

Abstract
Repetitive stimulation of Schaffer collaterals induces activity-dependent changes in the strength of polysynaptic inhibitory postsynaptic potentials (IPSPs) in hippocampal CA1 pyramidal neurons that are dependent on stimulation parameters. In the present study, we investigated the effects of two stimulation patterns, theta-burst stimulation (TBS) and 100 Hz tetani, on pharmacologically isolated monosynaptic GABAergic responses in adult CA1 pyramidal cells. Tetanization with 100 Hz trains transiently depressed both early and late IPSPs, whereas TBS induced long-term potentiation (LTP) of early IPSPs that lasted at least 30 min. Mechanisms mediating this TBS-induced potentiation were examined using whole-cell recordings. The paired-pulse ratio of monosynaptic inhibitory postsynaptic currents (IPSCs) was not affected during LTP, suggesting that presynaptic changes in GABA release are not involved in the potentiation. Bath application of the GABAB receptor antagonist CGP55845 or the group I/II metabotropic glutamate receptor antagonist E4-CPG inhibited IPSC potentiation. Preventing postsynaptic G-protein activation or Ca2+ rise by postsynaptic injection of GDP-β-S or BAPTA, respectively, abolished LTP, indicating a G-protein- and Ca2+-dependent induction in this LTP. Finally during paired-recordings, activation of individual interneurons by intracellular TBS elicited solely short-term increases in average unitary IPSCs in pyramidal cells. These results indicate that a stimulation paradigm mimicking the endogenous theta rhythm activates cooperative postsynaptic mechanisms dependent on GABABR, mGluR, G-proteins and intracellular Ca2+, which lead to a sustained potentiation of GABAA synaptic transmission in pyramidal cells. GABAergic synapses may therefore contribute to functional synaptic plasticity in adult hippocampus.
Long-term potentiation (LTP) of excitatory synaptic transmission has been examined extensively in the hippocampus. In contrast, much less is known about plasticity of GABAergic inhibitory transmission. Since hippocampal GABAergic interneurons powerfully control the excitability of CA1 pyramidal neurons (Lacaille & Schwartzkroin, 1988; Freund & Buzsáki, 1996), plasticity of inhibitory synaptic transmission may exert major influences on hippocampal excitability and function. Growing evidence suggests that the efficacy of GABAergic synapses can be persistently enhanced in neonatal rat hippocampus (reviewed by Gaïarsa et al. 2002). However, contradictory results have been reported at mature GABAergic synapses. Inhibitory postsynaptic currents (IPSCs) have been shown to be depressed during the expression of excitatory LTP in CA1 pyramidal cells (Stelzer et al. 1994) and this long-term depression (LTD) of GABAergic inhibition may underlie the EPSP-to-spike coupling associated with LTP (Lu et al. 2000).
In contrast, the activation of local inhibitory circuits in area CA1 generates polysynaptic inhibitory responses in pyramidal cells that showed potentiation following a stimulation paradigm that induces LTP at excitatory synapses (Haas & Rose, 1982). The long-lasting plasticity in polysynaptic inhibition is highly stimulus-dependent, showing long-term potentiation after theta-burst stimulation (TBS), but not following 100 Hz tetanization (Perez et al. 1999; see also Chapman et al. 1998). This activity-dependent enhancement in polysynaptic inhibition may result from changes occurring at several synaptic sites in the hippocampal circuit. First, an increase in synaptic drive onto pyramidal cells that results in an increased excitatory drive on feedback interneurons (Maccaferri & McBain, 1995), and/or a direct strengthening of excitatory synapses onto interneurons (Ouardouz & Lacaille, 1995; Perez et al. 2001) could both increase the synaptic activation of interneurons leading to an increase in the amount of GABA released. Second, the reports of lasting potentiation of miniature IPSCs (Kang et al. 1998) and monosynaptic inhibitory responses (Shew et al. 2000) in developing hippocampus, as well as a long-term increase of GABAergic transmission following repetitive stimulation in the nucleus of the solitary tract (Grabauskas & Bradley, 1999), cerebellum (Kano et al. 1992) or developing visual cortex (Komatsu, 1996), all suggest that a strengthening of GABAergic synapses themselves may also contribute to enhance inhibition, and therefore modulate neuronal excitability in parallel with plasticity at excitatory synapses.
Therefore, the aims of the present study were: (1) to examine whether long-term plasticity could occur directly at hippocampal GABAergic synapses by comparing monosynaptically evoked inhibitory responses in CA1 pyramidal neurons after TBS or 100 Hz tetani in the presence of ionotropic glutamate receptor antagonists; and if so, (2) to determine what mechanisms were responsible for such long-term changes. Our results show that TBS, but not 100 Hz trains, reliably induced LTP of monosynaptic GABAA responses. This stimulus-dependent potentiation was cooperative, requiring the activation of postsynaptic G-proteins, GABAB receptors, group I/II metabotropic glutamate receptors (mGluRs), and an increase in postsynaptic Ca2+. We also demonstrate that activation of these mechanisms is likely to require the simultaneous activation of several interneurons and/or excitatory afferents.
METHODS
Slice preparation
All experiments were carried out according to the guidelines laid down by our local Animal Care Committee at Université de Montréal. Adult male (4-6 weeks) Sprague-Dawley rats were anaesthetized with halothane (MTC Pharmaceuticals, Cambridge, Ontario, Canada) by inhalation (2 %). After decapitation, the brain was removed and placed in cold oxygenated (95 % O2-5 % CO2) artificial cerebrospinal fluid (ACSF) containing (mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3 and 10 dextrose (pH ≈7.4, 295-310 mosmol l−1). Hippocampal slices were prepared with a vibrotome (Campden Instruments Ltd, Loughborough, UK) and allowed to recover in oxygenated ACSF at room temperature for at least 1 h before experiments.
Intracellular recordings
Hippocampal slices (450 µm) were placed in a gas-fluid interface chamber and continuously perfused with ACSF (1.0 ml min−1) at room temperature while their upper surfaces were exposed to a humidified 95 % O2/5 % CO2 atmosphere. Intracellular recording electrodes were pulled from microfilament capillary glass on a Flaming-Brown micropipette puller (Sutter Instrument P-87, Novato, CA, USA) and filled with a solution containing 4 m potassium acetate and 0.01 m KCl. Recordings were performed at room temperature to improve the stability during long-term recordings. Membrane potential of CA1 pyramidal cells was held a few millivolts below spike threshold by constant depolarizing current injection to allow the recording of large-amplitude biphasic IPSPs. These IPSPs were amplified in bridge mode (Axoclamp 2A, Axon Instruments, Union City, CA, USA), displayed on a digital oscilloscope (model 1604; Gould, Ilford, UK), digitized using a data acquisition board (Axon Instruments TL-1-125) and stored on a microcomputer. Responses were also stored in digitized format using a video tape recorder (Neuro-Corder DR-886, Neuro Data Instruments Co., New York, USA) for later retrieval and analysis. Bridge balance was monitored regularly during recordings and adjusted as necessary.
