incremental reduction of extracellular acetate was used during the prepulse period (80, 40, 20 mm and zero) to induce a stepwise rise of pHi. Significantly in all cases, the step-wise change in pHi was larger at 27°C, suggesting that at a common pHi, βi was reduced at the lower temperature.
Figure 2. Ammonium prepulses at two temperatures.
Stepwise intracellular acid loads were induced sequentially by superfusing with 20 mm NH4Cl and then reducing extracellular ammonium concentration to 10, 5 and 0 mm, first at 27°C and then again at 37°C. All solutions contained 30 μm Hoe 642 to inhibit Na+−H+ exchange and were Hepes-buffered. (pHo 7.4). Each stepwise fall of pHi was used to calculate intracellular intrinsic buffering, βi. The resulting value is listed for the mid-point of each step fall in pHi.
Figure 3A shows estimates of βi from experiments similar to that shown in Fig. 2 using ammonium withdrawal. These have been pooled with results using acetate withdrawal. Sample measurements of βi performed at 37°C are in good agreement with the relationship for intrinsic buffering power determined previously at this temperature (continuous grey line; Leem et al. 1999; calculated using eqn (1) in Methods of present paper; see also Zaniboni et al. 2003). For any given pHi, the values of βi measured at 27°C are significantly lower. Intrinsic buffering at 27°C, expressed as a percentage of that at 37°C, is plotted in Fig. 3B. Over the pHi range 7.6–6.5, buffering capacity is reduced during hypothermia by about 40 %.
Figure 3. Temperature dependence of intracellular intrinsic buffering.
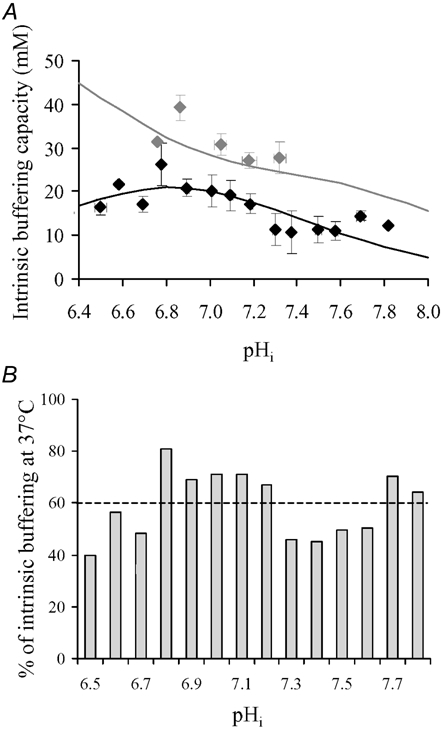
A, data pooled for intrinsic buffering measured at 37°C (grey diamonds) and 27°C (black diamonds) from cells prepulsed either with ammonium chloride or sodium acetate. Also shown (continuous grey line) is a previously determined relationship of the pHi dependence of intrinsic buffering at 37°C (from Leem et al. 1999), calculated using eqn (1) in Methods. This was cross-checked against the sample measurements of βi conducted at 37°C (grey diamonds). For convenience, the continuous black line drawn through the points at 27°C was fitted arbitrarily by a single buffer function (eqn (1)), concentration 36.5 mm and pKa 6.83. At 27°C, all data points averaged for n = 4–14, except at pHi 6.36 and 6.5 (each, n = 2), 6.58 and 7.82 (each, n = 1). At 37°C, data points averaged for n = 3, except at pHi 7.32 and 7.05 (each, n = 2) and 6.76 (n = 1). Note that for n = 2, the upper and lower s.e.m. represent the individual data points. B, intracellular buffering capacity measured at 27°C, expressed as a percentage of that computed at 37°C, has been plotted versus pHi. Over the whole pHi range tested, the mean reduction in buffering capacity during hypothermia is 40 % (indicated by horizontal dashed line).
Thermal sensitivity of Na+-H+ exchange
The effect of a 10°C decrease on the activity of Na+-H+ exchange was examined by observing pHi recovery following an intracellular acid load induced by 15 mm NH4Cl, first at 37°C and then at 27°C. The recovery in Hepes-buffered Tyrode (nominally free of CO2/HCO3−) is mediated by acid-extrusion through NHE (Lagadic-Gossmann et al. 1992). A representative experiment is shown in Fig. 4A. This demonstrates a significant slowing at 27°C, indicating a reduction in NHE activity.
Figure 4. Temperature dependence of Na+−H+ exchange.
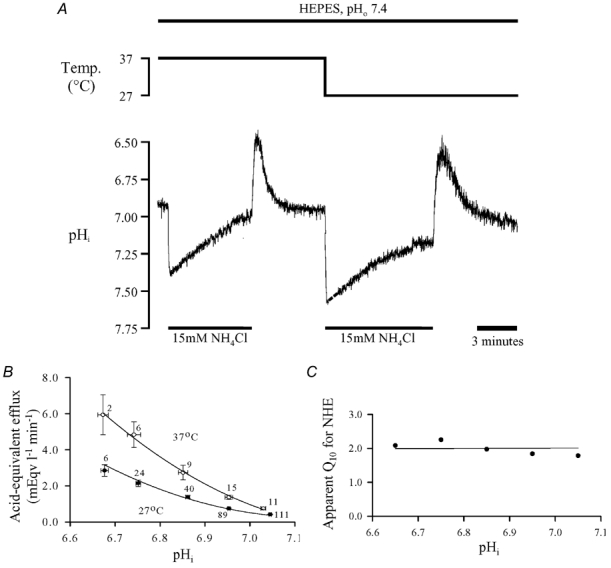
A, following an acid load (induced by 15 mm NH4Cl removal) in Hepes-buffered conditions, pHi recovery was monitored, first at 37°C and then again at 27°C. Extracellular pH was 7.4 throughout. B, acid-equivalent efflux (mean ±s.e.m.), pooled from experiments similar to that shown in A, plotted as a function of pHi at both 27°C (•) and 37°C (○). Numbers beside symbols denote the number of observations. C, apparent Q10 of Na+−H+ exchange, calculated as Q10= (acid-equivalent efflux at 37°C)/(acid-equivalent efflux at 27°C) over the pHi range shown in A.
