

Abstract
Renin is a central hormone in the control of blood pressure and various other physiological functions. In spite of the very early discovery of renin over 100 years ago, we have only recently gained a deeper understanding of the origin of renin-producing cells and of the mechanisms responsible for renin synthesis and secretion. The main source of renin is the juxtaglomerular cells (JGCs), which release renin from storage granules. Besides the renin-angiotensin system (RAS) in the JGCs, there exist local RASs in various tissues. JGCs originate in situ within the metanephric kidney from mesenchymal cells that are not related to smooth muscle lineages, as hitherto assumed. The previous notion that JGCs stem from vascular smooth muscle cells may be explained by JGC differentiation: they acquire smooth muscle markers that are maintained throughout adulthood. It has become clear that increasing intracellular free [Ca2+] inhibits renin secretion in JGCs. In contrast, cAMP stimulates renin release. Over the last decade, numerous studies on isolated JGCs and intact animals have provided contradictory results as to whether cGMP has a stimulatory or inhibitory action on renin release. More recent results strongly suggest that the effects of cGMP on renin release from JGCs involve the degradation of cAMP, which is modulated by cGMP. Finally, it has been found that not only is the production of renin modulated by enhancing or attenuating renin transcription, but renin mRNA stability is controlled by various proteins present in renin-producing cells.
Recent studies shed new light on renin production and release, thereby considerably modifying our view of this important hormone. Renin was one of the first hormones to be discovered: during an IUPS meeting in St Petersburg in 1898, the first data were presented indirectly suggesting the existence of a renally derived factor that increases blood pressure. This hormone enzyme initiates the enzymatic cascade generating the angiotensin peptides that regulate blood pressure, cell growth, apoptosis and electrolyte balance, to mention only some of the foremost-recognized functions. Renin is rate limiting in the production of angiotensin II (Ang II), a hormone that ultimately integrates cardiovascular and renal function in the control of blood pressure as well as salt and volume homeostasis. For instance, renin seems to be of vast importance for maintaining arterial blood pressure in the face of variations in salt intake: in mice, constant blood pressure is found during alterations in sodium intake, and this relies on controlling the activity of the renin-angiotensin system (RAS). Once the RAS is experimentally kept constant, salt sensitivity of blood pressure regulation becomes apparent (Cholewa & Mattson, 2001).
Recently we have gained a different view of the origin of renin-producing cells, renin secretion and the control of renin production at the level of transcription and translation. These issues will be discussed in the following sections.
The origin of renin-producing cells
The juxtaglomerular cells (JGCs) constitute the most important source of circulating renin.
Generally it is assumed that JGCs are metaplastically modified smooth muscle cells, because adult mammal JGCs contain myofilaments (Taugner & Hackenthal, 1989). However, only recently has a study has been performed to determine the lineage of these cells. In an extensive study, Sequeira Lopez et al. (2001) used single cell PCR and double immunostaining combined with lineage markers to define the lineage of JGCs. The authors transplanted embryonic kidneys between genetically marked and wild-type mice and labelled them for renin, smooth muscle and endothelial cells at different developmental stages. Normal cell culture systems cannot be applied, since no vessels form (under conventional conditions) and therefore renin cells do not assemble into arterioles. Thus, they remain dispersed in the interstitium. Sequeria Lopez et al. suggest that there are at least two distinct populations of cells, expressing either renin or smooth muscle markers, but never both. From E12 to E15, renin cells do not yet express smooth muscle or endothelial markers. Subsequently, the subpopulation of renin-expressing cells remarkably exhibit the capacity to express smooth muscle markers. Thus renin cells can give rise to smooth muscle cells rather than originating from them (Fig. 1) (Sequeira Lopez et al. 2001). In line with this interpretation, renin precursor cells are capable of assembling into the appropriate vessel types and segments. Moreover, smooth muscle cells derived from renin precursors seem to be those capable of undergoing metaplasia to renin cells when required.
Figure 1. The lineage of the JGC.
