

Abstract
The mechanism of renal glucose transport involves the reabsorption of filtered glucose from the proximal tubule lumen across the brush border membrane (BBM) via a sodium-dependent transporter, SGLT, and exit across the basolateral membrane via facilitative, GLUT-mediated, transport. The aim of the present study was to determine the effect of streptozotocin-induced diabetes on BBM glucose transport. We found that diabetes increased facilitative glucose transport at the BBM by 67.5 % (P < 0.05) – an effect that was abolished by overnight fasting. Western blotting and immunohistochemistry demonstrated GLUT2 expression at the BBM during diabetes, but the protein was undetectable at the BBM of control animals or diabetic animals that had been fasted overnight. Our findings indicate that streptozotocin-induced diabetes causes the insertion of GLUT2 into the BBM and this may provide a low affinity/high capacity route of entry into proximal tubule cells during hyperglycaemia.
The current model for renal glucose transport is that the sugar is actively taken up across the brush border membrane (BBM) of the proximal tubule by the sodium (Na+)-dependent transporters, SGLT1 and SGLT2 (Pajor et al. 1992; Kanai et al. 1994), a process driven by the membrane electrochemical gradient. Accumulated glucose then exits the cell across the basolateral membrane (BLM) via a Na+-independent mechanism, involving the facilitative glucose transporter isoforms GLUT1 and GLUT2 (Chin et al. 1993). Both active and facilitative glucose transporters have distinct distribution profiles along the proximal tubule related to their particular kinetic characteristics (Dominguez et al. 1992). This provides a proximal tubule environment in which the bulk of filtered glucose is reabsorbed in the early S1 segment by the low affinity/high capacity glucose transporters, SGLT2 and GLUT2, whereas the high affinity/low capacity transporters, SGLT1 and GLUT1, scavenge the remaining glucose that is presented to the later portions of the proximal tubule.
Streptozotocin-induced diabetes has a variety of effects on renal function, including changes in glucose transport (Debnam & Unwin, 1996). Although studies focusing on the effect of diabetes on SGLT-mediated glucose transport have yielded conflicting results (Harris et al. 1986; Blank et al. 1989; Yasuda et al. 1990), the effect on facilitative glucose transport seems to be more consistent: expression of GLUT2 and GLUT5 (the BBM fructose transporter) is increased at the basolateral and brush-border membranes, respectively (Dominguez et al. 1994; Kamran et al. 1997; Vestri et al. 2001), and is accompanied by corresponding increases in mRNA expression levels (Chin et al. 1997); in contrast, the levels of GLUT1 protein and its mRNA have been shown to decrease in diabetes (Chin et al. 1997; Vestri et al. 2001).
The importance of understanding how diabetes affects renal glucose handling is evident from the finding that renal glucose uptake plays a key role in reducing plasma glucose concentration during hyperglycaemia (Cersosimo et al. 1997). In addition, since the plasma glucose level can influence glucose handling and utilization by the kidney (Khandelwal et al. 1979; Biava et al. 1966), changes in glucose transport in diabetes may lead to tubule cell injury and the associated renal interstitial changes seen in diabetic kidneys (Larkins & Dunlop, 1992; Wolf & Thaiss, 1995). In this context, it is also known that hyperglycaemia can increase GLUT1 expression in mesangial cells, which can in turn increase transforming growth factor (TGF)-β production, a pathogenic factor in diabetic nephropathy (Heilig et al. 1995, 1997)
In intestinal enterocytes, where the transport process for glucose is similar to the kidney, diabetes increases BBM levels of GLUT1 (Boyer et al. 1996), GLUT2 (Corpe et al. 1996) and GLUT5 (Corpe et al. 1996). Recent reports have also demonstrated raised intestinal BBM levels of GLUT2 in response to high luminal concentrations of glucose (Kellett & Helliwell, 2000). Thus, the aim of the present study was to determine the effect of streptozotocin-induced diabetes on GLUT-mediated glucose transport at the renal BBM. Changes in glucose transport were assessed using BBM vesicles prepared from non-diabetic, diabetic, and overnight fasted diabetic animals, to determine the role of hyperglycaemia; Western blotting and immunohistochemistry were used to assess the contribution of the different GLUT isoforms to the diabetic response.
METHODS
Diabetes was induced in 230-260 g male Sprague-Dawley rats by administration of a single tail vein injection of streptozotocin (55 mg kg−1) 2-4 weeks prior to study. Streptozotocin was dissolved in freshly prepared 0.05 m citrate buffer (pH 4.5) and administered under light isoflurane anaesthesia. Animals were glycosuric 24 h after streptozotocin treatment. The weight of the control animals was matched to that of the 2-4 week diabetics. Animals were allowed ad libitum access to a standard rat chow (Diet RM1, SDS Ltd, Witham, Essex, UK) and water until the time of experimentation, with the exception of those subjected to an overnight fast. For all experimental procedures animals were terminally anaesthetized with intraperitoneal pentobarbitone sodium (Sagatal, Rhone-Merieux, Harlow, UK 90 mg kg−1) before removal of their kidneys. All procedures were carried out in accordance with the Animals (Scientific Procedures) Act 1986.