Figure 4B pools data obtained from several experiments similar to that shown in Fig. 4A. Using βi values appropriate for each temperature and pHi (obtained from the curves plotted in Fig. 3A), NHE-mediated acid extrusion (dpHi/dt×βi) was plotted versus pHi at 27°C (filled circles) and at 37°C (open circles). Inspection of the flux data in Fig. 4B indicates an approximate halving of NHE activity on reducing temperature. Calculating the quotient of the two activities at the two temperatures gives the apparent Q10 ((acid efflux at 37°C)/(acid efflux at 27°C)). This is plotted in Fig. 4C for the pHi range 6.6–7.1 and is ≈2.0. Over most of the pHi range, the Q10 appeared to be relatively constant.
Assuming that activation energy, Ea, for NHE is independent of temperature, the integrated Arrhenius equation is: ln(kT1/kT2) =−Ea/R(1/T1− 1/T2), where R is the gas constant and kT1 and kT2 are the rate constants at temperatures T1 and T2, respectively. With the calculated rates of acid efflux at 27 and 37°C, this gives an activation energy of 55.1 kJ mol−1.
Thermal sensitivity of Na+-HCO3− co-transport
Figure 5A illustrates an experiment similar to that shown in Fig. 4A, but performed in 5 % CO2/HCO3− rather than Hepes buffer. Under these conditions, the recovery from an acid load induced by 15 mm NH4Cl removal is mediated by both NHE and NBC (Lagadic-Gossmann et al. 1992; Dart & Vaughan-Jones, 1992). To examine NBC activity in isolation, NHE activity was eliminated by adding 30 μm cariporide to the superfusate. The pHi recovery rate following ammonium removal was significantly slower at 27°C, indicating a reduction in NBC activity.
Figure 5. Temperature dependence of Na+−HCO3− co-transport.
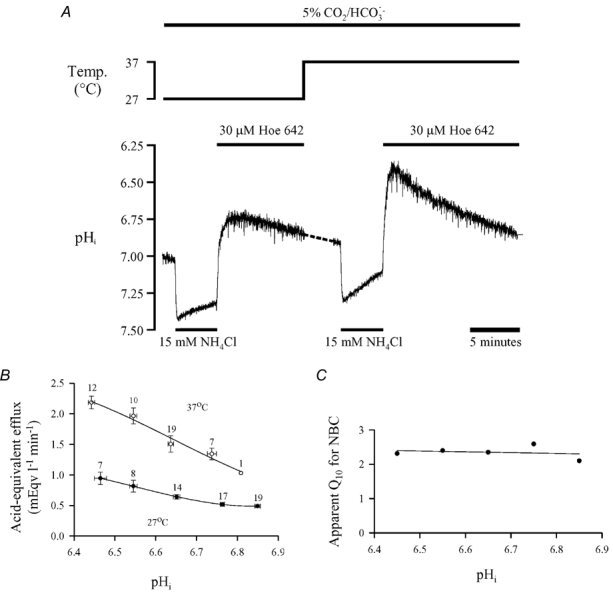
A, following an acid load (induced by 15 mm NH4Cl removal) in 5 % CO2/HCO3−-buffered conditions, pHi recovery was monitored, first at 37°C and then again at 27°C. Hoe 642 (30 μm) was added to inhibit Na+−H+ exchange and pHo was 7.35–7.40 throughout. B, acid-equivalent efflux (mean ±s.e.m.), pooled from experiments similar to that shown in A, plotted as a function of pHi at both 27°C (•) and 37°C (○). Numbers beside symbols denote the number of observations. C, apparent Q10 of Na+−HCO3− co-transport, calculated as Q10= (acid-equivalent efflux at 37°C)/(acid-equivalent efflux at 27°C) over the pHi range shown in A.
Figure 5B is a plot of data pooled from several experiments similar to those shown in Fig. 5A, including experiments where the order of the imposed temperatures was reversed, i.e. first 37°C then 27°C; there was no significant difference between the two protocols (data not shown). Acid-equivalent efflux has been plotted as a function of pHi at both 27°C (filled circles) and 37°C (open circles), and as with NHE activity, there was roughly a halving of activity on reducing temperature by 10°C. Figure 4C shows the apparent Q10 ((acid-equivalent efflux at 37°C)/(acid-equivalent efflux at 27°C)) for NBC over a range of pHi values. The apparent Q10 was, again, largely constant over the range and had a value of ≈2.4. Using the integrated Arrhenius equation above, these data give an activation energy for NBC of 65.8 kJ mol−1 of H+ extruded.