Whole-cell recordings
Hippocampal slices (300 µm) were placed in a recording chamber continuously perfused with oxygenated ACSF at a rate of 2.0-3.0 ml min−1. An upright microscope (Zeiss Axioskop, Thornwood, NY, USA) equipped with a long-range water immersion objective (×40), Nomarski optics and an infrared camera (Cohu 6500, San Diego, CA, USA) was used for visual identification of cells. Patch pipettes (3-9 MΩ) were filled with a solution containing (mm): 140 potassium gluconate, 5 NaCl, 2 MgCl2, 10 Hepes, 0.5 EGTA, 10 phosphocreatine, 2 ATP-tris, 0.4 GTP-tris and 0.1 % biocytin (pH adjusted to 7.2-7.3 with KOH, 275-290 mosmol l−1). After obtaining tight seals (> 1 GΩ), whole-cell recordings were obtained from CA1 pyramidal cells in current-clamp or voltage-clamp configurations using an Axoclamp 2A or an Axopatch 1D amplifier, respectively (Axon Instruments). Experiments obtained in current-clamp were carried at 32 °C (Fig. 2) and voltage-clamp experiments were performed at 22-24 °C (Figs 3-6). Inhibitory responses were low-pass filtered at 10 KHz (−3 dB), analog filtered at 1 KHz (model 900, 8-pole bessel filter; Frequency Devices, Haverhill, MA, USA) and digitized at 20 KHz on a microcomputer using a data acquisition board (Axon Instruments TL-1-125) and pCLAMP6/7 software (Axon Instruments). In voltage-clamp experiments, recordings were discarded if series resistances increased > 25 % or decreased > 20 %.
Figure 2. LTP requires postsynaptic G-protein activation.
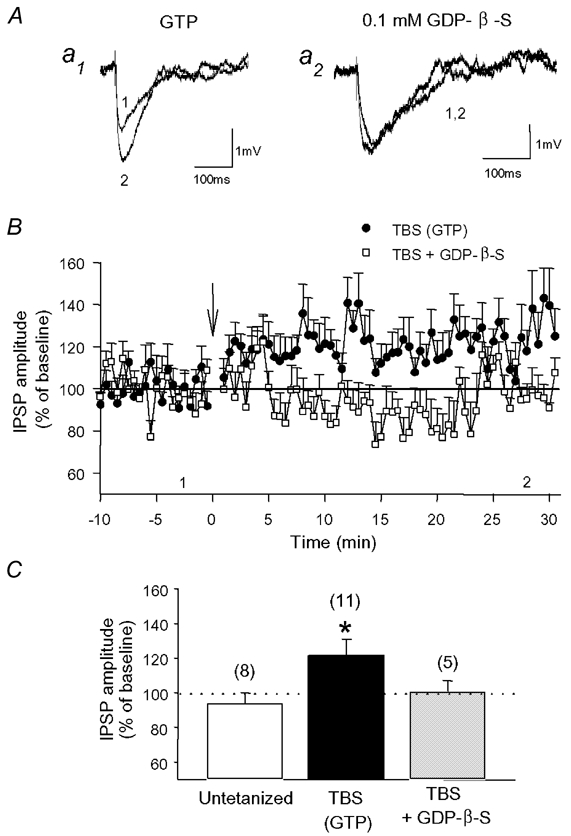
A, examples of average IPSPs obtained before (1) and after (2) TBS in whole-cell current-clamp recordings when the patch solution contained GTP (a1) or GDP-β-S (a2). B, graph of IPSP amplitude in cells containing GTP (•) or GDP-β-S (□). LTP of IPSPs induced by TBS was abolished in cells in which GDP-β-S was present to block the activation of G-proteins. C, histogram of average IPSP amplitude 30 min after TBS showing the unchanged IPSPs in untetanized cells, the significant potentiation of IPSPs in cells receiving TBS, and the abolition of LTP in cells loaded with GDP-β-S (* P < 0.05). Numbers in parentheses indicate the number of cells recorded in each condition.
Figure 3. Paired-pulse ratio of IPSCs is not affected following LTP induction.
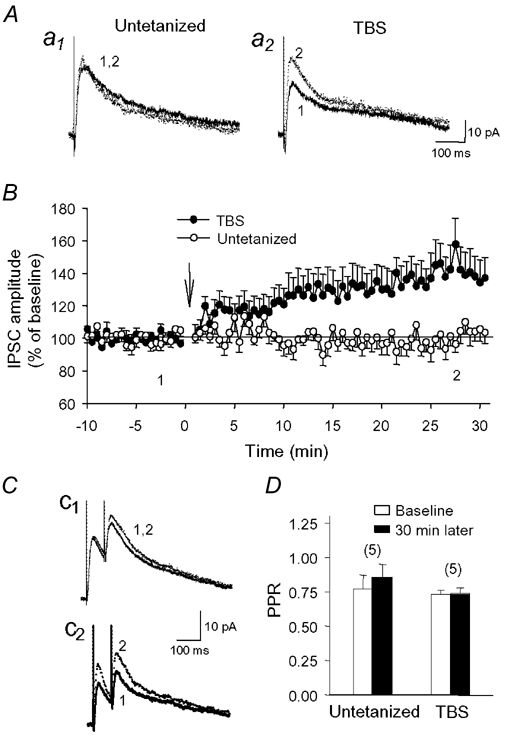
Representative IPSCs from an untetanized cell (a1) and a cell that received TBS (a2). B, mean amplitude of IPSCs in untetanized cells (○; n = 11) and cells that received TBS (•; n = 12), showing LTP of IPSCs following delivery of TBS. C, paired-stimulation given at a 75 ms interval produced similar paired-pulse depression of IPSCs in an untetanized cell (c1) and a cell that received TBS (c2). D, histogram of the paired-pulse ratio (PPR) during the control period (open bars) and 30 min later (filled bars) in untetanized cells, or during LTP in cells that received TBS. No change in PPR was found in either group of cells. Numbers in parentheses indicate the number of cells tested.
Figure 6. Short-term potentiation of unitary IPSCs during paired interneuron-pyramidal cell recordings.
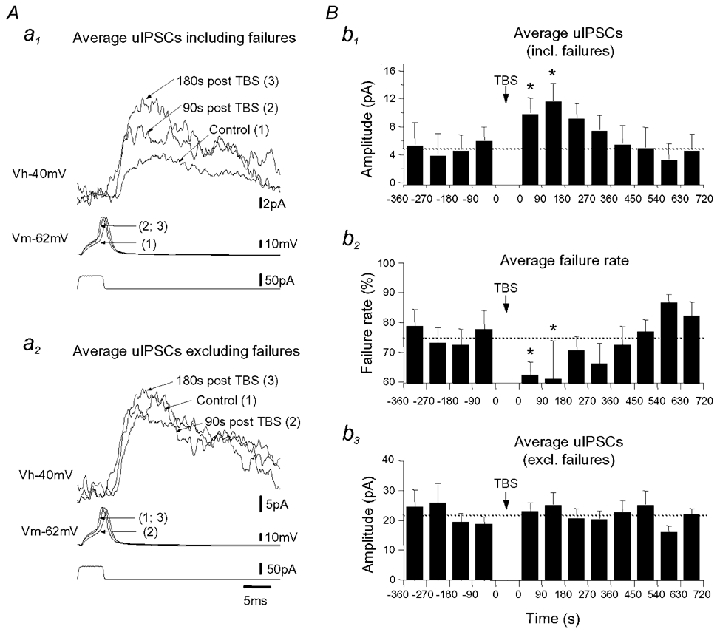
A, superimposed average uIPSCs including (a1) and excluding (a2) failures recorded from a representative lacunosum-moleculare interneuron-pyramidal cell pair (LM-PYR) in control, 90 s and 180 s after TBS. The pyramidal cell soma was held at −40 mV (holding potential (Vh), top traces). uIPSCs were generated by a single spike (membrane potential (Vm), middle traces) evoked by somatic current injection (bottom traces) in the interneuron in current-clamp mode. B, histograms of average uIPSCs including failures (b1), average failure rate (b2) and average uIPSCs excluding failures (b3) for all pairs tested (n = 3). The transient increase in uIPSCs following TBS is accompanied by a decrease in the failure rate of synaptic transmission (* P < 0.05).