Brush border membrane (BBM) vesicles
The methods used to prepare renal BBM vesicles have been described previously (Biber et al. 1981). In brief, both kidneys were removed from anaesthetized animals and placed in ice-cold 154 mm NaCl. After removal of the renal capsule, the kidneys were weighed and then sliced into 2 mm thick sections and the cortex dissected away. Cortical fragments from six kidneys were pooled, weighed and suspended in 30 ml of buffer containing (mm) 300 mannitol, 12 Tris HCl, 5 EGTA, pH 7.4. The resulting suspension was homogenized for 2 min (Ultra Turrax homogenizer, Janke & Kunkel, FRG) at half speed, followed by the addition of 42 ml of cold distilled water and MgCl2 to a concentration of 12 mm, and then stirred on ice for 15 min. The solution was then centrifuged at 2000 g for 15 min and the supernatant then re-centrifuged at 21 000 g for 30 min. The pellet was suspended in 20 ml buffer containing (mm): 150 mannitol, 6 Tris HCl, 2.5 EGTA, pH 7.4, using 10 cycles of a hand-operated glass-Teflon homogenizer. MgCl2 was added to a concentration of 12 mm and, after stirring on ice for 15 min, low and high-speed centrifugations described above were repeated. The pellet was then re-suspended in 20 ml buffer (mm: 300 mannitol, 12 Tris HCl, 2.5 EGTA; pH 7.4) and centrifuged at 21 000 g for 30 min. The purified BBM pellet was finally re-suspended in the same buffer to a protein concentration of 3-6 mg ml−1 using 5-6 passes through a syringe fitted with a 21-gauge needle. All steps were carried out at 4 °C. The concentration of protein (Bradford, 1976) and activity of alkaline phosphatase (Forstner et al. 1968) and Na+/K+-ATPase (Proverbio & Del Castillo, 1981) in the initial homogenate and BBM preparation were determined in order to derive values for enrichment of these marker enzymes.
Uptake studies were carried out at 20 °C on freshly prepared vesicles as described for intestinal BBM (Sharp & Debnam, 1994). In brief, the transport process was initiated by mixing equal volumes of vesicle suspension and uptake buffer consisting of (mm) 200 NaSCN, 20 Hepes, 0.1 MgSO4 containing d-[3H]glucose and such that the final concentration of glucose was 30-960 µm. Uptake was terminated after 4 s by the addition of 3 ml of 154 mm NaCl containing 0.5 mm phlorizin, followed by vacuum filtration through 0.45 µm nitrocellulose filters (Sartorius, Germany). Three further washes were carried out. Parallel uptakes using buffer containing 1 mm phlorizin to block SGLT-mediated transport indicated that uptake was > 96 % phlorizin-sensitive. In order to assess GLUT-mediated transport, a higher glucose concentration (20 mm) was used in the presence of 1 mm phlorizin - this value is consistent with the low affinity of GLUT transporters for glucose binding (Debnam & Unwin, 1996). In addition, uptake of l-glucose at both 100 µm and 20 mm was performed to establish the contribution of passive glucose transport. Scintillation counting of the filters was used to calculate glucose accumulation and this was expressed as nmol (mg protein)−1 (4 s)−1. The kinetic parameters of Vmax (maximum transport capacity) and Kt (glucose concentration at half Vmax) for phlorizin-sensitive uptake were derived using a non-linear least squares curve-fitting program.
Western blotting
Antiserum against GLUT1, GLUT2 and GLUT5 were raised in rabbits with synthetic peptides corresponding to residues 477-492 (GLUT1) or 507-522 (GLUT2) or 490-502 (GLUT5) conjugated with an additional N-terminal cysteine residue to ovalbumin. GLUT antibodies were affinity purified by passage of the antiserum through a column of peptide immobilized on Sulfollink gel (Pierce, Chester, UK) followed by elution in 50 mm diethylamine-HCl and dialysis into PBS. For Western blotting, BBM samples (20 µg protein) were solubilized in Laemmli sample buffer containing 5 % SDS and electrophoresed on a 10 % SDS polyacrylamide gel. The proteins were transferred to nitrocellulose membranes by electrophoretic blotting for 1 h at a constant current of 1 mA cm−2. Non-specific protein-binding sites were blocked with PBS-T (PBS containing 0.1 % Tween 20) and 5 % fat-free milk for 3 h at room temperature. The filters were incubated with GLUT1 (1:1000 dilution), GLUT2 (1:1500) or GLUT5 (1:1000) antibodies for 16 h at 4 °C. The filters were washed (2 × 15 min) with PBS-T containing 1 % fat-free milk, and incubated with a swine anti-rabbit IgG antibody conjugated to horseradish peroxidase for 2 h at room temperature and finally washed again with PBS-T. Bound antibodies were detected by an enhanced chemiluminescence system (Amersham International plc, Amersham, Bucks, UK) and visualized using a Fluor-S MultiImager system (Biorad, Hertfordshire, UK).