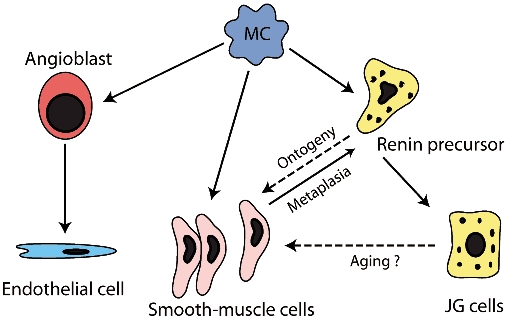
Metanephric mesenchymal cells (MC) are the origin of angioblasts, from which the endothelial cells stem. MCs are also the origin of vascular smooth muscle cells and of renin precursor cells. During ontogeny, these renin precursor cells can give rise to JGCs and to a subset of arteriolar smooth muscle cells. The smooth muscle cells originating from the renin precursors seem to be the cells capable of undergoing metaplasia to renin cells. Reproduced with permission from Sequeira Lopez et al. (2001).
The functioning of a fetal and early postnatal RAS is a prerequisite for normal nephrogenesis in the rat. In the fetal kidney, the renin-angiotensin system is markedly activated. The metanephros at embryonic day 14 (E14), contains renin and Ang II and both the Ang II receptors (AT1 and AT2). At this stage, renin is found in cells scattered within the mesenchyme (Norwood et al. 2000). Insulin-like growth factor (IGF)-I seems to be pivotal in this process, as underlined by the observation that angiotensin-converting enzyme inhibition suppresses renal IGF-I expression. Interestingly, treatment with IGF-I normalizes renal function and histology after early ACE inhibition (Nilsson et al. 2000). Moreover, IGF-1 stimulates renin production in the late gestation fetal sheep (Marsh et al. 2001).
Besides the circulating RAS, there exist several local ones within various tissues including the heart, brain and adrenal glands. Even though all components of the RAS are expressed in these tissues, the mode of action can be quite different (Peters & Clausmeyer, 2002), as underscored by the remarkable discovery of renin in inclusion bodies of the mitochondria. Although the exact function of mitochondrial renin remains to be fully elucidated, the fact that aldosterone production takes place in adrenal mitochondria indicates a potential role of mitochodrial renin in its control. This functional link between mitochondrial renin and aldosterone has been shown experimentally after bilateral nephrectomy (Peters et al. 1999), where the amount of mitochodria containing renin increases with the augmented aldosterone production.
It seems that only a truncated form of prorenin can be imported into the mitochondria. This truncated prorenin is synthesized by an alternative transcript characterized by an alternative exon 1A. Interestingly, this is the only renin transcript expressed in the heart. The classical mRNA coding for preprorenin does not respond to stimuli of the cardiac RAS such as hypertrophy or myocardial ischaemia. Conversely, exon 1A renin trancript increases promptly (Peters et al. 2002). In addition to renin located in the mitochondria, the cardiac cells can also internalize circulating renin. There are two proposed mechanisms by which this can occur, one by mannose-6-phosphate receptor-mediated endocytosis (Saris et al. 2002), the other by the internalization of nonglycosylated prorenin (Peters & Clausmeyer, 2002). The mannose-6-phosphate receptor-mediated uptake may simply constitute a clearance mechanism, i.e. it inactivates the circulating RAS. The second form of internalization may be crucial for the intracardiac RAS and its various functions (Peters & Clausmeyer, 2002).
Renin secretion
The regulation of renin release from the kidney is complex (Skott, 2002; Todorov et al. 2002; Cheng et al. 2002; Kammerl et al. 2002). Among the manifold controllers is the macula densa mechanism, which couples the tubular chloride concentration inversely to the plasma renin concentration in the rat. This occurs in the thick ascending limb of the tubule of Henle. Changes in regional renin release in the kidney help to determine the sensitivity of the tubuloglomerular feedback mechanism, consequently modifying the set point for autoregulation of renal blood flow.