Synaptic responses and data analysis
Synaptic responses were evoked by constant monophasic current pulses (50 µs, 25-300 µA) delivered through a stimulus isolation unit (WPI-A360, World Precision Instruments, Sarasota, FL, USA) to a stimulating electrode (fine monopolar, resin-coated tungsten electrode or concentric bipolar platinum-iridium electrode; Frederick Haer Co., Bowdoinham, ME, USA) placed in stratum radiatum of CA1 region. Monosynaptic inhibitory responses were pharmacologically isolated using 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and (±)-2-amino-5-phosphopentanoic acid (AP5) to block non-NMDA and NMDA receptors, respectively. Stimulation intensity was adjusted so that the amplitude of the fast GABAA-mediated response was about 50 % of maximum.
Inhibitory responses were evoked every 30 s and cells were kept for further analysis only if recordings were stable over a 10 min baseline period. Slices then received either three trains of high-frequency stimulation (HFS) or three episodes of theta-burst stimulation (TBS) delivered at 30 s intervals. HFS consisted of 50 µs duration pulses repeated at 100 Hz for 1 s and an individual episode of TBS was composed of five bursts separated by 200 ms intervals (5 Hz), each burst consisting of four 100 µs pulses at 100 Hz. Following HFS or TBS, inhibitory responses were recorded for 30 min, and untetanized control cells were recorded for a similar 40 min period. Tests in various conditions were interleaved with control experiments.
The peak amplitude of early and late GABAergic responses was measured relative to the pre-stimulus baseline using Clampfit 6/7 (Axon Instruments). Potentiation of early and late responses in intracellular recordings with sharp electrodes was assessed by comparing the average responses recorded during the last 5 min of baseline period (n = 10 traces) to those acquired after 30 s, 5-10 min, 15-20 min and 25-30 min post-tetanus (n = 10 traces) using repeated measures ANOVAs followed by Newman-Keuls tests (Fig. 1). In whole-cell recordings, early IPSP/C amplitudes were compared between the last 5 min of baseline period and 25-30 min post-TBS using Student's t test. Data are expressed as means ± s.e.m. and differences were considered significant when P < 0.05.
Figure 1. Long-term potentiation (LTP) of monosynaptic early IPSPs is induced by theta-burst stimulation (TBS) but not by high-frequency stimulation (HFS) in CA1 pyramidal cells.
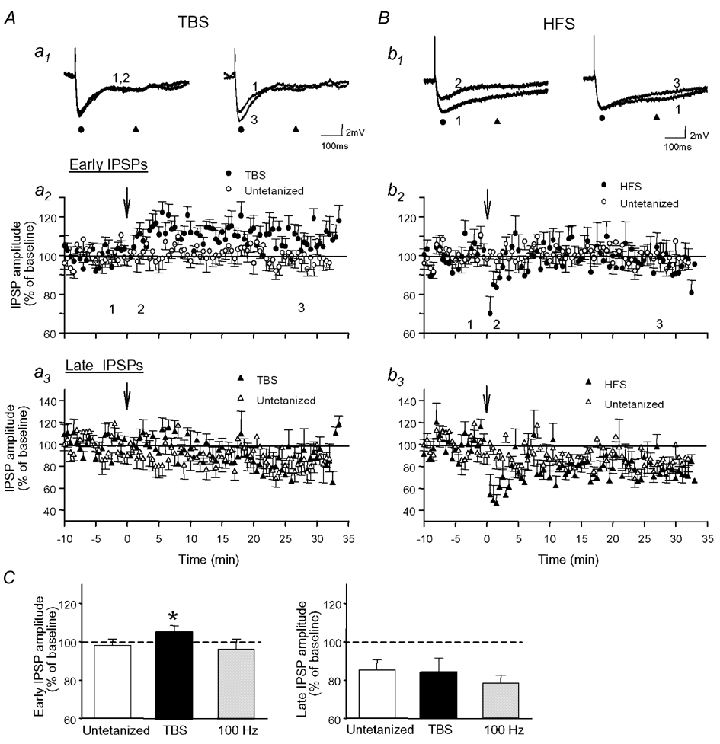
Cells recorded intracellularly received either TBS (A) or HFS (B). A, average monosynaptic IPSPs from a representative cell (a1) and graphs of the early (a2) and late (a3) IPSP amplitude for all cells tested prior to (1) and after (2, 3) delivery of TBS in stratum radiatum. Fast GABAA-mediated responses (•), but not late GABAB-mediated IPSPs (▴), were gradually increased following TBS. B, results for cells that received HFS. Early (b2) and late (b3) IPSPs were depressed immediately after HFS and returned to control levels within 5 min. Arrows indicate the times that TBS or HFS were delivered. In this and subsequent figures, numbers 1–3 indicate times at which traces were taken. C, summary histogram of average IPSP amplitude in untetanized cells, following TBS and 100 Hz tetani (n = 10 per group), illustrating the selective potentiation of early IPSPs 30 min post-TBS.
Paired-recordings of GABAA unitary IPSCs
Unitary inhibitory postsynaptic currents (uIPSCs) were evoked during paired whole-cell recordings from a synaptically connected interneuron, located in stratum lacunosum-moleculare near its border with stratum radiatum, and a CA1 pyramidal cell, as previously described (Bertrand & Lacaille, 2001). Briefly, interneurons were recorded in whole-cell current-clamp mode using an Axoclamp 2A amplifier and pyramidal cells were voltage clamped at -40 mV with an Axopatch 200B amplifier (Axon Instruments). uIPSCs were elicited by a single presynaptic action potential triggered by somatic depolarization of the interneuron every 5-7 s. To investigate the plasticity of the interneuron-pyramidal cell synapse, the TBS was delivered to the interneuron intracellularly using current injection. During TBS, the postsynaptic pyramidal cell was held in current-clamp mode. uIPSCs were analysed as previously described (Bertrand & Lacaille, 2001). In summary plots, data from 13 to 18 consecutive IPSCs were binned and averaged across experiments.
Pharmacology
Concentrated stock solutions of CNQX, AP5 (Tocris Cookson, Ellisville, MO, USA), CGP55845 (Novartis, Basel, Switzerland), acetazolamide and (RS)-α-ethyl-4-carboxyphenylglycine (E4-CPG; Tocris Cookson) were prepared in advance, frozen at -20 °C, and diluted to their final concentrations on the day of experiment. These agents were bath-applied throughout the recording period, except acetazolamide which was perfused starting 20 min before the beginning of experiment. For experiments with blocking activation of postsynaptic G-proteins, GTP was replaced by 100 µm guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) in the intracellular solution. In experiments where the Ca2+ chelator BAPTA was added to the patch solution, potassium gluconate was reduced to 110 mm and EGTA was removed. All chemicals were purchased from Sigma (St Louis, MO, USA) unless otherwise indicated.
Histology
In most whole-cell recordings, biocytin was added to the patch solution to confirm morphologically that cells were pyramidal neurons. After recordings, slices containing biocytin-filled cells were fixed at 4 °C overnight with 4 % paraformaldehyde in 0.1 m phosphate buffer (PB) and were then washed and stored in 0.1 m PB. To reveal biocytin, the slices were processed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) as previously described (Chapman & Lacaille, 1999a), air-dried for 24 h, mounted with DPX (Electron Microscopy Sciences, Fort Washington, PA, USA) and observed under a light microscope.