Immunohistochemistry
The abdominal aorta of anaesthetized control and 3 week streptozotocin-treated animals was cannulated and the left kidney perfused with Hanks balanced salt solution (Sigma, Dorset, UK) to remove blood, followed by perfusion with 40 ml of 2 % periodate-lysine-paraformaldehyde (PLP) (McLean & Nakane, 1974). The left kidney was removed, decapsulated, embedded in OCT compound (BDH, Poole, UK) and then frozen in isopentane pre-cooled in liquid nitrogen. Samples were stored at -80 °C until use. Cryostat sections of 7 µm were mounted onto polysine-coated slides (BDH, Poole, UK) and air-dried for 1 h. Sections were washed for 5 min in 0.1 m PBS (pH 7.4) followed by treatment with 0.3 % methanolic H2O2 for 15 min to eliminate endogenous peroxidase activity. Sections were washed (3 × 10 min) with 0.1 m PBS and then blocked for 30 min with 10 % normal goat serum (NGS) (Sigma, Poole, UK) containing 1 % BSA and 1 % Triton X-100. Sections were briefly washed and incubated overnight at 4 °C with GLUT2 antibody at 1:100 dilution in 1 % NGS, 0.1 % BSA and 0.1 % Triton X-100 in 0.1 m PBS (PBGST). Following overnight incubation, sections were washed (3 × 10 min, 0.1 m PBS) and then incubated with biotin-conjugated goat anti-rabbit IgG (Vector Laboratory, Burlingame, CA, USA) at 1:500 dilution in PBGST for 1 h at room temperature. Subsequently, sections were washed (3 × 10 min, 0.1 m PBS) and then incubated with ExtrAvidin (Sigma, Poole, UK) at 1:1000 dilution in PBGST for 1 h at room temperature. Sections were washed (3 × 10 min) using 0.5 m Tris/HCl buffer (pH 7.4) and then incubated in a mixture of 0.1 mg ml−1 3,3′-diaminobenzidine tetrahydrochloride (DAB), 0.5 m Tris buffer and 0.1 % H2O2. The reaction was stopped by three brief washes with cold Tris/HCl. Slides were dehydrated, cleared and cover-slipped using DPX (BDH, Poole, UK).
Confocal imaging of GLUT2
In vivo fixation and tissue handling were as described earlier; 7 µm cryostat sections were allowed to reach room temperature and then washed for 5 min in 0.1 m PBS (pH 7.4), followed by blocking for 30 min with 10 % normal goat serum (NGS) in 0.1 m PBS (Sigma, Poole, UK), containing 1 % bovine serum albumin (BSA) and 1 % Triton X-100. Sections were briefly washed and incubated overnight at 4 °C with GLUT2 antibody at 1:100 dilution, in 0.1 m PBS containing 1 % NGS, 0.1 % BSA and 0.1 % Triton X-100 (PBGST). Following overnight incubation, sections were washed (3 × 10 min, 0.1 m PBS) and then incubated with Cy2- labelled goat anti-rabbit IgG (Jackson Immunoresearch Laboratories) at a 1:100 dilution in PBGST for 30 min at room temperature. Subsequently, sections were washed (3 × 10 min, 0.1 m PBS) and mounted with Mowoil overnight at 4 °C. Cell fluorescence was recorded by excitation at 488 nm with an Omnichrome series 43 (PerkinElmer) argon ion laser system using an Olympus IX70 microscope fitted with a × 100 oil immersion objective. Images were acquired with a charged coupled device camera (PerkinElmer) cooled to -35 °C and controlled with the Ultraview 4.0 software (PerkinElmer). Z-Scans (5.0 µm thickness) were performed with 0.5 µm spacing between each slice. The images obtained were produced as an overlay of one to three images selected from the Z-stack as the most representative of the entire section.
Statistics
Values were expressed as mean ± s.e.m.; n values represent numbers per treatment group. Differences between groups were tested by Student's unpaired t test with a significance of P < 0.05.
Chemicals
d-[3H]glucose was obtained from Amersham International (Amersham, Buckinghamshire, UK). Unless otherwise stated all chemicals were purchased from Sigma (Poole, Dorset, UK) or Merck Ltd (Poole, Dorset, UK) and were of analytical grade.
RESULTS
Blood glucose levels were 3 to 4-fold higher in diabetic animals compared with control (Table 1). Diabetic animals lost an average of 37.2 ± 3.6 g (n = 12) during the period of streptozotocin treatment, although kidney and cortex weights were significantly increased by 37 and 38 %, respectively (P < 0.001, unpaired t test) (Table 1). Overnight fasting of diabetic animals reduced blood glucose levels to that of control, but had no significant effect on whole kidney or cortex weight (Table 1).
Table 1.
Effect of diabetes on plasma glucose levels and weights of whole kidney and cortex
| Treatment group | Plasma glucose (mM) | Kidney weight (g kidney−1) | Cortex weight (g kidney−1) |
|---|---|---|---|
| Control | 10.3 ± 0.35 | 0.97 ± 0.04 | 0.44 ± 0.02 |
| 2–4 week diabetic | 36.8 ± 1.87** | 1.33 ± 0.06* | 0.62 ± 0.03* |
| 2–4 week diabetic + fast | 11.5 ± 0.6† | 1.28 ± 0.05* | 0.59 ± 0.03* |
Results are expressed as means ± s.e.m. Values were obtained from six animals per treatment group.
P < 0.001
P < 0.00001 compared to control
P < 0.00001 compared with 2–4 week diabetic.