A second control element is sympathetic nervous discharge to the kidney, which stimulates renin secretion through β-adrenergic receptors on the JGCs (DiBona, 2000). Increased renin secretion in response to β-adreneroceptor stimulation is mediated via the formation of cAMP. Cyclic nucleotides are critical second messengers that determine renin secretory rate and there seems to exist a common pathway of stimulating renin secretion via cAMP. Hormones, neurotransmitter, and autacoids that raise the intracellular production of cAMP enhance renin secretion and augment renin mRNA levels. Thus, it is unequivocally held that cAMP stimulates renin secretion. In contrast, the effects of cGMP on renin secretion have been discussed controversially. Some find a stimulatory action; others have reported an inhibitory effect of cGMP on renin release. Chiu and Reid demonstrated an important interaction between cAMP and cGMP for the secretion of renin by hydrolysis of cAMP that occurs in response to various phosphodiesterase (PDE) isoforms (Chiu & Reid, 1996; Chiu et al. 1996). Among these, PDE 3 may play a particular role since it is inhibited by cGMP. As shown by Chiu and Reid, cGMP can stimulate renin release by decreasing cAMP breakdown via attenuated PDE 3-activity. Accordingly, inhibition of PDE 3 also augments the renin secretory response to β-adrenoceptor stimulation (Chiu & Reid, 1996). Friis and colleagues very recently employed an elegant technique to further test the concept that cGMP enhances renin release through the blunting of cAMP degradation (Fig. 2; Friis et al. 2002). Two PDE 3 subtypes (A and B) have been suggested to mediate cross-talk between cAMP and cGMP pathways in JGCs. The PDE 3 enzymes are specific for cAMP and, as mentioned above, they are inhibited by cGMP. In the study by Friis et al., patch clamp experiments were made on isolated JGCs. Increases in membrane capacitance indicate cell surface area increase, and this is an accepted measure of exocytosis at the level of the single cell. cGMP had similar effects on membrane capacitance to cAMP, although 10-fold higher concentrations were required. This effect of cGMP on membrane capacitance was inhibited by blocking PKA, thus underscoring that the cAMP-PKA pathway takes part in cGMP-mediated responses of JGCs. Therefore renin release from JGCs is significantly regulated by degradation of cAMP, which is modulated by cGMP inhibition of PDEs.
Figure 2.
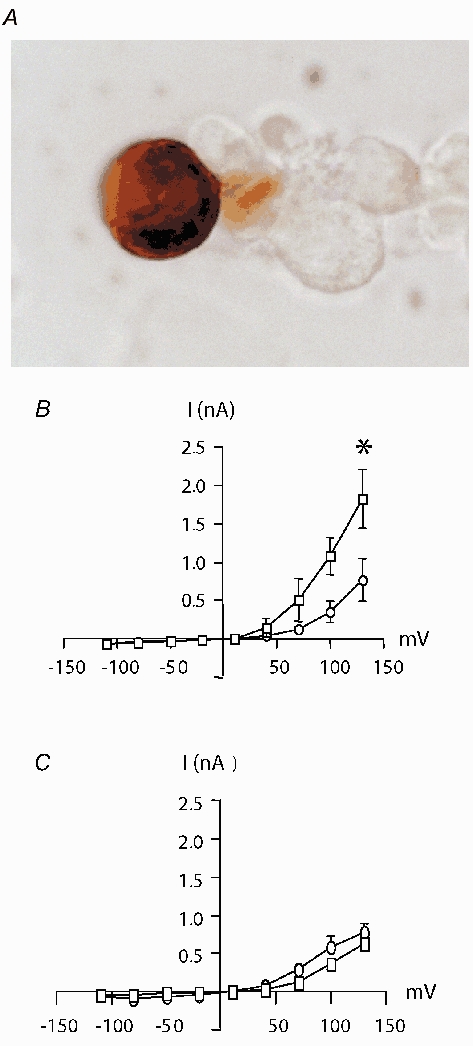
A, immunolabelling of renin protein in a JGC using rabbit anti-mouse renin antibody, B, effects of cGMP on whole-cell currents and Cm of isolated JGCs. I-V relationships before (•) and after (▪) JGCs were dialysed with 10 μmol l−1 cGMP. C, as before, but JGC were dialysed with cGMP (10 μmol l−1) together with the PKA blocker Rp-cAMP (25 μmol l−1). Adapted from Friis et al. (2002).
There is also a potent pressure-sensitive mechanism for renin release; its stimulation is associated with activation of the sympathetic nervous system and release of hormones, such as oxytocin, which stimulate renin release in rats via a β-adrenergic receptor-dependent mechanism (Huang et al. 2001).
As mentioned before, the release of active renin from JGCs is considered to be the main rate-limiting step in providing circulating renin. Remarkably, the stimuli that inhibit renin secretion, e.g. increased arterial pressure or Ang II, increase intracellular free calcium. This is in contrast to the other secretory cells of the organism, in which augmented free calcium levels lead to enhanced depletion of secretory granules. Thus, this unique feature of renin secretion is commonly referred to as the calcium paradox. The reason for the opposite effect of calcium on renin secretion can be found in the origin of the renin storage granules that appear to be modified lysosomes (for review see Peters & Clausmeyer, 2002).