RESULTS
TBS induces LTP at GABA synapses
Stable intracellular recordings were obtained with sharp electrodes from 30 CA1 pyramidal cells that had a mean resting membrane potential of -56.9 ± 0.9 mV, a mean input resistance of 63 ± 3 MΩ and a mean action potential amplitude of 85.6 ± 1.4 mV. When cells were held at subthreshold membrane potentials (−52.2 ± 0.8 mV), Schaffer collateral stimulation in the presence of CNQX (20 µm) and AP5 (50 µm) evoked biphasic IPSPs composed of an early GABAA-mediated component (peak latency: 27.5 ± 1.1 ms) and a late component dependent on GABAB receptors (peak latency: 269.0 ± 5.6 ms). In untetanized cells, the amplitude of early IPSPs was stable for the 40 min recording period (99 ± 3 % of baseline), but the amplitude of late IPSPs slowly decreased to 86 ± 5 % of baseline after 40 min (Fig. 1A and C; n = 10). A similar small ‘run-down’ of late IPSPs was also observed previously (Perez et al. 1999). When TBS was delivered, the amplitude of early IPSPs increased gradually to 114 ± 3 % of baseline at 5-10 min post-TBS, remained elevated at 15-20 min post-TBS (110 ± 2 %; P < 0.05, repeated measures ANOVA) and at 25-30 min post-TBS (106 ± 3 % of baseline; P < 0.05, one-tailed; Fig. 1A and C), whereas the amplitude of late IPSPs declined to 87 ± 7 % of baseline, a level similar to that observed in untetanized cells. In contrast, both early and late IPSPs were depressed to 68 ± 8 and 52 ± 17 % of baseline, respectively, immediately after high-frequency stimulation (HFS) (Fig. 1B; P < 0.05). IPSPs then returned to control levels within 5 min post-HFS and remained constant through 30 min (early: 97 ± 5 % of baseline, late: 79 ± 4 % of baseline; Fig. 1C). Thus, while a selective LTP of early GABAA-mediated IPSPs was induced by TBS, HFS resulted in a transient depression of both early and late IPSPs and did not exert long-term changes.
Postsynaptic G-protein activation is required for LTP
We next investigated the mechanisms of TBS-induced potentiation of early GABAA responses using whole-cell recordings. To determine whether LTP could be elicited at a temperature closer to physiological conditions, we carried out current-clamp experiments at 32 °C. Similar to that observed with intracellular recordings using sharp microelectrodes at room temperature, the amplitude of early IPSPs increased progressively and was potentiated to 122 ± 9 % of baseline levels 30 min after TBS (n = 11; Fig. 2), suggesting that LTP of GABAA synaptic transmission can be expressed at a near-physiological temperature. Since G-protein-coupled receptors are involved in tetanization-induced LTP at inhibitory synapses in visual cortex (Komatsu, 1996), we next examined whether blocking postsynaptic G-proteins affected the potentiation. This was done by replacing GTP in the patch solution with GDP-β-S (0.1 mm). This substitution did not alter the baseline IPSPs (Fig. 2A) but abolished TBS-induced LTP (101 ± 6 % of baseline 30 min after TBS; n = 5), indicating that activation of postsynaptic G-proteins in CA1 pyramidal cells is required for the induction of LTP of GABAA synaptic responses.
LTP of synaptic currents does not involve changes in GABA release
Whole-cell voltage-clamp recordings were next conducted to verify that GABAA inhibitory postsynaptic currents (IPSCs) could also be potentiated (holding potential (Vh) = -55 to -60 mV). Cells (n = 59) had electrophysiological properties (input resistance, spike accommodation) characteristic of pyramidal cells and for 44 of them, biocytin-labelling confirmed their pyramidal nature. Monosynaptic early IPSCs were completely blocked by bath application of bicuculline (25 µm), confirming that they were mediated by GABAA receptors (data not shown). Consistent with current-clamp results, we found that early IPSCs gradually increased in the first 2-5 min following TBS (Fig. 3B) and were significantly potentiated to 141 ± 13 % of baseline levels 30 min post-TBS (n = 12). This potentiation was not attributed to changes in intrinsic properties of pyramidal cells since input resistance (158 ± 11 MΩ during baseline vs. 163 ± 18 MΩ post-TBS) and holding current (−31 ± 11 pA during baseline vs. -40 ± 14 pA post-TBS) were not affected by LTP (P > 0.3). The reversal potential of IPSCs (EIPSC) was shifted from -64.9 ± 0.5 mV during baseline to -69.1 ± 0.6 mV during LTP (P < 0.05; n = 5). However, it is unlikely that displacement in EIPSC contributed to LTP because EIPSC was also significantly shifted towards hyperpolarized potentials at the end of recording in untetanized cells (baseline: -64.6 ± 1.2 mV; 30 min later: -66.8 ± 1.3 mV; P < 0.05; n = 7), in which IPSCs were not potentiated (102 ± 5 % of baseline; n = 11). Although the shift in EIPSC appears more pronounced after TBS than in untetanized cells (−4.2 mV for TBS cells vs. -2.2 mV for untetanized cells), this difference was not significant (t test; P > 0.1). Therefore, these results argue against a modification of Cl− gradient in LTP, and suggest that the potentiation is mediated by an increase in GABAA receptor activity.
We wondered whether the potentiation of IPSCs was accompanied by presynaptic changes in GABA release. To examine this question, we tested whether changes in the paired-pulse ratio (PPR), which is representative of changes in the probability of transmitter release (Zucker, 1989), could be observed during LTP. Paired-pulse stimulation given at a 75 ms interval generated two successive IPSCs that showed paired-pulse depression. In untetanized cells, IPSC amplitude and PPR were both stable throughout the recording period (Fig. 3Cc1 and D; PPR during baseline: 0.77 ± 0.10; 30 min later: 0.86 ± 0.09; P > 0.1; n = 5). During LTP, the increase in IPSC amplitude was not accompanied by a change in PPR (Fig. 3Cc2 and D; baseline ratio: 0.73 ± 0.03; 30 min post-TBS: 0.74 ± 0.04; n = 5), suggesting that presynaptic mechanisms governing the release of GABA were not affected during LTP.
GABAB receptor activation in LTP
G-protein-coupled receptors play important roles in the modulation of GABA synaptic transmission (Cartmell & Schoepp, 2000). Notably, GABAB receptors have been reported to regulate inhibitory synaptic transmission following repeated stimulation and/or postsynaptic depolarization (Komatsu, 1996; Kawaguchi & Hirano, 2000; Kotak et al. 2001). We therefore wanted to determine whether G-protein-dependent mechanisms in LTP involved GABAB receptors. The application of the GABAB receptor antagonist CGP55845 (1-5 µm) affected GABA transmission in two ways. First, the amplitude of IPSCs was transiently depressed to 85 ± 8 % of baseline immediately after TBS in the presence of CGP55845 (n = 10), as compared to a small increase to 109 ± 5 % of baseline in its absence (Fig. 4B; n = 12; t test, P < 0.05). Second, CGP55845 produced variable effects on the potentiation of IPSCs, blocking LTP in half of cells tested (5/10) but not in the remaining ones. Overall, however, TBS did not produce significant LTP in the presence of CGP55845 (118 ± 12 % of baseline; Fig. 4B and C; P > 0.1), suggesting that LTP of GABA synapses depends on GABAB receptor activation.
Figure 4. TBS-induced LTP of IPSCs depends on GABAB receptor activation.