Control BBM vesicles were enriched in alkaline phosphatase (7.32 ± 0.40-fold, n = 6), but not Na+/K+-ATPase (0.71 ± 0.15-fold, n = 6). Diabetes had no significant effect on these values (results not shown). Glucose uptake studies revealed the expected time-dependent overshoot, which was blocked by phlorizin (Fig. 1), the remaining component being a mixture of facilitated and passive (non-carrier mediated) transport. Passive transport was measured using l-[3H]glucose and was shown to contribute < 1 % to phlorizin-insensitive transport (0.02 nmol (mg protein)−1 (4 s)−1) and < 2 % of phlorizin-sensitive transport (0.013 nmol (mg protein)−1 (4 s)−1). Vesicle-trapped space, as determined by incubation of BBM vesicles with 100 µm [3H]glucose for 15 min, was also unaffected by diabetes (in µl (mg protein)−1: control: 1.96 ± 0.18 vs. diabetic: 1.76 ± 0.15, P < 0.1, n = 6). The maximum capacity for SGLT-mediated glucose transport was unchanged by diabetes (in nmol (mg protein)−1 (4 s)−1: control: 3.54 ± 0.41 vs. diabetic: 3.93 ± 0.49, P > 0.5, n = 6) (Table 2). In contrast, 2-4 week diabetes increased significantly the rate of GLUT-mediated glucose uptake by 67.5 % (control: 1.14 ± 0.18 vs. diabetic: 1.91 ± 0.21 nmol (mg protein)−1 (4 s)−1, P < 0.05, n = 6) (Table 2). Overnight fasting of diabetic animals had no effect on SGLT-mediated glucose uptake, but returned GLUT-mediated uptake to that of control (diabetic: 1.91 ± 0.21 vs. fasted-diabetic: 1.21 ± 0.15 nmol (mg protein)−1 (4 s)−1, P < 0.05, n = 6) (Table 2). Addition of cytochalasin B, believed to be an inhibitor of facilitative glucose transport, abolished GLUT-mediated glucose transport in control BBM vesicles, incubated with 20 mm glucose, by 24 ± 3.2 % (n = 4), and by 25.25 ± 5.6 % in diabetic BBM vesicles (n = 4).
Figure 1. Time dependency of glucose uptake by BBM vesicles prepared from renal cortex of normal rats.
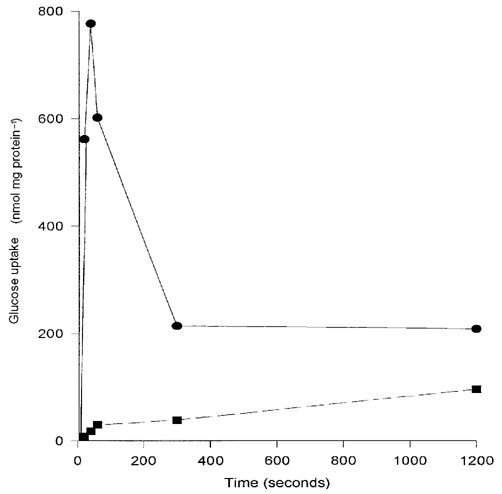
Uptake was measured at room temperature using buffer containing 200 mm NaSCN and 100 µmd-[3H]glucose with (▪, dashed line) or without (•, continuous line) 1 mm phlorizin (PZ) to block SGLT-mediated transport.
Table 2.
Kt and Vmax of SGLT-mediated glucose transport derived from uptakes measured at 30–960 μm glucose and GLUT-mediated glucose uptake measured at 20 mm glucose
| Treatment group | Kt | Vmax | GLUT mediated |
|---|---|---|---|
| Control | 434.3 ± 68.8 | 3.54 ± 0.41 | 1.14 ± 0.18 |
| 2–4 week diabetic | 585.0 ± 54.8 | 3.93 ± 0.49 | 1.91 ± 0.21* |
| 2–4 week diabetic + fast | 504.4 ± 134.2 | 3.61 ± 0.75 | 1.21 ± 0.15† |
Kt is expressed as μm and Vmax and GLUT-mediated uptake as nmol (mg protein)−1(4s)−1. Results are expressed as means ± s.e.m. of six vesicle preparations per treatment group.
P < 0.05 compared to control
P < 0.05 compared to 2–4 week diabetic.
Western blotting for detection of GLUT1, GLUT2 and GLUT5 was carried out in order to define the transport proteins responsible for the increased GLUT-mediated glucose uptake. All three transporters were detectable in BBM vesicles at 54, 58 and 49 kDa, respectively (Fig. 2A). Diabetes had no significant effect on the level of GLUT1, but resulted in enhanced expressions of GLUT2 and GLUT5 (P < 0.001 and P < 0.05 respectively, n = 6) (Fig. 2B). Overnight fasting of diabetic animals abolished the diabetes-induced increase in GLUT2 protein expression (P < 0.001 compared with diabetes, n = 6) (Fig. 2B). There was a tendency for decreased GLUT5 protein expression in fasted diabetic compared to diabetic BBM but this did not reach statistical significance.
Figure 2. Detection and quantification of renal BBM facilitative glucose transporters.
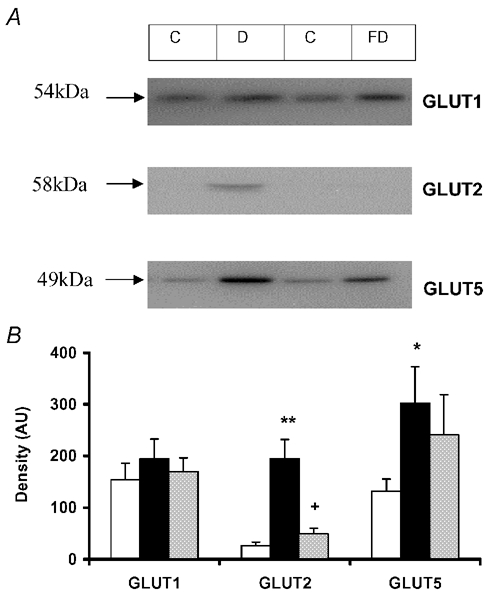
A, detection of GLUT1, GLUT2 and GLUT5 by Western blotting using BBM vesicles prepared from renal cortex of control (C), 2–4 week diabetic (D) and 2–4 week diabetic rats subjected to an overnight fast (FD). B, quantification of levels of GLUT1, GLUT2 and GLUT5 from bands produced by Western blotting of cortical BBM vesicles prepared from normal (open bars), 2–4 week diabetic (black bars) and overnight fasted, 2–4 week diabetic rats (grey bars). Results are expressed as mean ± s.e.m. Values were obtained from Western blots for each GLUT isoform carried out on six vesicle preparations. * P < 0.05, ** P < 0.001 compared to control; + P < 0.001 compared to 2–4 week diabetic.