The membrane potential of JGCs acts in concert with the various mechanisms controlling renin secretion. Again, calcium seems to play an important role in this context. For instance, voltage-dependent L-type calcium channels and calcium-sensitive voltage-gated calcium channels have been identified in JGCs (Friis et al. 2003). The latter channels are sensitive to cAMP as they belong to the ZERO variant, and play an important role in determining membrane potential. However, they are not directly responsible for renin secretion, as their blockade does not affect renin secretion. Conversely, activating the L-type calcium channels inhibits cAMP-mediated renin secretion, thus providing evidence for a functional link between these channels and the control of renin secretion (Friis et al. 2003).
Control of renin synthesis
Transcriptional control depends on the DNA located immediately upstream of the gene itself. As for other peptides, transcription of renin RNA requires the binding of RNA polymerase II to the basic promoter region of the gene. For the renin gene, there are additional regulatory elements considerably further upstream of the cap site which activate or repress transcription (Petrovic et al. 1996). These regulatory elements are found in areas where remarkable interspecies homology can occur (Mrowka et al. 2003; Persson et al. 2003). One of the homologous sequences has been a matter of extensive investigation (Shi et al. 2001a,b). It was termed the ‘renin enhancer’ before it was recognized that this sequence is a compound regulatory element with several stimulatory and inhibitory activities. In the renin enhancer, cAMP response elements have been identified. For murine renin gene transcription, the steroid hormone receptors LXR and retinoic acid receptor-RXR complex, transcriptional factors CREB/CREM and USF1/USF2, and finally HOX gene family members are known to be important (Pan et al. 2001a,b). In order to exert a specific action, the transcription factors binding at the enhancer region must interact with other transcription factors acting in the closer vicinity of the site of transcription, e.g. the proximal promoter element. Interestingly, vitamin D3 and its receptor seem to play an important role in the complex regulation of renin transcription (Li et al. 2002). The vitamin D receptor belongs to the thyroid hormone (T3) subfamily of nuclear hormone receptor transcription factors. These also include the retinoic acid receptor. The latter shares a binding site with the vitamin D receptor. As an example of the clinical importance of this binding site, it has been shown by observations in patients that there is an inverse relationship between plasma vitamin D3 levels and plasma renin activity as well as with blood pressure (Burgess et al. 1990). Moreover, it has been known for some time that vitamin D3 supplementation lowers blood pressure in hypertensive subjects (Lind et al. 1989). Thus, the vitamin D3 receptor may constitute an important negative regulator of renin expression.
These are examples of the significance of the hitherto identified renin enhancer region, yet there may exist further regions of importance. Indeed, the interspecies homology that has been found in two more upstream areas of this enhancer sequence underscores the potental existence of more regulatory sites (Mrowka et al. 2003). Their functional importance, however, remains to be quantified.
Finally, in addition to the control of renin via transcriptional regulation, strong evidence points to the paramount role of post-transcriptional modulation. In particular, renin mRNA stability can be modified by cAMP (Chen et al. 1993; Sinn & Sigmund, 1999). Currently, light is being shed on possible mechanisms behind this effect: beyond the coding region of about 200 nucleotides, renin mRNA contains an untranslated region (UTR). The homology of this region is striking between sheep, rats, mice and humans (Skalweit et al. 2003). Several RNA-binding regulatory proteins, which were identified by MALDI-TOF, bind to this UTR. Remarkably, as shown in in vitro experiments, adding these proteins to cell lysates markedly protects renin mRNA from degradation, thereby increasing the amounts of renin being synthesized (Skalweit et al. 2003).