Note that in both Fig. 4B and Fig. 5B, the ordinates refer to net acid-equivalent efflux through NHE and NBC respectively. The calculated efflux values were not corrected for possible simultaneous acid influx through sarcolemmal acid-loading transporters such as Cl−-OH− exchange (CHE) and the Cl−-HCO3− anion exchanger (AE). Previous work at 37°C has shown that acid loading through these latter two transporters is sufficiently small at pHi < 7.0 that it may conveniently be ignored (Leem et al. 1999).
Thermal sensitivity of resting pHi
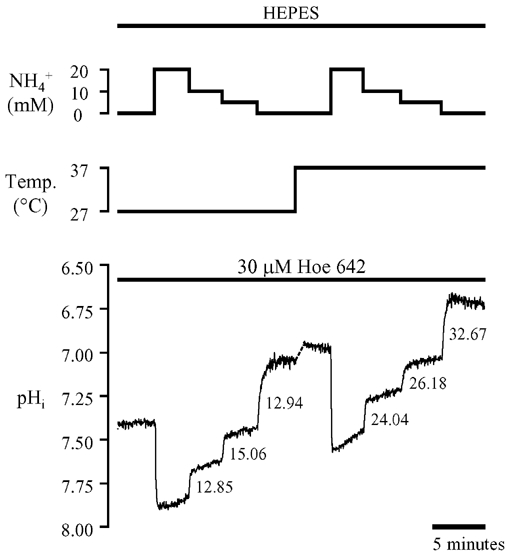
The trace shown in Fig. 6A demonstrates the typical effect on resting pHi of restoring the perfusion temperature from 27 to 37°C under 5 % CO2-buffered conditions (pHo 7.4). The middle trace shows that the raw SNARF ratio decreased within about 1 min, coinciding with the time course of rise of temperature in the bath (see Methods). Calibration of the ratiometric signal indicated that pHi became more acidic by 0.15 u within the 1 min period. Thereafter it remained constant for several minutes. On average, pHi fell by 0.13 u as shown in the histogram plotted in Fig. 6B (filled columns). Note that the discontinuity in the pHi trace in Fig. 6A corresponds to the time taken for the bath temperature to reach 37°C (see above); given the temperature dependence of SNARF, the ratio could not be properly calibrated during this period. Quantitatively similar changes of resting pHi occurred in Hepes-buffered conditions (effect not illustrated; pHo 7.4). The mean pHi values measured at the two temperatures are also plotted in Fig. 6B (open columns).
Figure 6. Temperature dependence of resting intracellular pH.
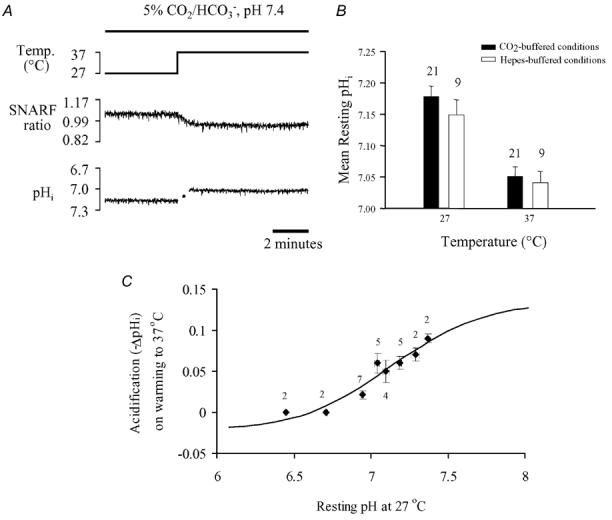
A, effect of raising bath temperature from 27 to 37°C on carboxy-SNARF-1 fluorescence ratio (middle trace) and corresponding pHi (bottom trace) in 5 % CO2/HCO3−-buffered conditions, pHo 7.35–7.40 at 37°C. B, mean resting pHi measured at 27 and 37°C for 5 % CO2/HCO3−-buffered (filled columns) and Hepes-buffered (open columns) conditions. C, pooled data (mean ±s.e.m.) of the change in pHi (-ΔpHi) following an increase in bath temperature from 27 to 37°C in Hepes-buffered conditions. Numbers beside symbols denote the number of observations. Note that for n = 2, the upper and lower s.e.m. values represent the individual data points. Low initial pHi levels were generated by pre-pulsing with 15 mm NH4Cl in the presence of 30 μm Hoe 642 (see text for details). The continuous line shows predictions of the dual-buffer model described in the Appendix.
In another set of experiments, the thermally induced fall of pHi (i.e. on raising temperature from 27 to 37°C) was examined for various starting levels of pHi, ranging from 7.35 to 6.45. This range was achieved partly by using cells not pre-pulsed with ammonium chloride where resting pHi at 27°C was 7.1–7.4, and partly by using cells pre-pulsed for varying intervals (3–6 min) with 15 mm ammonium chloride in the presence of 30 μm Hoe 642. For these experiments, only Hepes-buffered superfusates were used. The results, pooled in Fig. 6C, show that the magnitude of the thermally induced acidosis was smaller at lower starting pHi values and was absent at pHi≈6.45.
DISCUSSION
The present work shows that activity of both Na+−H+ exchange (NHE) and Na+−HCO3− co-transport (NBC) in mammalian ventricular myocytes is strongly temperature dependent, a cooling of 10°C reducing acid efflux by about 50 %. This is accompanied by a significant reduction of intrinsic buffering and a rise of resting pHi.