Immunohistochemistry using kidneys that had been fixed in vivo using 2 % periodate-lysine-paraformaldehyde (PLP) showed the expected basolateral staining of GLUT2 (Fig. 3A) in the S1 segment of the proximal convoluted tubule (PCT) in control animals (Fig. 3E). However, staining on both basolateral and brush border membranes of the same region was evident in diabetic animals (Fig. 3B). In agreement with vesicle uptake and Western blotting data, GLUT2 was not seen at the BBM of proximal tubules from diabetic animals subjected to an overnight fast (Fig. 3C). Antibody specificity was checked using sections probed with antibody pre-absorbed with an excess of antigenic peptide (Fig. 3D). Confocal imaging confirmed these findings (Fig. 4); in addition it revealed that following the overnight fast, GLUT2 protein was present not only at the BLM, but also intracellularly, near the basolateral region of proximal tubular cells (Fig. 4C). In light of these findings it appears that transcytosis of GLUT2 protein occurs in response to fluctuations in plasma glucose concentrations. Indeed, Kellett et al. have demonstrated previously that GLUT2 is rapidly translocated into and out of the enterocyte BBM in response to changes in plasma or luminal glucose concentrations (Kellett & Helliwell, 2000), and that even during euglycaemia, the transporter is still present at the BBM (Kellett et al. 2002). However, in contrast, immunohistochemistry and Western blotting of renal tissue have failed to detect this protein at the BBM, except during hyperglycaemia. To ensure that removal of blood, and therefore of circulating glucose, from the kidneys prior to fixation did not result in redistribution of BBM GLUT2, the Hanks balanced salt solution was supplemented with 7 mm glucose. This glucose concentration is representative of the circulating blood concentration in control rats. GLUT2 staining was only detected at the BLM of control proximal tubules following perfusion with the supplemented Hanks buffer (results not shown), confirming that GLUT2 is normally localized to the renal BLM during euglycaemia.
Figure 3. Localization of GLUT2 protein in kidneys from control (A and E), 2–4 week diabetic (B and D) and overnight fasted, 2–4 week-diabetic rats (C).
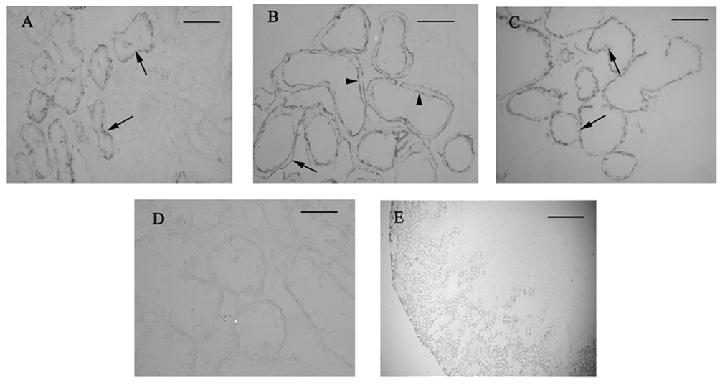
GLUT2 is localized at the BLM in control and overnight fasted kidneys (arrows), but can be detected at both the BLM (arrows) and BBM (arrowheads) of diabetic kidneys. Antibody specificity was confirmed using sections probed with antibody pre-absorbed with an excess of antigenic peptide (D). E, low power image demonstrating that GLUT2 protein is expressed in the outer cortex of the kidney. Scale bars: A-D = 50 µm, E = 500 µm.
Figure 4. Confocal imaging of GLUT2 protein in kidneys from control, 2-4 week diabetic and overnight fasted, 2-4 week diabetic rats.
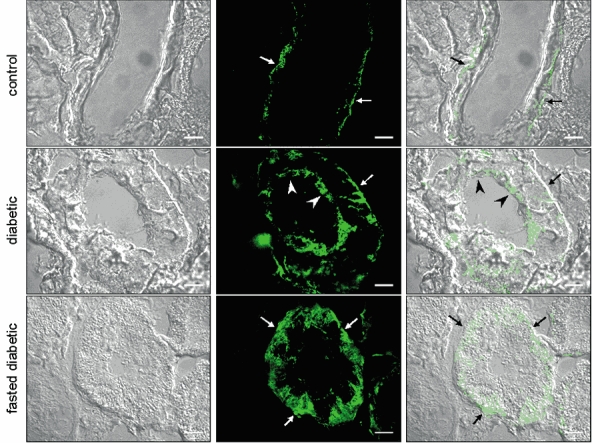
The images obtained were produced as an overlay of one to three images selected from the Z-stack as the most representative of the entire section. GLUT2 was localized at the BLM (arrows) in control kidneys, but was detected at both the BLM and BBM (arrowheads) of diabetic kidneys. Overnight fasting of diabetic rats shows intracellular and BLM staining of GLUT2. Scale bars = 10 µm.