REFERENCES
- Burgess ED, Hawkins RG, Watanabe M. Interaction of 1,25-dihydroxyvitamin D and plasma renin activity in high renin essential hypertension. Am J Hypertens. 1990;3:903–905. doi: 10.1093/ajh/3.12.903. [DOI] [PubMed] [Google Scholar]
- Chen M, Schnermann J, Smart AM, Brosius FC, Killen PD, Briggs JP. Cyclic AMP selectively increases renin mRNA stability in cultured juxtaglomerular granular cells. J Biol Chem. 1993;268:24138–24144. [PubMed] [Google Scholar]
- Cheng HF, Wang SW, Zhang MZ, McKanna JA, Breyer R, Harris RC. Prostaglandins that increase renin production in response to ACE inhibition are not derived from cyclooxygenase-1. Am J Physiol Regul Integr Comp Physiol. 2002;283:R638–646. doi: 10.1152/ajpregu.00150.2002. [DOI] [PubMed] [Google Scholar]
- Chiu N, Park I, Reid IA. Stimulation of renin secretion by the phosphodiesterase IV inhibitor rolipram. J Pharmacol Exp Ther. 1996;276:1073–1077. [PubMed] [Google Scholar]
- Chiu T, Reid IA. Role of cyclic GMP-inhibitable phosphodiesterase and nitric oxide in the beta adrenoceptor control of renin secretion. J Pharmacol Exp Ther. 1996;278:793–799. [PubMed] [Google Scholar]
- Cholewa BC, Mattson DL. Role of the renin-angiotensin system during alterations of sodium intake in conscious mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R987–993. doi: 10.1152/ajpregu.2001.281.3.R987. [DOI] [PubMed] [Google Scholar]
- Dibona GF. Neural control of the kidney: functionally specific renal sympathetic nerve fibers. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1517–1524. doi: 10.1152/ajpregu.2000.279.5.R1517. [DOI] [PubMed] [Google Scholar]
- Friis UG, Jensen BL, Sethi S, Andreasen D, Hansen PB, Skott O. Control of renin secretion from rat juxtaglomerular cells by cAMP-specific phosphodiesterases. Circulation Research. 2002;90:996–1003. doi: 10.1161/01.res.0000017622.25365.71. [DOI] [PubMed] [Google Scholar]
- Friis UG, Jorgensen F, Andreasen D, Jensen BL, Skott O. Molecular and functional identification of cyclic AMP-sensitive BKCa potassium channels (ZERO variant) and L-type voltage-dependent calcium channels in single rat juxtaglomerular cells. Circ Res. 2003;93:213–220. doi: 10.1161/01.RES.0000085041.70276.3D. [DOI] [PubMed] [Google Scholar]
- Huang W, Sjoquist M, Skott O, Stricker EM, Sved AF. Oxytocin antagonist disrupts hypotension-evoked renin secretion and other responses in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2001;280:R760–765. doi: 10.1152/ajpregu.2001.280.3.R760. [DOI] [PubMed] [Google Scholar]
- Kammerl MC, Richthammer W, Kurtz A, Kramer BK. Angiotensin II feedback is a regulator of renocortical renin, COX-2, and nNOS expression. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1613–1617. doi: 10.1152/ajpregu.00464.2001. [DOI] [PubMed] [Google Scholar]
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind L, Wengle B, Wide L, Ljunghall S. Reduction of blood pressure during long-term treatment with active vitamin D (alphacalcidol) is dependent on plasma renin activity and calcium status. A double-blind, placebo-controlled study. Am J Hypertens. 1989;2:20–25. doi: 10.1093/ajh/2.1.20. [DOI] [PubMed] [Google Scholar]
- Marsh AC, Gibson KJ, Wu J, Owens PC, Owens JA, Lumbers ER. Insulin-like growth factor I alters renal function and stimulates renin secretion in late gestation fetal sheep. J Physiol. 2001;530:253–262. doi: 10.1111/j.1469-7793.2001.0253l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowka R, Steinhage K, Patzak A, Persson PB. An evolutionary approach for identifying potential transcription factor binding sites: the renin gene as an example. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1147–1150. doi: 10.1152/ajpregu.00448.2002. [DOI] [PubMed] [Google Scholar]
- Nilsson AB, Nitescu N, Chen Y, Guron GS, Marcussen N, Matejka GL, Friberg P. IGF-I treatment attenuates renal abnormalities induced by neonatal ACE inhibition. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1050–1060. doi: 10.1152/ajpregu.2000.279.3.R1050. [DOI] [PubMed] [Google Scholar]