Thermal sensitivity of intracellular buffering power
Intrinsic buffering
At 27°C, βi is about 60 % of that at 37°C, as determined by ammonium and acetate prepulse techniques that have been validated extensively in cardiac cells (Vaughan-Jones & Wu, 1990; Bountra et al. 1990). In contrast, two other reports on reducing temperature in the heart have suggested a rise of intrinsic buffering; these were for a multicellular ventricular preparation in vitro (Do et al. 1996) and rat isolated ventricular myocytes (Hoshino & Avkiran, 2001). In non-cardiac tissues, either a decrease (Graber et al. 1992: opossum kidney cells) or no change (Aickin & Thomas, 1977b: skeletal muscle; Kiang et al. 1990: human A431 cells) has been reported.
Intrinsic buffering is attributable to the net effect of a range of constitutive, intracellular molecules, including taurine, amino acids, inorganic phosphate, proteins and dipeptides (e.g. homocarnosine, acetyl anserine). Many of these molecules contain histidine, the principal intracellular buffer (see e.g. Vaughan-Jones et al. 2002). Depending on which molecules are present, their concentrations and the temperature dependence of their respective pKa values, the overall effect of temperature on total intrinsic buffering power may be expected to differ among cell types. This serves to emphasise that, for any given preparation, changes in βi must be measured directly in situ. Values for βi should, of course, be viewed with caution if plasmalemmal acid extruders were active during the experimental measurements as this leads to an overestimate of βi (Bountra et al. 1990). In recent temperature studies, however, (ferret, rat and guinea-pig heart; Do et al. 1996; Hoshino & Avkiran, 2001; and present work, respectively) acid extrusion via NHE was pharmacologically inhibited. It should also be noted that, at 37°C, βi appears very similar among ventricular myocytes from e.g. rat, rabbit and guinea-pig (Zaniboni et al. 2003). The cause of the different thermal sensitivity of ventricular βi reported here for guinea-pig and that reported previously for ferret and rat therefore remains unexplained.
CO2-dependent buffering
When computing total intracellular buffering power in 5 % CO2-buffered conditions, a second component of intracellular buffering, βCO2, must be added to βi. It can be shown that (see Methods):
| (2) |
Provided the cell remains an open system for CO2, the value of βCO2 at a given pHi, [HCO3−]i and [HCO3−]o is thus temperature-insensitive.
Thermal sensitivity of NHE and NBC
Both NHE and NBC display a high temperature-sensitivity, their activities declining with temperature, indicating a Q10 of about 2 and 2.4, respectively. In other species and cell types, a Q10 for NHE of between 1.05 and 2.65 is evident (e.g. Aickin & Thomas, 1977a; Ives, 1985; Ellis & Macleod, 1985; Graber et al. 1992; Do et al. 1996; Hoshino & Avkiran, 2001). In some of these cases, however, Q10 values were determined from changes in dpHi/dt rather than acid efflux (Je), while in others they were determined without correcting for temperature effects on buffering power or the pH-sensitive fluorophore, or without allowing for significant contributions to Je from NBC. In the present work we have attempted to account for these factors when computing the Q10 for NHE. In addition we have provided the first estimate for the Q10 of NBC.
The Q10 for NHE and NBC appears relatively constant over the pHi range (≈6.60–7.50). This ensures that NHE and NBC operate optimally at 37°C for a wide range of acid challenges. It is of interest that NHE activity has been shown to decrease on raising temperature above 40°C (in A431 cells; Kiang et al. 1990; and in proximal tubule brush border membrane vesicles, Ives, 1985). This is consistent with mammalian proteins having an optimal operating temperature close to 37°C. While protein denaturation is likely to be a factor at very high temperatures, modulation of NHE and NBC has also been suggested to occur through either an amphoteric amino acid on the transporter, e.g. histidine, which is selectively affected by the temperature change, or via an internal modulator such as a kinase that has temperature sensitivity (Graber et al. 1992).
Note that there are two reports of increased NHE activity on decreasing temperature below 37°C. In guinea-pig erythrocytes, hypothermia stimulated amiloride-sensitive Na+ uptake, suggested to be via NHE (Zhou & Willis, 1989); similar results were found in cultured chick embryonic myocytes using the NHE inhibitor EIPA (Knerr & Lieberman, 1993). The latter study, however, used neonatal cardiac cells that have unusually high NHE activity (Meno et al. 1989). From the present work, it is clear that in adult cells, hypothermia reduces both NHE and NBC activity.
Thermal sensitivity of resting pHi
Resting pHi at 37°C was similar in both Hepes- and CO2-buffered conditions (7.04 and 7.05, respectively) and was comparable to that reported previously in guinea-pig myocytes (7.07 in Hepes and 7.04 in 5 % CO2; Leem et al. 1999), as well as in ferret ventricle (7.03 in Hepes at 35°C; Do et al. 1996). Resting pHi was ≈0.1 pH units more alkaline at 27°C but was again similar in the two buffer conditions.
Role of intrinsic buffer
The thermal decrease of pHi in guinea pig ventricular myocytes (27–37°C) seems paradoxical given that NHE and NBC activity is enhanced on warming. Intuitively, a rise in pHi would be expected. One possibility is that temperature-elevation simultaneously stimulates sarcolemmal acid influx transporters (CHE and AE), thus effectively neutralising the influence on pHi of enhanced acid extrusion. An assessment of the temperature-sensitivity of acid loading transporters will be required to test this possibility. Effects on sarcolemmal acid-base transport, however, cannot account for the observed thermally induced fall of pHi, as it is too rapid. Figure 6A shows that pHi falls by 0.15 u within 50 s of raising temperature by 10°C. If caused by enhanced sarcolemmal acid-loading, this would be equivalent to an influx of 5 mm min−1 (influx = dpHi/dt×βi). At a pHi of 7.1 at 37°C, CHE and AE each mediate an acid influx about 330-fold lower than this (≈0.15 mm min−1; Leem et al. 1999). The fall of pHi is thus likely to be mediated at intracellular buffer sites rather than at sarcolemmal transport sites.