Figure 5 demonstrates the staining pattern of GLUT1 and GLUT5 in control and diabetic kidneys fixed with 2 % PLP. Under both control (Fig. 5A) and diabetic (Fig. 5B) conditions, GLUT1 displayed intracellular as well as membrane staining in the S3 segment of the proximal tubule; whereas GLUT5 protein was located exclusively at the BBM of the S3 segment under both conditions (Fig. 5C and D). In accordance with the Western blotting data, GLUT5 staining appeared more intense in the diabetic proximal tubules (Fig. 5D). In order to confirm the segment-specific localization of the GLUT proteins, sections were probed with Vector Red (Vector Labs LTD, Peterborough, UK) to stain for endogenous alkaline phosphatase, a marker of the BBM, which allows identification of the proximal tubules (results not shown).
Figure 5. Localization of GLUT1 (A and B) and GLUT5 (C and D) protein in kidneys from control (A and C) and diabetic rats (B and D).
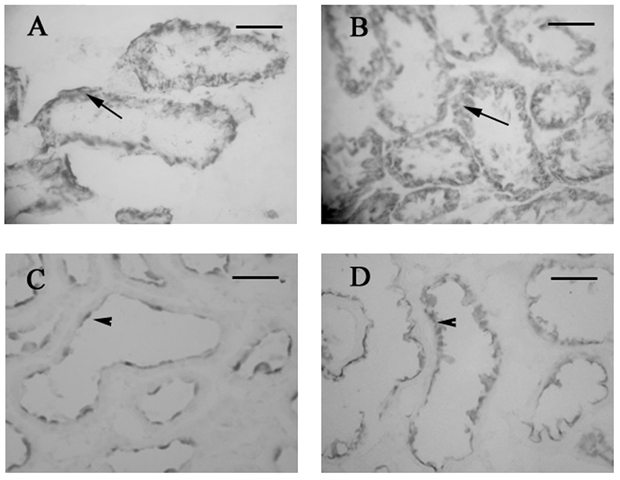
In both control and diabetic kidneys, GLUT1 displays intracellular as well as membrane staining, whilst, GLUT5 can be detected exclusively at the BBM. Scale bars = 50 µm.
DISCUSSION
Glucose reabsorption across the proximal tubule involves SGLT1- and SGLT2-mediated uptake across the BBM and facilitated efflux across the BLM mediated by GLUT1 and GLUT2. Another facilitative glucose transporter, GLUT5, is located at the BBM, where it transports fructose (Sugawara-Yokoo et al. 1999). In vitro, this isoform has a low affinity for glucose (Rand et al. 1993), and therefore in states of hyperglycaemia, such as in diabetes, when the filtered load of glucose is high, this isoform may contribute to BBM glucose uptake.
Previous studies have yielded conflicting results on the effects of streptozotocin-induced diabetes on SGLT-mediated glucose transport (Harris et al. 1986; Blank et al. 1989; Yasuda H et al. 1990). Our present results support the view that diabetes has no effect on SGLT-mediated transport, which is also in keeping with the observation that levels of SGLT1 protein are unchanged in diabetes (Dominguez et al. 1994). However, the effect of diabetes on SGLT2-mediated transport is unknown and our kinetic data for phlorizin-sensitive glucose transport could obscure any mediation of glucose uptake by this isoform. The lack of a suitable antibody to SGLT2 means that it is not possible, as yet, to study the protein expression levels of this transporter in diabetes.
The most significant finding of the present study is that facilitative transport represents an important route for glucose uptake across the BBM during hyperglycaemia. Overnight fasting of diabetic rats reduced blood glucose levels to those of controls, and abolished the increase in GLUT-mediated transport. The increase in glucose uptake during diabetes was also accompanied by significantly raised levels of GLUT2 and GLUT5 protein at the BBM, but not GLUT1, as demonstrated by Western blotting. Overnight fasting completely abolished the increase in GLUT2 protein expression, yet only had a small effect on the levels of GLUT5. This finding implies that GLUT2 is responsible for the increased glucose uptake seen during diabetes, with raised levels of glucose in the plasma or tubular fluid being the stimulus to increased GLUT2 expression. The disproportionate increase in GLUT2 expression at the BBM in relation to the degree of stimulation of glucose transport suggests that hyperglycaemia also affects the intrinsic activity of this transporter. Indeed, changes in the activity of GLUT2 protein have been reported for the intestinal BBM in response to altered luminal glucose concentration (Kellett & Helliwell, 2000) and systemic infusion of glucagon-like peptide 2 (Au et al. 2002).
Immunohistochemistry performed on unfixed tissue has previously failed to localize GLUT2 protein to the proximal tubule BBM (authors’ unpublished observations). However, immunohistochemistry performed on kidneys fixed in vivo with periodate-lysine-paraformaldehyde (PLP) revealed that GLUT2 is expressed at the BBM during hyperglycaemia. In addition, this fixation method demonstrated that GLUT1 protein displays extensive intracellular as well as BLM and BBM staining, the pattern of which is unaltered in diabetes. In contrast, GLUT5 is localized exclusively to the BBM, with a more intense staining pattern in diabetic proximal tubules. This important finding supports our vesicle uptake and Western blotting data and demonstrates that contamination by BLM is unlikely to be responsible for our results. PLP fixation has been reported to preserve antigenicity and cellular ultrastructure, as well as paraformaldehyde and glutaraldehyde, respectively. McLean & Nakane (1974) proposed that stabilization of carbohydrate structures occurs as a result of the interactions of periodate and lysine. Where periodate oxidizes carbohydrates to form aldehyde groups and lysine, a divalent amine then cross-links the carbohydrate-containing molecules by interacting with the aldehyde groups and fixing the protein in the membrane. Therefore, this in vivo fixation method is critical for the detection of GLUT2 at the proximal tubule BBM.