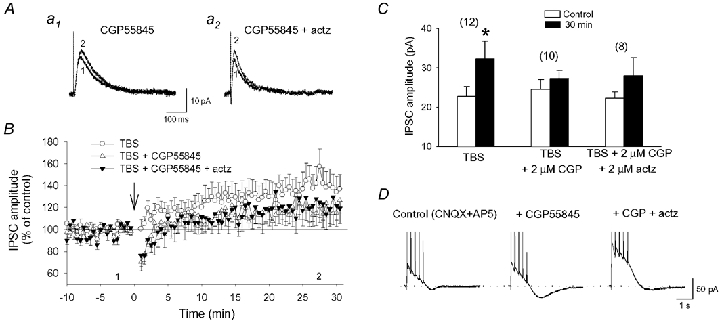
A, examples of IPSCs obtained from a cell in the presence of 2 µm CGP55845 (a1) and 2 µm CGP55845 + 2 µm acetazolamide (actz) (a2). B, graph of average IPSC amplitude for cell groups in CGP55845 (▵) and CGP55845 + acetazolamide (▾). Notice the similar changes in IPSCs in both conditions. IPSCs in normal ACSF (TBS alone; ○) are illustrated for comparison (same as in Fig. 3). C, summary histogram of average IPSC amplitudes showing that the LTP induced by TBS was inhibited by CGP55845 alone and by CGP55845 co-applied with acetazolamide. D, mean responses from three episodes of TBS obtained from a cell in control ACSF (left), in presence of CGP55845 (middle) and CGP55845 + acetazolamide (right). Note that the slow inward current generated during the application of the GABAB receptor antagonist disappeared when acetazolamide was added. The dotted line corresponds to the baseline level before the delivery of TBS.
The transient depression of IPSCs following TBS in the presence of the GABAB receptor antagonist may result from an alteration of synaptic transmission during the episodes of TBS because a slow inward current reaching -23.0 ± 5.9 pA (n = 4) was generated in presence of CGP55845, but not in its absence (−3.3 ± 1.1 pA; n = 5; Fig. 4D). A similar inward current has been described in CA1 pyramidal cells following intense stimulation and prolonged activation of GABAA receptors (Kaila et al. 1997). It has been attributed to bicarbonate (HCO3−) efflux through GABAA receptors (Grover et al. 1993) or HCO3−-dependent increase in extracellular K+ concentration (Smirnov et al. 1999). Analogous mechanisms appear responsible for the generation of the inward current during TBS because this current almost completely disappeared in the presence of acetazolamide (2 µm), an inhibitor of carbonic anhydrase, the enzyme synthesizing HCO3− (Fig. 4D; -6.7 ± 2.4 pA; n = 8). It is therefore possible that CGP55845, by blocking GABAB receptor-mediated presynaptic inhibition, may increase GABA release and prolong GABAA receptor activation. This may in turn result in dendritic Cl− accumulation and collapse of Cl− gradient (Staley et al. 1995; Staley & Proctor, 1999), which may promote HCO3−-dependent currents.
To determine whether this inward current is responsible for the transient depression of IPSCs and inhibition of LTP, we examined whether simultaneous application of CGP55845 and acetazolamide would eliminate the depression and restore LTP. While the inward current disappeared, the transient depression (83 ± 7 % of baseline) and inhibition of LTP (122 ± 13 % of baseline; n = 8) persisted in the presence of CGP55845 and acetazolamide (Fig. 4B and C). The results indicate that inhibition of presynaptic GABAB receptors generate a HCO3−-dependent inward current during TBS, but this is not responsible for the inhibition of LTP of IPSCs. Thus, GABAB receptors located postsynaptically, rather than presynaptically, may be those critical for LTP at GABA synapses.
Group I/II mGluR antagonist attenuates LTP
As described above, GABAB receptor blockade did not always inhibit LTP, suggesting that other mechanisms might also participate in this potentiation. Because fast excitatory transmission by ionotropic glutamate receptors was blocked, but metabotropic glutamate receptors (mGluRs) could still be activated, we tested whether mGluR-dependent mechanisms could be involved in LTP. The delivery of TBS during the application of 500 µm E4CPG, a group I/II mGluR antagonist, resulted in an initial gradual increase in IPSCs that was similar to that observed in control (Fig. 5B). However, this potentiation reached a plateau about 10-15 min after TBS such that IPSCs were only enhanced to 115 ± 7 % of baseline 30 min post-TBS (n = 7), a level that was not significantly different from baseline (Fig. 5B and C). This inhibition of LTP therefore indicates that group I/II mGluRs may also participate in LTP of inhibitory synaptic transmission.
Figure 5. LTP of IPSCs is reduced by a group I/II mGluR antagonist and is abolished by postsynaptic Ca2+ chelation.
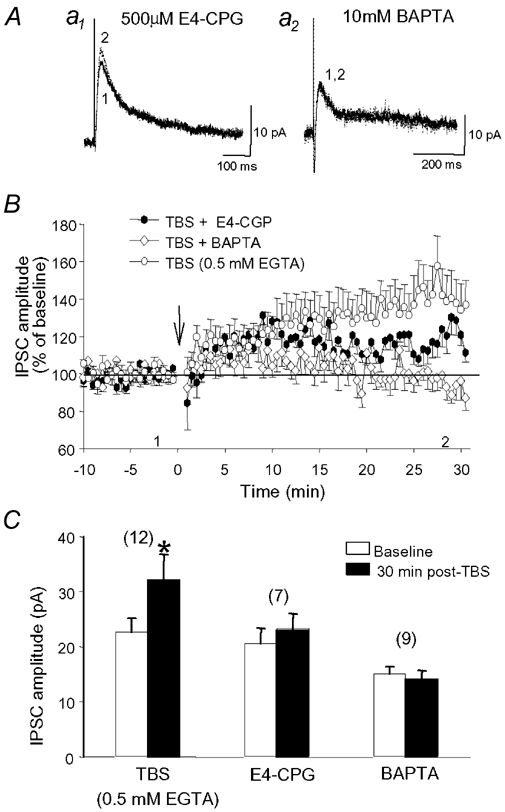
A, representative IPSCs recorded before and after TBS during perfusion with 500 µm E4-CPG (a1) and in a cell loaded with 10 mm BAPTA (a2). B, the average IPSC amplitudes show a considerable decrease in the potentiation of IPSCs in E4-CPG (•) and the prevention of LTP when cells were loaded with BAPTA (⋄). Data for cells recorded with a standard patch solution (TBS alone ○; 0.5 mm EGTA; see Methods) has been added for comparison (same as in Fig. 3). C, summary histogram illustrating that TBS-induced potentiation of IPSCs is inhibited by 500 µm E4-CPG and blocked by 10 mm BAPTA.
A postsynaptic Ca2+ rise is needed at GABA synapses to elicit LTP
Given that mGluRs can trigger postsynaptic Ca2+ elevations in hippocampal neurons (Murphy & Miller, 1988), we next wanted to test whether an increase in postsynaptic Ca2+ is required to elicit LTP of IPSCs. As shown in Fig. 5B, LTP was induced when the basal postsynaptic Ca2+ level was only moderately buffered by a low concentration (0.5 mm) of the Ca2+ chelator EGTA present in the patch pipette (see Methods; same as in Fig. 3B). In contrast, when a high concentration of the Ca2+ chelator BAPTA (10 mm) was substituted for EGTA to prevent Ca2+ elevation, LTP was abolished (Fig. 4B and C; 95 ± 6 % of baseline; n = 9). These results therefore indicate that, as found for plasticity at excitatory synapses (Malenka, 1991), a rise in postsynaptic Ca2+ is also required for the induction of LTP at GABA synapses onto CA1 pyramidal cells.