A 10°C rise in temperature acidifies most cells of most organisms by 0.1 to 0.2 pH units (Roos & Boron, 1981), including ectothermic and endothermic animals, hibernators and also plants (Marjanovic et al. 1998). Comparable changes in pHi have been found in mouse skeletal muscle (−0.0178 u°C−1, Aickin & Thomas, 1977a; −0.015 u°C−1, Westerblad et al. 1997), sheep Purkinje fibres (−0.015 u°C−1; Ellis & Macleod, 1985), the right ventricle of ferret heart (−0.012 u°C−1; Do et al. 1996), and Hepes-buffered rat myocytes (−0.015 u°C−1; Hoshino & Avkiran, 2001). An effect of temperature on histidine residues, the primary intrinsic buffer, may partly underlie this phenomenon since, for a 10°C rise, the pKa of imidazole in aqueous solution falls by 0.17 u (Reeves, 1972). This observation has led to the so-called ‘alphastat hypothesis’, where the fractional dissociation of imidazole histidine remains constant on changing temperature. As a consequence, ionic charge on intracellular proteins and other buffers is also proposed to be maintained (Reeves, 1972).
An intriguing observation is that thermal shifts of resting pHi did not occur at pHi 6.4 but were clearly evident at 7.4 (see Fig. 6C). The reason for this is not known, but one possibility is that the intrinsic buffers that dominate at pHi 6.4 differ from those at 7.4, and that the former are less temperature sensitive. Leem et al. (1999) have provided evidence for at least two populations of intrinsic buffer in the ventricular myocyte, differing in their concentration and principal pKa value (pKa values of about 6.0 and 7.6 were suggested; see eqn (1) of present Methods). More recently, Zaniboni et al. (2003) have suggested that the low pKa population comprises intracellular proteins, the remaining population comprising a mixture of lower molecular weight buffers with higher pKa values, such as dipeptides and inorganic phosphate. In the Appendix to the present paper we show that a simple model that utilises the above two categories of buffer predicts a thermally induced acidification of resting pHi (-ΔpHi), accompanied by an increase of βi, as observed experimentally. The model indicates that the −ΔpHi is consistent with an average 0.15 unit fall in pKa for the higher-pKa buffers, with little or no change in the lower-pKa population. The predictions for −ΔpHi have been included (continuous line) with the experimental measurements plotted in Fig. 6C.
The predictive behaviour of the dual-buffer model strongly suggests that a heterogeneous population of intracellular intrinsic buffers displaying different temperature-sensitive pKa values may account for the thermal sensitivity of resting pHi and intracellular buffering power. Given that most intrinsic buffering is attributable to imidazole histidine, the above hypothesis implies that the microenvironment around such groups on, say, intracellular proteins (the proposed lower-pKa category of buffer; Zaniboni et al. 2003) may possibly confer a lower thermal sensitivity on pKa than that for imidazole located on dipeptides such as homocarnosine and acetylanserine that have been proposed to comprise the higher-pKa category.
Role of carbonic buffer
Thermal intracellular acidosis is the same in the presence and absence of CO2/HCO3−. While the explanation for this is not entirely clear, it is notable that the pH of a 5 % CO2/22 mm HCO3−-buffered Tyrode solution is largely temperature-insensitive: its pH falls from 7.40 to 7.35 on cooling from 37 to 27°C. This lack of major temperature sensitivity can be attributed to a rise in the pKa of carbonic acid at lower temperature being offset by an increase in the solubility of CO2 (Seldin & Giebisch, 1989). Thermal sensitivity of pHi therefore depends largely on intrinsic rather than carbonic buffer.
Hypothermia and cardiac physiology and pathophysiology
Low temperature experiments
The present findings reinforce the need to interpret results of experiments performed at low temperature with some caution. For example, the amplitude of rapid cooling contractures associated with release Ca2+ from the sarcoplasmic reticulum (e.g. Bers, 1987) may in part be due to an alkaline shift on cooling, as intracellular alkalosis increases myofilament Ca2+-sensitivity (Fabiato & Fabiato, 1978). In the case of protocols performed at room temperature, pHi regulation will be slowed considerably, and so manoeuvres that displace pHi will produce larger and more sustained effects. Given the important link between H+i and Ca2+i in heart, such abnormalities in pHi control will be expected to distort the normal physiological control of Ca2+i.