The observation that GLUT2 is not detectable at the BBM of proximal tubules from control or diabetic animals after an overnight fast also suggests that the protein is rapidly shuttled into and out of the BBM in response to changes in plasma or luminal glucose concentrations. The fact that GLUT2 has not been localized previously to this membrane might be because this shuttling process is both rapid and dependent on ambient glucose concentrations. What mechanism underlies this process has yet to be elucidated.
It is now accepted that diabetic-induced glomerular and tubular cell damage occur as a consequence of prolonged exposure to high glucose concentrations (Larkins & Dunlop, 1992; Wolf & Thaiss, 1995; Nishikawa et al. 2000). Mesangial cells grown under normal glucose conditions, but made to express increased levels of GLUT1 protein, display the pathological features of cells grown in high glucose concentrations, which include increased collagen and fibronectin synthesis (Heilig et al. 1995). It has also been suggested that increased GLUT1 expression in these cells is a consequence of hyperglycaemia and that this contributes to the glomerular damage seen in diabetes (Heilig et al. 1995, 1997). Since levels of GLUT2, but not GLUT1, are increased in BBM prepared from diabetic kidneys, GLUT2, like GLUT1 in mesangial cells, might be responsible for increased glucose uptake in proximal tubular cells, which sets in process a series of biochemical events leading to diabetic nephropathy. Indeed, it is intriguing to note that an inherited metabolic disorder known as the Fanconi-Bickel syndrome, in which glycogen accumulates in the proximal tubule and a diabetic-like nephropathy develops, has been linked recently to mutations of the GLUT2 gene (Berry et al. 1995), emphasizing the potential importance of this facilitative glucose transporter in renal tubular pathophysiology.
In conclusion, we have demonstrated for the first time the involvement of facilitative glucose transport across the renal BBM in a model of diabetes mellitus and that this process is most likely to be mediated by the GLUT2 isoform. In addition, GLUT2 protein expression at this membrane depends on the blood glucose level. This alters our current view of the process of glucose transport across the renal BBM during hyperglycaemia, in that GLUT2 is probably a major contributor to glucose reabsorption in diabetes. Finally, of relevance to our findings in the kidney, is that the conventional model of intestinal glucose uptake has also been challenged following the observation that there is increased expression of GLUT2 at the enterocyte BBM in diabetes (Boyer et al. 1996; Corpe et al. 1996) and in response to elevated glucose levels in the intestinal lumen (Kellett & Helliwell, 2000).
Acknowledgments
The Wellcome Trust (grant 060789/Z/00) and the St Peter's Trust supported this work. We thank Mr M. Adams for excellent technical assistance and Miss L. Churchill for advice on the immunohistochemistry.
REFERENCES
- Au A, Gupta A, Schembri P, Cheeseman CI. Rapid insertion of GLUT2 into rat Jejunal brush-border membrane promoted by glucagon-like peptide 2. Biochem J. 2002;397:247–254. doi: 10.1042/BJ20020393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry GT, Baker L, Kaplan FS, Witzleben CL. Diabetes-like renal glomerular disease in Fanconi-Bickel syndrome. Pediatr Nephrol. 1995;9:287–291. doi: 10.1007/BF02254185. [DOI] [PubMed] [Google Scholar]
- Biava C, Grossman A, West M. Ultrastructural observations on renal glycogen in normal and pathologic human kidneys. Lab Invest. 1966;15:330–356. [PubMed] [Google Scholar]
- Biber J, Stieger B, Haase W, Murer H. A high yield preparation for rat kidney brush border membranes. Different behaviour of lysosomal markers. Biochim Biophys Acta. 1981;647:169–176. doi: 10.1016/0005-2736(81)90243-1. [DOI] [PubMed] [Google Scholar]
- Blank ME, Bode F, Baumann K, Diedrich DF. Computer analysis reveals changes in renal Na+-glucose cotransporter in diabetic rats. Am J Physiol. 1989;257:C385–396. doi: 10.1152/ajpcell.1989.257.2.C385. [DOI] [PubMed] [Google Scholar]
- Boyer S, Sharp PA, Debnam ES, Baldwin SA, Srai SK. Streptozotocin diabetes and the expression of GLUT1 at the brush border and basolateral membranes of intestinal enterocytes. FEBS Lett. 1996;396:218–222. doi: 10.1016/0014-5793(96)01102-7. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cersosimo E, Ajmal M, Naukam RJ, Molina PE, Abumrad NN. Role of the kidney in plasma glucose regulation during hyperglycemia. Am J Physiol. 1997;272:E756–761. doi: 10.1152/ajpendo.1997.272.5.E756. [DOI] [PubMed] [Google Scholar]
- Chin E, Zamah AM, Landau D, Gronbcek H, Flyvbjerg A, Leroith D, Bondy CA. Changes in facilitative glucose transporter messenger ribonucleic acid levels in the diabetic rat kidney. Endocrinology. 1997;138:1267–1275. doi: 10.1210/endo.138.3.5015. [DOI] [PubMed] [Google Scholar]