Short-term plasticity elicited by TBS in interneuron-pyramidal cell pairs
The results described so far were obtained using extracellular stimulation in stratum radiatum which activates several inhibitory/excitatory fibres. To determine whether LTP could also be induced by activation of a single interneuron, we obtained paired whole-cell recordings from synaptically connected interneurons in stratum lacunosum-moleculare (LM) and pyramidal cells (PYR). Five of 93 LM-PYR pairs obtained were synaptically connected, which corresponds to 5.4 % of all cell pairs tested (Bertrand & Lacaille, 2001). The intracellular delivery of TBS to a single LM interneuron resulted in short-term potentiation (STP) of average unitary IPSC (uIPSC) amplitude in three of the five pairs tested (Fig. 6), and no changes in the remaining two pairs. In the three pairs with changes, the average amplitude of uIPSCs (including failures) was significantly increased until 180 s post-TBS and returned to control values 360 s post-TBS (Fig. 6Aa1 and Bb1). This STP of uIPSCs was associated with a significant decrease in failure rate, as the percentage of failures decreased from 75.3 ± 3 to 62.2 ± 4 % 90 s post-TBS and further to 60.7 ± 13 % 180 s post-TBS, before returning to control level 360-450 s after TBS (Fig. 6Bb2). In contrast, the amplitude of uIPSC was not affected when failures of synaptic transmission were excluded (Fig. 6Aa2 and Bb3). These results demonstrate that at synapses between individual LM interneuron and pyramidal cells, TBS activation elicits solely short-term changes, which are likely to involve presynaptic mechanisms. Thus, cooperativity among inhibitory and excitatory inputs appears to be required for LTP.
DISCUSSION
The major findings of the present study are that tetanization at 100 Hz produced short-term depression of both GABAA and GABAB responses of CA1 pyramidal cells, whereas theta-patterned stimulation induced selective long-term potentiation of monosynaptic GABAA responses. This is markedly different from plasticity at excitatory synapses of CA1 pyramidal cells, where both tetanic and theta-patterned stimulation reliably induce LTP (Larson et al. 1986; Davies et al. 1991). The examination of the signalling pathways mediating TBS-induced LTP of inhibition revealed that postsynaptic G-protein-dependent mechanisms are necessary for LTP induction, probably through activation of postsynaptic GABAB receptors and group I/II mGluRs, and that a rise in postsynaptic Ca2+ is also required. Moreover, we found that concomitant activation of several interneurons/ presynaptic fibres appears necessary to induce LTP since the intracellular delivery of TBS to a single interneuron resulted only in a transient increase in GABAA responses at individual interneuron-pyramidal cell synapses.
Synaptic inhibition regulated at several sites in the hippocampal circuit
Perez et al. (1999) previously reported that both early GABAA and late GABAB components of polysynaptic IPSPs, evoked in pyramidal cells by stimulation of local inhibitory circuits, displayed long-lasting enhancement following theta-patterned stimulation. A long-lasting enhancement was present following 100 Hz tetanization only when postsynaptic Ca2+ elevation was prevented. This activity-dependent plasticity of polysynaptic responses was also prevented by NMDA receptor blockade. In contrast, the present study of pharmacologically isolated monosynaptic GABAergic responses has determined that only GABAA-mediated monosynaptic IPSPs are enhanced after theta-patterned stimulation by NMDA receptor independent processes. The different mechanisms involved in the long-term plasticity of polysynaptic versus monosynaptic responses indicate that these phenomena may involve changes at distinct sites in the circuit. For monosynaptic responses, direct modulation of GABA synapses may be induced by TBS. For polysynaptic responses, since feedforward and feedback excitatory inputs to interneurons are functional, TBS-induced LTP at excitatory synapses of pyramidal cells may passively propagate to interneurons (Maccaferri & McBain, 1995) and/or LTP may occur directly at excitatory synapses of interneurons (Ouardouz & Lacaille, 1995; Perez et al. 2001). These changes may lead to an increase in synaptic activation of interneurons, resulting in an enhancement of GABA release and potentiation of inhibition in pyramidal cells. Thus, theta-patterned activation may influence inhibition of pyramidal neurons by regulating synaptic transmission at many levels in the hippocampal circuit. Moreover, the activation of multiple processes by theta-patterned stimulation could explain why a potentiation of late IPSPs was observed only for polysynaptic and not for monosynaptic responses, since changes leading to enhanced activation of interneurons and GABA release are observed solely in polysynaptic recording conditions.
Frequency-dependent plasticity of monosynaptic GABAA responses
The modulation of GABAergic synapses in the adult hippocampal CA1 region appears to be highly dependent on tetanization parameters and may switch from a sustained enhancement of GABAA synaptic function to a transient depression of both GABAA and GABAB responses with increased frequency of repetitive stimulation. The depression following 100 Hz tetani could involve presynaptic mechanisms because both early and late components of IPSPs were affected. A transient depression of early IPSCs was also observed when TBS was given in presence of a GABAB receptor antagonist. Although we did not investigate the underlying mechanisms, we believe that a shift in the Cl− gradient leading to dendritic inward current and suppression of GABA responses is unlikely to contribute since the depression persisted even though the inward current was prevented with the carbonic anhydrase inhibitor acetazolamide. Alternatively, a depletion of releasable vesicles caused by increased GABA release during sustained tetanization (HFS), as well as during TBS when GABAB receptor-mediated presynaptic inhibition was removed, could account for the short-term depression (Jensen et al. 1999b). In turn, this depletion of releasable vesicles may have precluded the activation of postsynaptic mechanisms, which could explain the absence of LTP of monosynaptic GABAA responses following HFS.
The effectiveness of theta-patterned stimulation in inducing LTP at excitatory synapses has been linked to a transient reduction in GABAergic transmission via presynaptic GABAB autoreceptors (Davies et al. 1991). For the potentiation of GABAA transmission, however, TBS may result in LTP because of an effective activation of postsynaptic GABAB receptors, mGluRs and Ca2+-dependent processes, while not activating other mechanisms that contribute to depression (Stelzer et al. 1994; Stelzer & Shi, 1994).
LTP involves postsynaptic mechanisms
The suggestion that postsynaptic changes are involved in LTP is supported by the observations that: (1) TBS potentiates selectively GABAA, but not GABAB responses, (2) PPR is unaffected by LTP, (3) the reduction of LTP by a GABAB receptor antagonist persisted after the elimination of the slow inward current resulting from presynaptic GABAB receptor blockade, (4) blocking activation of postsynaptic G-proteins prevents LTP and (5) the LTP is also prevented by postsynaptic injection of BAPTA. GABAA receptors contains phosphorylation sites for protein kinases and their function may be regulated depending on their phosphorylation/dephosphorylation state (Smart, 1997). Since GABAB receptors are linked to cyclic AMP (cAMP) production (Cunningham & Enna, 1996), signalling via cAMP/protein kinase A (PKA) pathways could participate in LTP. Although reports concerning the potentiation of hippocampal GABAA receptor function by PKA-dependent phosphorylation remain controversial (Kapur & Macdonald, 1996; Poisbeau et al. 1999), such effects on GABAA synaptic transmission need to be explored in detail in CA1 region. Alternatively, modifications in protein phosphatases activity might also be involved (Wang et al. 2003).
A displacement of EIPSC toward hyperpolarized potentials was observed during whole-cell recordings, which should increase the Cl− driving force. Activity-dependent shifts in Cl− reversal potential have been reported, but these changes were toward positive membrane potentials which decreased inhibitory transmission (Sun et al. 2000; Gusev & Alkon, 2001). During development, the action of GABA switches from excitatory to inhibitory because of an increased expression of the K+-Cl− cotransporter 2 (KCC2), which shifts the Cl− reversal potential toward more hyperpolarized potentials (Clayton et al. 1998). However, it is unlikely that the shifts in EIPSC were critically involved in the potentiation of IPSCs in the present study since similar shifts were also present in untetanized cells that did not show potentiation of IPSCs.