Myocardial ischaemia and reperfusion
The thermal sensitivity of NHE and NBC may be relevant to cardiac injury and arrhythmogenesis during pathophysiological conditions. Na+ influx coupled to acid efflux plays an important role in the generation of Na+i-driven Ca2+i overload during conditions of low pHi, such as those found during ischaemia and early post-ischaemic reperfusion. Intracellular Ca2+ overload impairs cardiac function (e.g. Piper et al. 1996) and may be arrhythmogenic (e.g. Ch'en et al. 1998). In animal and whole heart models of ischaemia, pharmacological NHE inhibitors such as Hoe 694 and cariporide (Hoe 642) that suppress Na+-coupled acid efflux appear to reduce the incidence of reperfusion injury and arrhythmia, and are thus potentially useful cardioprotective agents (Karmazyn, 1999; Hartmann & Decking, 1999; Xiao & Allen, 2000). Hypothermia offers another potential cardioprotective route as it decreases not only NHE (Hoshino & Avkiran, 2001), but also NBC activity, both of which are sources of Na+ influx. Notably, hypothermia (4–10°C) is often combined with cardioplegia to minimise myocardial damage during cardiac surgery. If the high Q10 of the transporters extends to these low temperatures, then they will operate at about 10 % of their rate at 37°C. Na+ influx during acid overload, such as that associated with post-ischaemic reperfusion, would thus be severely attenuated, especially when combined with pharmacological NHE inhibitors; indeed this strategy appears to offer additional cardioprotection during reperfusion in animals (Shipolini et al. 1997). In conclusion, the thermal sensitivity of NHE and NBC is consistent with suggestions that inhibition of these transporters may contribute to some of the cardioprotective effects of cooling during ischaemia and reperfusion.
Acknowledgments
The excellent technical assistance of Mrs Anna Skyrme & Ms Neelam Banger is gratefully acknowledged. The work was funded by a British Heart Foundation Programme Grant (RDV-J), a British Heart Foundation Studentship (F.F.-T.C.), Wellcome-Trust Prize Studentships (E.D. and P.S.) and a Medical Research Council Studentship (R.S.G.). Cariporide was the gift of Dr H.-W. Kleemann (Aventis, Germany).
APPENDIX
Modelling the thermal sensitivity of pHi and intracellular intrinsic buffering
For simplicity, intracellular, intrinsic buffering capacity is assumed to be a function of the concentration and dissociation constant (K) of two populations of buffer (A and B), as suggested previously (Leem et al. 1999; Zaniboni et al. 2003). A change of temperature is assumed to affect K only (e.g. Reeves, 1972; Seldin & Giebisch, 1989). Assuming the total concentration of intracellular buffer remains constant, numerical minimalisation algorithms (fminsearch, MATLAB, Mathworks Corporation) may then be used to find the equilibrium concentrations of H+, and the protonated and unprotonated forms of the buffers, after a change in their dissociation constants.
We assume a temperature dependence of the dissociation of water, Kw, such that the ratio, γ,
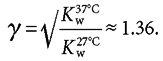 |
Next we define a function f that must equal zero at equilibrium,
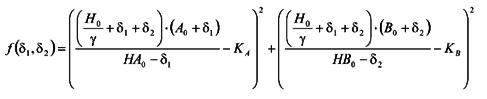 |
where H0, A0, HA0, B0 and HB0 are the initial concentrations of protons and the unprotonated and protonated forms of buffers A and B, before the temperature change. Constants KA and KB are the dissociation constants for the two buffers at the new temperature. We minimise f with respect to variables δ1 and δ2. New equilibrium concentrations are given as:
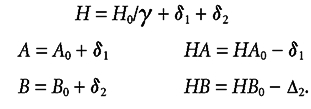 |
We then use data from Fig. 6C to quantify the temperature dependence of KA and KB. We assume the values for KA and KB determined previously at 37°C (pK values of 6.03 and 7.57 respectively; Leem et al. 1999). Least squares fitting suggests that the pHi dependence of the acidosis during warming from 27 to 37°C (-ΔpHi) can be reproduced if pKA is slightly lower at 27°C (6.005), and pKB is higher (7.72) than at 37°C. This latter 0.15 u shift is similar to that reported previously for the temperature sensitivity of imidazole (Reeves, 1972). The pHi sensitivity of −ΔpHi predicted by these changes of KA and KB has been superimposed (continuous line) on the experimental data shown in Fig. 6C.
Changes in KA and KB will alter the pHi sensitivity of total intrinsic buffering power (βi). At any given pHi, intracellular buffering equals the sum (βA+βB) of buffer capacities for A and B. The thin line in Fig. 7 shows total βivs. pHi determined previously at 37°C by Leem et al. (1999). The thick line shows the relationship at 27°C, predicted from the KA and KB values at this temperature (relationship calculated using eqn (1) in Methods). It is notable that over the pHi range 7.6–6.6, total βi is significantly lower at 27°C than at 37°C, in agreement with the experimentally observed decrease of βi during hypothermia (Fig. 3). The predicted decrease is, however, ≈20 % rather than the 40 % decrease actually measured, suggesting that the dual buffer model is not entirely successful in its predictions. Nevertheless, the model illustrates well the principle that the observed rise of resting pHi and fall of intrinsic buffering capacity during hypothermia may occur as a result of increases in the pK of selected intracellular buffers.
Figure 7. Modelling temperature dependence of intrinsic buffering power.
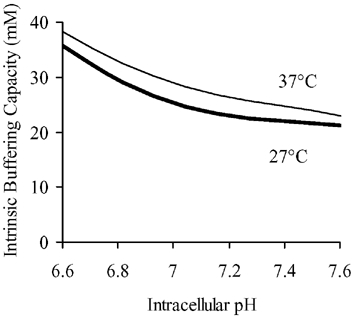
The curve at 37°C was computed from eqn (1) in Methods, i.e. for the sum of the two buffers, A and B, of concentration 84.22 and 29.38 mm respectively, and pK 6.03 and 7.57 respectively. The curve at 27°C was computed for the same concentrations of A and B, but with pKA= 6.005 and pKB= 7.72.