- Chin E, Zhou J, Bondy C. Anatomical and developmental patterns of facilitative glucose transporter gene expression in the rat kidney. J Clin Invest. 1993;91:1810–1815. doi: 10.1172/JCI116392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpe CP, Basaleh MM, Affleck J, Gould G, Jess TJ, Kellett GL. The regulation of GLUT5 and GLUT2 activity in the adaptation of intestinal brush-border fructose transport in diabetes. Pflugers Arch. 1996;432:192–201. doi: 10.1007/s004240050124. [DOI] [PubMed] [Google Scholar]
- Debnam ES, Unwin RJ. Hyperglycemia and intestinal and renal glucose transport: implications for diabetic renal injury. Kidney Int. 1996;50:1101–1109. doi: 10.1038/ki.1996.416. [DOI] [PubMed] [Google Scholar]
- Dominguez JH, Camp K, Maianu L, Feister H, Garvey WT. Molecular adaptations of GLUT1 and GLUT2 in renal proximal tubules of diabetic rats. Am J Physiol. 1994;266:F283–290. doi: 10.1152/ajprenal.1994.266.2.F283. [DOI] [PubMed] [Google Scholar]
- Dominguez JH, Camp K, Maianu L, Garvey WT. Glucose transporters of rat proximal tubule: differential expression and subcellular distribution. Am J Physiol. 1992;262:F807–812. doi: 10.1152/ajprenal.1992.262.5.F807. [DOI] [PubMed] [Google Scholar]
- Forstner GG, Sabesin SM, Isselbacher KJ. Rat intestinal microvillus membranes. Purification and biochemical characterization. Biochem J. 1968;106:381–390. doi: 10.1042/bj1060381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RC, Brenner BM, Seifter JL. Sodium-hydrogen exchange and glucose transport in renal microvillus membrane vesicles from rats with diabetes mellitus. J Clin Invest. 1986;77:724–733. doi: 10.1172/JCI112367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig CW, Brosius FC, Henry DN. Glucose transporters of the glomerulus and the implications for diabetic nephropathy. Kidney Int Suppl. 1997;60:S91–99. [PubMed] [Google Scholar]
- Heilig CW, Concepcion LA, Riser BL, Freytag SO, Zhu M, Cortes P. Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin Invest. 1995;96:1802–1814. doi: 10.1172/JCI118226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran M, Peterson RG, Dominguez JH. Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J Am Soc Nephrol. 1997;8:943–948. doi: 10.1681/ASN.V86943. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for d-glucose. J Clin Invest. 1994;93:397–404. doi: 10.1172/JCI116972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellett GL, Helliwell PA. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem J. 2000;350:155–162. [PMC free article] [PubMed] [Google Scholar]
- Kellett GL, Helliwell PA, Affleck J, Cheeseman CI. The facilitated component of intestinal glucose absorption. J Physiol. 2002;60:539P. doi: 10.1111/j.1469-7793.2001.0585h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal RL, Zinman SM, Knull HR. The effect of streptozotocin-induced diabetes on glycogen metabolism in rat kidney and its relationship to the liver system. Arch Biochem Biophys. 1979;197:310–316. doi: 10.1016/0003-9861(79)90250-9. [DOI] [PubMed] [Google Scholar]
- Larkins RG, Dunlop ME. The link between hyperglycaemia and diabetic nephropathy. Diabetologia. 1992;35:499–504. doi: 10.1007/BF00400475. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl. 2000;77:S26–30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- Pajor AM, Hirayama BA, Wright EM. Molecular evidence for two renal Na+/glucose cotransporters. Biochim Biophys Acta. 1992;1106:216–220. doi: 10.1016/0005-2736(92)90241-d. [DOI] [PubMed] [Google Scholar]
- Proverbio F, Del Castillo JR. Na+-stimulated ATPase activities in kidney basal-lateral plasma membranes. Biochim Biophys Acta. 1981;646:99–108. doi: 10.1016/0005-2736(81)90276-5. [DOI] [PubMed] [Google Scholar]
- Rand EB, Depaoli AM, Davidson NO, Bell GI, Burant CF. Sequence, tissue distribution, and functional characterization of the rat fructose transporter GLUT5. Am J Physiol. 1993;264:G1169–1176. doi: 10.1152/ajpgi.1993.264.6.G1169. [DOI] [PubMed] [Google Scholar]
- Sharp PA, Debnam ES. The role of cyclic AMP in the control of sugar transport across the brush-border and basolateral membranes of rat jejunal enterocytes. Exp Physiol. 1994;79:203–214. doi: 10.1113/expphysiol.1994.sp003753. [DOI] [PubMed] [Google Scholar]
- Sugawara-Yokoo M, Suzuki T, Matsuzaki T, Naruse T, Takata K. Presence of fructose transporter GLUT5 in the S3 proximal tubules in the rat kidney. Kidney Int. 1999;56:1022–1028. doi: 10.1046/j.1523-1755.1999.00635.x. [DOI] [PubMed] [Google Scholar]
- Vestri S, Okamoto MM. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182:105–112. doi: 10.1007/s00232-001-0036-y. [DOI] [PubMed] [Google Scholar]
- Wolf G, Thaiss F. Hyperglycaemia-pathophysiological aspects at the cellular level. Nephrol Dial Transplant. 1995;10:1109–1112. [PubMed] [Google Scholar]
- Yasuda H, Kurokawa T, Fujii Y, Yamashita A, Ishibashi S. Decreased d-glucose transport across renal brush-border membrane vesicles from streptozotocin-induced diabetic rats. Biochim Biophys Acta. 1990;1021:114–118. doi: 10.1016/0005-2736(90)90022-g. [DOI] [PubMed] [Google Scholar]