While long-term GABAergic plasticity has been reported to result from postsynaptic changes in layer V neurons of the visual cortex (Komatsu, 1996) and in layer II/III neocortical pyramidal cells (Holmgren & Zilberter, 2001), LTP of GABAergic transmission in the hippocampus has mainly been linked to presynaptic changes. Shew et al. (2000) demonstrated that 100 Hz tetanization induces a GABAB receptor-dependent LTP of early IPSPs in rat CA1 pyramidal neurons during development. The GABAB receptors that mediate this effect are thought to be located presynaptically because irreversible activation of postsynaptic G-proteins by GTP-γ-S did not prevent the potentiation. In addition, Kang et al. (1998) described a potentiation of GABAA synaptic transmission induced by repetitive activation of interneurons that triggers GABAB receptor-dependent intracellular Ca2+ elevation in astrocytes. This potentiation was also induced by direct stimulation of astrocytes and blocked by preventing astrocytic Ca2+ rises. However, since ionotropic glutamate receptor antagonists blocked this astrocyte-mediated potentiation, it is unlikely that such mechanisms contribute to the TBS-induced potentiation of monosynaptic IPSP/Cs observed here. Thus, the present results are, to our knowledge, the first indication that long-term potentiation at adult hippocampal GABA synapses involves postsynaptic GABAB receptor activation. These results are consistent with a growing body of evidence reflecting the importance of GABAB receptors for the long-term regulation of inhibitory synaptic transmission in different brain areas (Komatsu, 1996; Kawaguchi & Hirano, 2000, 2002; Kotak et al. 2001).
Cooperativity requirement for LTP
The intracellular delivery of TBS to a single interneuron resulted in a short-term increase in uIPSCs in pyramidal cells. A similar transient IPSC increase has been observed following tetanic stimulation in paired recordings in culture (Jensen et al. 1999a). The absence of long-term increase during paired recordings suggests that the activation of a single interneuron is insufficient for LTP induction at GABA synapses. This may result from the fact that simultaneous stimulation of several interneurons is necessary to activate postsynaptic GABAB receptors (Scanziani, 2000; Bertrand et al. 2001) or that mechanisms other than those activated by GABA are involved. Interestingly, Komatsu (1996) reported that tetanization-induced LTP of inhibitory transmission in the visual cortex required GABAB receptor activation as well as postsynaptic Ca2+ rise linked to α1-adrenoceptor and/or 5-HT2 receptor activation. The implication of GABA-independent mechanisms in TBS-induced LTP is supported by the observation that the potentiation of IPSCs is prevented by a group I/II mGluR antagonist. mGluRs play important roles in excitatory LTP in area CA1 (Bashir et al. 1993) as well as in LTP at excitatory synapses of oriens-alveus interneurons (Perez et al. 2001). However, while it is well known that mGluRs can decrease the release of GABA (Desai et al. 1994; Gereau & Conn, 1995), information concerning their role in long-term regulation of inhibitory transmission is scarce (Liu et al. 1993; Chevaleyre & Castillo, 2003).
How can mGluRs affect the transmission at GABA synapses? TBS also triggers the release of glutamate from excitatory fibres, which may spillover and activate perisynaptic mGluRs located at proximity of GABAergic synapses on pyramidal cells (Baude et al. 1993; Lujan et al. 1997). Given that group I mGluRs are linked to inositol 1,4,5-trisphosphate (IP3) formation and release of Ca2+ from internal stores (Murphy & Miller, 1988), they could trigger Ca2+ signals leading to LTP. Because in some cells, a residual LTP persisted in presence of the GABAB receptor antagonist, it is possible that the strength of stimulation of Schaffer collateral fibres which impinged on those cells may have been sufficient to provide appropriate mGluR-mediated signalling to induce potentiation. Further, it remains to be examined if, as reported for astrocytes (Kang et al. 1998), GABAB receptors can generate Ca2+ rises in CA1 pyramidal cells which may be sufficient for LTP induction. Thus, we suggest that the co-activation of GABAergic and glutamatergic mechanisms, either in parallel or in synergy (Hirono et al. 2001), may cooperatively lead to LTP of GABAA synaptic transmission in adult pyramidal cells.
Functional significance
The hippocampal theta rhythm may contribute to learning and memory by promoting plasticity at excitatory synapses. Our work indicates that theta-patterned activity also induces plasticity at GABAergic synapses. What is the physiological relevance of this strengthening of GABA synapses ? LTP can be induced at Schaffer collateral-pyramidal cell synapses by either TBS or 100 Hz tetani. However, LTP of field EPSPs (fEPSPs) was of larger magnitude following TBS than 100 Hz tetani when GABAA inhibition was blocked by bicuculline (Chapman et al. 1998). This unmasking of LTP of fEPSPs by bicuculline may reflect a sustained increase in inhibition induced by TBS, opposing the LTP at excitatory synapses (Chapman et al. 1998). Thus, the physiological relevance of LTP at GABA synapses may be an activity-dependent regulation of excitability of pyramidal cells. In addition, since inhibitory interneurons contribute to pacing of theta oscillations in the hippocampus (Chapman & Lacaille, 1999a,b), theta-dependent strengthening of GABA synapses may serve to reinforce their role in hippocampal theta activity. Finally, rhythmic activation of excitatory and inhibitory inputs to pyramidal cells appears necessary to cooperatively promote plasticity of GABA synapses. Such cooperative induction of plasticity at inhibitory synapses may provide a novel mechanism through which hippocampal excitability can be modulated adaptively in an activity-dependent manner, and thus participate in the processes mediating learning and memory.
Acknowledgments
This research was funded by grants to J.-C.L. from the Canadian Institutes of Health Research (CIHR; MT-10848), Fonds de la Recherche en Santé du Québec, and Fonds pour la Formation de Chercheurs et l'Aide à la Recherche. J.-C.L. is the recipient of a Canada Research Chair in Cellular and Molecular Neurophysiology, and a member of the CIHR Group on Synaptic Transmission and Plasticity.