REFERENCES
- Aickin CC, Thomas RC. An investigation of the ionic mechanism of intracellular pH regulation in mouse soleus muscle fibres. J Physiol. 1977a;273:295–316. doi: 10.1113/jphysiol.1977.sp012095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aickin CC, Thomas RC. Microelectrode measurement of the intracellular pH and buffering power of mouse soleus muscle fibres. J Physiol. 1977b;267:791–810. doi: 10.1113/jphysiol.1977.sp011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Ryanodine and the calcium content of cardiac SR assessed by caffeine and rapid cooling contractures. Am J Physiol. 1987;253:C408–415. doi: 10.1152/ajpcell.1987.253.3.C408. [DOI] [PubMed] [Google Scholar]
- Blank PS, Silverman HS, Chung OY, Hogue BA, Stern MD, Hansford RG, Lakatta EG, Capogrossi MC. Cytosolic pH measurements in single cardiac myocytes using carboxy-seminaphthorhodafluor-1. Am J Physiol. 1992;263:H276–284. doi: 10.1152/ajpheart.1992.263.1.H276. [DOI] [PubMed] [Google Scholar]
- Bountra C, Powell T, Vaughan-Jones RD. Comparison of intracellular pH transients in single ventricular myocytes and isolated ventricular muscle of guinea-pig. J Physiol. 1990;424:343–365. doi: 10.1113/jphysiol.1990.sp018071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bountra C, Vaughan-Jones RD. Effect of intracellular and extracellular pH on contraction in isolated, mammalian cardiac muscle. J Physiol. 1989;418:163–187. doi: 10.1113/jphysiol.1989.sp017833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Application of a new pH-sensitive fluoroprobe (carboxy-SNARF-1) for intracellular pH measurement in small, isolated cells. Pflugers Arch. 1990;417:234–239. doi: 10.1007/BF00370705. [DOI] [PubMed] [Google Scholar]
- Ch'en FF-T, Goddard RS, Vaughan-Jones RD. Temperature dependence of cardiac Na+−H+ exchange and Na+−HCO3− co-transport. J Physiol. 2000;527.P:66–67P. doi: 10.1113/jphysiol.2003.051888. abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'en FF-T, Goddard RS, Vaughan-Jones RD. Temperature sensitivities of Na+−H+ exchange and Na+−HCO3− co-transport in guinea-pig ventricular myocytes. Biophys J. 2001;80:2645. abstract. [Google Scholar]
- Ch'en FF-T, Vaughan-Jones RD, Clarke K, Noble D. Modelling myocardial ischaemia and reperfusion. Prog Biophys Mol Biol. 1998;69:515–538. doi: 10.1016/s0079-6107(98)00023-6. [DOI] [PubMed] [Google Scholar]
- Dart C, Vaughan-Jones RD. Na+−HCO3− symport in the sheep cardiac Purkinje fibre. J Physiol. 1992;451:365–385. doi: 10.1113/jphysiol.1992.sp019169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do E, Ellis D, Noireaud J. Intracellular pH and intrinsic H+ buffering capacity in normal and hypertrophied right ventricle of ferret heart. Cardiovasc Res. 1996;31:729–738. doi: 10.1016/0008-6363(96)00024-7. [DOI] [PubMed] [Google Scholar]
- Ellis D, Macleod KT. Sodium-dependent control of intracellular pH in Purkinje fibres of sheep heart. J Physiol. 1985;359:81–105. doi: 10.1113/jphysiol.1985.sp015576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. J Physiol. 1978;276:233–255. doi: 10.1113/jphysiol.1978.sp012231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber M, Barry C, Dipaola J, Hasagawa A. Intracellular pH in OK cells. 2. Effects of temperature on cell pH. Am J Physiol. 1992;262:F723–730. doi: 10.1152/ajprenal.1992.262.5.F723. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Decking UKM. Blocking Na+−H+ exchange by cariporide reduces Na+-overload in ischaemia and is cardioprotectiv e. J Mol Cell Cardiol. 1999;31:1985–1995. doi: 10.1006/jmcc.1999.1029. [DOI] [PubMed] [Google Scholar]
- Hearse DJ, Braimbridge MV, Jynge P. Protection of the Ischaemic Myocardium: Cardioplegia. New York: Blackwell Science Inc; 1981. [Google Scholar]
- Hoshino K, Avkiran M. Effects of moderate hypothermia on sarcolemmal Na+−H+ exchanger activity and its inhibition by cariporide in cardiac ventricular myocytes. Br J Pharm. 2001;134:1587–1595. doi: 10.1038/sj.bjp.0704405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives HE. Proton hydroxyl permeability of proximal tubule brush-border vesicles. Am J Physiol. 1985;248:F78–86. doi: 10.1152/ajprenal.1985.248.1.F78. [DOI] [PubMed] [Google Scholar]
- Karmazyn M. Expression and role of the Na+/H+ exchanger in myocardial injury: From the ischaemic and reperfused myocardium to congestive heart failure. Eur Heart J Suppl. 1999;1:K38–K44. [Google Scholar]
- Kiang JG, McKinney LC, Gallin EK. Heat induces intracellular acidification in human A-431 cells – role of Na+−H+ exchange and metabolism. Am J Physiol. 1990;259:C727–737. doi: 10.1152/ajpcell.1990.259.5.C727. [DOI] [PubMed] [Google Scholar]
- Knerr SMM, Lieberman M. Ion-transport during hypothermia in cultured heart-cells – implications for protection of the immature myocardium. J Mol Cell Cardiol. 1993;25:277–288. doi: 10.1006/jmcc.1993.1034. [DOI] [PubMed] [Google Scholar]