- Norwood VF, Garmey M, Wolford J, Carey RM, Gomez RA. Novel expression and regulation of the renin-angiotensin system in metanephric organ culture. Am J Physiol Regul Integr Comp Physiol. 2000;279:R522–530. doi: 10.1152/ajpregu.2000.279.2.R522. [DOI] [PubMed] [Google Scholar]
- Pan L, Black TA, Shi Q, Jones CA, Petrovic N, Loudon J, Kane C, Sigmund CD, Gross KW. Critical roles of a cyclic AMP responsive element and an E-box in regulation of mouse renin gene expression. J Biol Chem. 2001a;276:45530–45538. doi: 10.1074/jbc.M103010200. [DOI] [PubMed] [Google Scholar]
- Pan L, Xie Y, Black TA, Jones CA, Pruitt SC, Gross KW. An Abd-B class HOX. PBX recognition sequence is required for expression from the mouse Ren-1c gene. J Biol Chem. 2001b;276:32489–32494. doi: 10.1074/jbc.M011541200. [DOI] [PubMed] [Google Scholar]
- Persson PB, Skalweit A, Mrowka R, Thiele B. Control of renin synthesis. Am J Physiol Regul Integr Comp Physiol. 2003;285:R491–497. doi: 10.1152/ajpregu.00101.2003. [DOI] [PubMed] [Google Scholar]
- Peters J, Clausmeyer S. Intracellular sorting of renin: Cell type specific differences and their consequences. J Mol Cell Cardiol. 2002;34:1561–1568. doi: 10.1006/jmcc.2002.2079. [DOI] [PubMed] [Google Scholar]
- Peters J, Farrenkopf R, Clausmeyer S, Zimmer J, Kantachuvesiri S, Sharp MG, Mullins JJ. Functional significance of prorenin internalization in the rat heart. Circulation Research. 2002;90:1135–1141. doi: 10.1161/01.res.0000019242.51541.99. [DOI] [PubMed] [Google Scholar]
- Peters J, Obermuller N, Woyth A, Peters B, Maser-Gluth C, Kranzlin B, Gretz N. Losartan and angiotensin II inhibit aldosterone production in anephric rats via different actions on the intraadrenal renin-angiotensin system. Endocrinology. 1999;140:675–682. doi: 10.1210/endo.140.2.6489. [DOI] [PubMed] [Google Scholar]
- Petrovic N, Black TA, Fabian JR, Kane C, Jones CA, Loudon JA, Abonia JP, Sigmund CD, Gross KW. Role of proximal promoter elements in regulation of renin gene transcription. J Biol Chem. 1996;271:22499–22505. doi: 10.1074/jbc.271.37.22499. [DOI] [PubMed] [Google Scholar]
- Saris JJ, Van Den Eijnden MM, Lamers JM, Saxena PR, Schalekamp MA, Danser AH. Prorenin-induced myocyte proliferation: no role for intracellular angiotensin II. Hypertension. 2002;39:573–577. doi: 10.1161/hy0202.103002. [DOI] [PubMed] [Google Scholar]
- Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA. Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol. 2001;281:F345–356. doi: 10.1152/ajprenal.2001.281.2.F345. [DOI] [PubMed] [Google Scholar]
- Shi Q, Gross KW, Sigmund CD. NF-Y antagonizes renin enhancer function by blocking stimulatory transcription factors. Hypertension. 2001a;38:332–336. doi: 10.1161/01.hyp.38.3.332. [DOI] [PubMed] [Google Scholar]
- Shi Q, Gross KW, Sigmund CD. Retinoic acid-mediated activation of the mouse renin enhancer. J Biol Chem. 2001b;276:3597–3603. doi: 10.1074/jbc.M008361200. [DOI] [PubMed] [Google Scholar]
- Sinn PL, Sigmund CD. Human renin mRNA stability is increased in response to cAMP in Calu-6 cells. Hypertension. 1999;33:900–905. doi: 10.1161/01.hyp.33.3.900. [DOI] [PubMed] [Google Scholar]
- Skalweit A, Doller A, Huth A, Kahne T, Persson PB, Thiele BJ. Posttranscriptional control of renin synthesis: identification of proteins interacting with renin mRNA 3’-untranslated region. Circulation Research. 2003;92:419–427. doi: 10.1161/01.RES.0000059300.67152.4E. [DOI] [PubMed] [Google Scholar]
- Skott O. Renin. Am J Physiol Regul Integr Comp Physiol. 2002;282:R937–939. doi: 10.1152/ajpregu.00625.2001. [DOI] [PubMed] [Google Scholar]
- Taugner R, Hackenthal E. The Juxtaglomerular Apparatus. Heidelberg: Blackwell Science Inc; 1989. [Google Scholar]
- Todorov V, Muller M, Schweda F, Kurtz A. Tumor necrosis factor-alpha inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1046–1051. doi: 10.1152/ajpregu.00142.2002. [DOI] [PubMed] [Google Scholar]