REFERENCES
- Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Lacaille JC. Unitary synaptic currents between lacunosum-moleculare interneurones and pyramidal cells in rat hippocampus. J Physiol. 2001;532:369–384. doi: 10.1111/j.1469-7793.2001.0369f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chapman CA, Lacaille JC. Cholinergic induction of theta-frequency oscillations in hippocampal inhibitory interneurons and pacing of pyramidal cell firing. J Neurosci. 1999a;19:8637–8645. doi: 10.1523/JNEUROSCI.19-19-08637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CA, Lacaille JC. Intrinsic theta-frequency membrane potential oscillations in hippocampal CA1 interneurons of stratum lacunosum-moleculare. J Neurophysiol. 1999b;81:1296–1307. doi: 10.1152/jn.1999.81.3.1296. [DOI] [PubMed] [Google Scholar]
- Chapman CA, Perez Y, Lacaille JC. Effects of GABAA inhibition on the expression of long-term potentiation in CA1 pyramidal cells are dependent on tetanization parameters. Hippocampus. 1998;8:289–298. doi: 10.1002/(SICI)1098-1063(1998)8:3<289::AID-HIPO10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: A novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation-Cl- cotransporter expression in rat neocortex. Brain Res Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Cunningham MD, Enna SJ. Evidence for pharmacologically distinct GABAB receptors associated with cAMP production in rat brain. Brain Res. 1996;720:220–224. doi: 10.1016/0006-8993(96)00120-5. [DOI] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABAB autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- Desai MA, McBain CJ, Kauer JA, Conn PJ. Metabotropic glutamate receptor-induced disinhibition is mediated by reduced transmission at excitatory synapses onto interneurons and inhibitory synapses onto pyramidal cells. Neurosci Lett. 1994;181:78–82. doi: 10.1016/0304-3940(94)90564-9. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Gaïarsa JL, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. TINS. 2002;25:564–570. doi: 10.1016/s0166-2236(02)02269-5. [DOI] [PubMed] [Google Scholar]
- Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabauskas G, Bradley RM. Potentiation of GABAergic synaptic transmission in the rostral nucleus of the solitary tract. Neuroscience. 1999;94:1173–1182. doi: 10.1016/s0306-4522(99)00379-6. [DOI] [PubMed] [Google Scholar]
- Grover LM, Lambert NA, Schwartzkroin PA, Teyler TJ. Role of HCO3- ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus. J Neurophysiol. 1993;69:1541–1555. doi: 10.1152/jn.1993.69.5.1541. [DOI] [PubMed] [Google Scholar]
- Gusev PA, Alkon DL. Intracellular correlates of spatial memory acquisition in hippocampal slices: Long-term disinhibition of CA1 pyramidal cells. J Neurophysiol. 2001;86:881–899. doi: 10.1152/jn.2001.86.2.881. [DOI] [PubMed] [Google Scholar]
- Haas HL, Rose G. Long-term potentiation of excitatory synaptic transmission in the rat hippocampus: the role of inhibitory processes. J Physiol. 1982;329:541–552. doi: 10.1113/jphysiol.1982.sp014318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M, Yoshioka T, Konishi S. GABAB receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat Neurosci. 2001;4:1207–1216. doi: 10.1038/nn764. [DOI] [PubMed] [Google Scholar]
- Holmgren CD, Zilberter Y. Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J Neurosci. 2001;21:8270–8277. doi: 10.1523/JNEUROSCI.21-20-08270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Jensen MS, Lambert JDC. Post-tetanic potentiation at GABAergic IPSCs in cultured rat hippocampal neurones. J Physiol. 1999a;519:71–84. doi: 10.1111/j.1469-7793.1999.0071o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Lambert JDC, Jensen MS. Activity-dependent depression of GABAergic IPSCs in cultured hippocampal neurons. J Neurophysiol. 1999b;82:42–49. doi: 10.1152/jn.1999.82.1.42. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Cyclic AMP-dependent protein kinases enhances hippocampal dentate granule cell GABAA receptor currents. J Neurophysiol. 1996;76:2626–2634. doi: 10.1152/jn.1996.76.4.2626. [DOI] [PubMed] [Google Scholar]
- Kawaguchi SY, Hirano T. Suppression of inhibitory synaptic potentiation by presynaptic activity through postsynaptic GABAB receptors in a Purkinje neuron. Neuron. 2000;27:339–347. doi: 10.1016/s0896-6273(00)00041-6. [DOI] [PubMed] [Google Scholar]
- Kawaguchi SY, Hirano T. Signaling cascade regulating long-term potentiation of GABAA receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu Y. GABAB receptors, monoamine receptors, and postsynaptic inositol triphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci. 1996;16:6342–6352. doi: 10.1523/JNEUROSCI.16-20-06342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Dimattina C, Sanes DH. GABAB and Trk receptor signaling mediates long-lasting inhibitory synaptic depression. J Neurophysiol. 2001;86:536–540. doi: 10.1152/jn.2001.86.1.536. [DOI] [PubMed] [Google Scholar]
- Lacaille J-C, Schwartzkroin PA. Stratum lacunosum-moleculare interneurons of hippocampal CA1 region. II. Intrasomatic and intradendritic recordings of local circuit synaptic interactions. J Neurosci. 1988;8:1411–1424. doi: 10.1523/JNEUROSCI.08-04-01411.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–350. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- Liu YB, Disterhoft JF, Slater NT. Activation of metabotropic glutamate receptors induces long-term depression of GABAergic inhibition in hippocampus. J Neurophysiol. 1993;69:1000–1004. doi: 10.1152/jn.1993.69.3.1000. [DOI] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron. 2000;26:197–205. doi: 10.1016/s0896-6273(00)81150-2. [DOI] [PubMed] [Google Scholar]
- Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1α, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat. 1997;13:219–241. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. Passive propagation of LTD to stratum oriens-alveus inhibitory neurons modulates the temporoammonic input to the hippocampal CA1 region. Neuron. 1995;15:137–145. doi: 10.1016/0896-6273(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Malenka RC. The role of postsynaptic calcium in the induction of long-term potentiation. Mol Neurobiol. 1991;5:289–295. doi: 10.1007/BF02935552. [DOI] [PubMed] [Google Scholar]
- Murphy SN, Miller RJ. A glutamate receptor regulates Ca2+ mobilization in hippocampal neurons. Proc Natl Acad Sci U S A. 1988;85:8737–8741. doi: 10.1073/pnas.85.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouardouz M, Lacaille J-C. Mechanisms of selective long-term potentiation of excitatory synapses in stratum oriens/alveus interneurons of rat hippocampal slices. J Neurophysiol. 1995;73:810–819. doi: 10.1152/jn.1995.73.2.810. [DOI] [PubMed] [Google Scholar]
- Perez Y, Chapman CA, Woodhall G, Robitaille R, Lacaille J-C. Differential induction of long-lasting potentiation of inhibitory postsynaptic potentials by theta patterned stimulation versus 100 Hz tetanization in hippocampal pyramidal cells in vitro. Neuroscience. 1999;90:747–757. doi: 10.1016/s0306-4522(98)00531-4. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Lacaille JC. A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc Natl Acad Sci U S A. 2001;98:9401–9406. doi: 10.1073/pnas.161493498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J Neurosci. 1999;19:674–683. doi: 10.1523/JNEUROSCI.19-02-00674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABAB receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Shew T, Yip S, Sastry BR. Mechanisms involved in tetanus-induced potentiation of fast IPSCs in rat hippocampal CA1 neurons. J Neurophysiol. 2000;83:3388–3401. doi: 10.1152/jn.2000.83.6.3388. [DOI] [PubMed] [Google Scholar]
- Smart TG. Regulation of excitatory and inhibitory neurotransmitter-gated ion channels by protein phosphorylation. Curr Opin Neurobiol. 1997;7:358–367. doi: 10.1016/s0959-4388(97)80063-3. [DOI] [PubMed] [Google Scholar]
- Smirnov S, Paalasmaa P, Uusisaari M, Voipio J, Kaila K. Pharmacological isolation of the synaptic and nonsynaptic components of the GABA-mediated biphasic response in rat CA1 hippocampal pyramidal cells. J Neurosci. 1999;19:9252–9260. doi: 10.1523/JNEUROSCI.19-21-09252.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl- and HCO3- transport. J Physiol. 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Stelzer A, Shi H. Impairment of GABAA receptor function by N-methyl-d-aspartate-mediated calcium influx in isolated CA1 pyramidal cells. Neuroscience. 1994;62:813–828. doi: 10.1016/0306-4522(94)90479-0. [DOI] [PubMed] [Google Scholar]
- Stelzer A, Simon G, Kovacs G, Rai R. Synaptic disinhibition during maintenance of long-term potentiation in the CA1 hippocampal subfield. Proc Natl Acad Sci U S A. 1994;91:3058–3062. doi: 10.1073/pnas.91.8.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK, Nelson TJ, Alkon DL. Functional swithching of GABAergic synapses by ryanodine receptor activation. Proc Natl Acad Sci U S A. 2000;97:12300–12305. doi: 10.1073/pnas.210396697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu SH, Haditsch U, Tu WH, Cochrane K, Ahmadian G, Tran L, Paw J, Wang YT, Mansuy IM, Salter MM, Lu YM. Interaction of calcineurin and type-A GABA receptor γ2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci. 2003;23:826–836. doi: 10.1523/JNEUROSCI.23-03-00826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]