- Lagadic-Gossmann D, Buckler KJ, Vaughan-Jones RD. Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. J Physiol. 1992;458:361–384. doi: 10.1113/jphysiol.1992.sp019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD. Characterisation of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol. 1999;517:159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic M, Elliott AC, Dawson MJ. The temperature dependence of intracellular pH in isolated frog skeletal muscle: Lessons concerning the Na+−H+ exchanger. J Membr Biol. 1998;161:215–225. doi: 10.1007/s002329900328. [DOI] [PubMed] [Google Scholar]
- Meno H, Jarmakani JM, Philipson KD. Developmental-changes of sarcolemmal Na+−H+ exchange. J Mol Cell Cardiol. 1989;21:1179–1185. doi: 10.1016/0022-2828(89)90694-9. [DOI] [PubMed] [Google Scholar]
- Orchard CH, Cingolani HE. Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res. 1994;28:1312–1319. doi: 10.1093/cvr/28.9.1312. [DOI] [PubMed] [Google Scholar]
- Piper HM, Balser C, Ladilov YV, Schafer M, Siegmund B, Ruiz Meana M, Garcia Dorado D. The role of Na+−H+ exchange in ischaemia-reperfusion. Basic Res Cardiol. 1996;91:191–202. doi: 10.1007/BF00788905. [DOI] [PubMed] [Google Scholar]
- Reeves RB. An imidazole alphastat hypothesis for vertebrate acid-base regulation: Tissue carbon dioxide content and body temperature in bullfrogs. Respir Physiol. 1972;14:219–236. doi: 10.1016/0034-5687(72)90030-8. [DOI] [PubMed] [Google Scholar]
- Richmond PH, Vaughan-Jones RD. Assessment of evidence for K+−H+ exchange in isolated type-1 cells of neonatal rat carotid body. Pflugers Arch. 1997;434:429–437. doi: 10.1007/s004240050417. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Schafer C, Ladilov YV, Siegmund B, Piper HM. Importance of bicarbonate transport for protection of cardiomyocytes against reoxygenation injury. Am J Physiol Heart Circ Physiol. 2000;278:H1457–1463. doi: 10.1152/ajpheart.2000.278.5.H1457. [DOI] [PubMed] [Google Scholar]
- Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, Weichert A, Scholkens BA. Protective effects of Hoe 642, a selective sodium–hydrogen exchange subtype-1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–268. [PubMed] [Google Scholar]
- Seldin DW, Giebisch GH. Regulation of Acid-Base Balance. New York: Blackwell Science Inc; 1989. [Google Scholar]
- Shipolini AR, Galinanes M, Edmondson SJ, Hearse DJ, Avkiran M. Na+−H+ exchanger inhibitor Hoe 642 improves cardioplegic myocardial preservation under both normothermic and hypothermic conditions. Circulation. 1997;96(suppl. II):266–273. [PubMed] [Google Scholar]
- Sun B, Leem CH, Vaughan-Jones RD. Novel chloride-dependent acid loader in the guinea-pig ventricular myocyte: Part of a dual acid-loading mechanism. J Physiol. 1996;495:65–82. doi: 10.1113/jphysiol.1996.sp021574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk PLM, Hardewig I, Portner HO. Temperature-dependent shift of pHi in fish white muscle: Contributions of passive and active processes. Am J Physiol. 1997;41:R84–89. doi: 10.1152/ajpregu.1997.272.1.R84. [DOI] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Peercy B, Keener JP, Spitzer KW. Intrinsic H+ ion mobility in the mammalian ventricular myocyte. J Physiol. 2002;541:139–158. doi: 10.1113/jphysiol.2001.013267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Wu ML. pH dependence of intrinsic H+ buffering power in the sheep cardiac Purkinje fibre. J Physiol. 1990;425:429–448. doi: 10.1113/jphysiol.1990.sp018112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H, Bruton JD, Lannergren J. The effect of intracellular pH on contractile function of intact, single fibres of mouse muscle declines with increasing temperature. J Physiol. 1997;500:193–204. doi: 10.1113/jphysiol.1997.sp022009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao XH, Allen DG. Activity of the Na/H exchanger is critical to repperfusion damage and preconditioning in the isolated rat heart. Cardiovasc Res. 2000;48:244–253. doi: 10.1016/s0008-6363(00)00166-8. [DOI] [PubMed] [Google Scholar]
- Xue YX, Aye NN, Hashimoto K. Anti-arrhythmic effects of Hoe 642, a novel Na+−H+ exchange inhibitor, on ventricular arrhythmias in animal hearts. Eur J Pharm. 1996;317:309–316. doi: 10.1016/s0014-2999(96)00755-8. [DOI] [PubMed] [Google Scholar]
- Zaniboni M, Swietach P, Rossini A, Yamamoto T, Spitzer KW, Vaughan-Jones RD. Intracellular proton mobility and buffering power in cardiac ventricular myocytes from rat, rabbit and guinea-pig. Am J Physiol Heart Circ Physiol. 2003;285:H1236–1246. doi: 10.1152/ajpheart.00277.2003. [DOI] [PubMed] [Google Scholar]
- Zhou ZQ, Willis JS. Differential effects of cooling in hibernator and non-hibernator cells – Na+ permeation. Am J Physiol. 1989;256:R49–55. doi: 10.1152/ajpregu.1989.256.1.R49. [DOI] [PubMed] [Google Scholar]