

Abstract
Background K+ channels exert control over neuronal excitability by regulating resting potential and input resistance. Here, we show that GABAB receptor-mediated activation of a background K+ conductance modulates transmission at rat carotid body chemosensory synapses in vitro. Carotid body chemoreceptor (type I) cells expressed GABAB(1) and GABAB(2) subunits as well as endogenous GABA. The GABAB receptor agonist baclofen activated an anandamide- and Ba2+-sensitive TASK-1-like background K+ conductance in chemoreceptor cell clusters, but was without effect on voltage-gated Ca2+ channels. Hydroxysaclofen (50 μm), 5-aminovaleric acid (100 μm) and CGP 55845 (100 nm), selective GABAB receptor blockers, potentiated the hypoxia-induced receptor potential; this effect was abolished by pre-treatment with pertussis toxin (PTX; 500 ng ml−1), an inhibitor of Gi, or by H-89 (50 μm), a selective inhibitor of protein kinase A. The protein kinase C inhibitor chelerythrine chloride (100 μm) was without effect on this potentiation. GABAB receptor blockers also caused depolarisation of type I cells in clusters, and enhanced spike discharge in spontaneously firing cells. In functional co-cultures of type I clusters and petrosal sensory neurones, GABAB receptor blockers potentiated hypoxia-induced postsynaptic chemosensory responses mediated by the fast-acting transmitters ACh and ATP. Thus GABAB receptor-mediated activation of TASK-1 or a related channel provides a presynaptic autoregulatory feedback mechanism that modulates fast synaptic transmission in the rat carotid body.
The carotid body responds to decreases in arterial PO2 (hypoxia) by increasing afferent chemosensory discharge of the carotid sinus nerve, initiating changes in ventilation (Gonzalez et al. 1994). A popular hypothesis concerning the mechanism of hypoxic chemotransduction involves depolarisation of chemoreceptive type I cells due to hypoxic inhibition of both large-conductance Ca2+-activated (Peers, 1990) and voltage-independent background (Buckler et al. 2000) K+ channels, leading to activation of voltage-gated Ca2+ channels. The resultant influx of extracellular Ca2+ through these channels elevates intracellular Ca2+ levels, which stimulates Ca2+-dependent neurotransmitter release onto sensory afferent nerve endings (Buckler & Vaughan-Jones, 1994; Gonzalez et al. 1994; Urena et al. 1994; Lopez-Barneo, 1996; Zhang et al. 2000). Support for this hypothesis is not universal however, particularly since in some studies inhibitors of K+ channels fail to mimic the effects of hypoxia (Donnelly, 1997; Osanai et al. 1997; Lahiri et al. 1998). In contrast, the application of K+ channel blockers such as TEA or iberiotoxin was sufficient to evoke extracellular Ca2+-dependent release of catecholamines from thin slices of the rat carotid body (Pardal et al. 2000) and cultured type I clusters (Jackson & Nurse, 1997). To advance our knowledge of the mechanisms underlying carotid body function, we have developed a model of the rat carotid body in which functional synapses form de novo between isolated type I clusters and petrosal neurones (PNs) in co-culture (Zhong et al. 1997). This chemosensory complex transduces hypoxia and transmits the chemosensory response via the co-release of the excitatory neurotransmitters ACh and ATP (Zhang et al. 2000) onto postsynaptic nicotinic and purinergic receptors (Zhang et al. 2000; Prasad et al. 2001), giving rise to an increased discharge rate or depolarisation in the adjacent postsynaptic PN.
γ-Aminobutyric acid (GABA) is a well-characterised inhibitory neurotransmitter which acts at ionotropic (GABAA and GABAC) and metabotropic (GABAB) receptors in the central nervous system. The effects of GABAB receptor stimulation are slow and result in modulation of synaptic transmission via G-proteins and intracellular effector systems (Mott & Lewis, 1994; Kerr & Ong, 1995; Couve et al. 2000; Greengard, 2001) linked to Ca2+ and K+ channels (Bowery & Enna, 2000). GABA is co-localised with catecholamines and 5-HT in type I cells of the mouse carotid body (Oomori et al. 1994). GABA-immunoreactivity has also been demonstrated in the type I (glomus) cells of other species, including chipmunk and bat (Ohtomo et al. 2000), and also in neurosecretory chromaffin cells of the adrenal medulla (Oomori et al. 1993) where their activation is linked to the regulation of catecholamine secretion (Castro et al. 1989).
In carotid body type I cells, hypoxia-induced depolarisation is attributable in part to inhibition of a background K+ channel with characteristics of a member of the tandem-pore-domain family of K+ channels, TASK-1 (Buckler et al. 2000). These channels regulate resting membrane potential and help control neuronal excitability (Goldstein et al. 2001). Their activity is under strict regulation by several neurotransmitters, including 5-HT, noradrenaline, substance P, glutamate, thyrotropin releasing hormone (TRH) and ACh, acting at G-protein-coupled receptors (Millar et al. 2000; Talley et al. 2000; Goldstein et al. 2001). Since GABA is present in the carotid body, and G-protein-coupled GABAB receptors participate in autoreceptor feedback in other neurosecretory cell types (Castro et al. 1989), we tested the hypothesis that GABAB receptors participate in an autoregulatory feedback mechanism to regulate secretion in the rat carotid body. We found that selective inhibitors of G-protein coupled metabotropic GABAB receptors enhanced type I cell receptor potential via a Gi-and PKA-dependent pathway. Additionally, voltage-clamp experiments revealed that GABAB receptor stimulation activates a voltage-independent K+ conductance with pharmacological and biophysical properties similar to TASK-1. Taken together, these data suggest a novel GABA-mediated autoregulatory feedback mechanism in the carotid body that modulates synaptic efficacy by converging two separate regulatory influences onto the same background K+ conductance.
METHODS
Cell culture
Details of methods used in preparing co-cultures or separate cultures of dissociated type I cell clusters or petrosal neurones have been described previously (Zhong et al. 1997). Briefly, dissociated cells were obtained from carotid bodies or petrosal ganglia of 7- to 14-day-old rat pups (Wistar; Charles River, QC, Canada). Following humane killing by stunning with a blow to the head and decapitation, the carotid bifurcation and attached nodose-petrosal complex were excised. Procedures for animal handling and tissue removal were carried out in accordance with the guidelines of the Canadian Council on Animal Care (CCAC). To produce dissociated type I cells, carotid bodies were incubated for ≈45 min at 37 °C in an enzymatic solution containing 0.1 % collagenase-0.1 % trypsin (GibcoBRL Life Technologies, Burlington, ON, Canada). The tissues were mechanically dissociated with forceps, and triturated to yield a suspension of dispersed single cells and type 1 cell clusters, consisting of a few to 20 or more cells. The cell suspension was plated onto a thin layer of collagen or Matrigel (Collaborative Research, Bedford, MA, USA) that was previously applied to the central wells of 35 mm tissue culture dishes. For co-cultures, petrosal neurones were obtained by enzymatically and mechanically dissociating the petrosal ganglion, as described above. Dissociated petrosal neurones were then overlaid onto cultures of carotid body type I cells, after the latter had been in culture for 3-5 days. All cultures were grown at 37 °C in a humidified atmosphere of 95 % air-5 % CO2 in F-12 nutrient medium (GibcoBRL) supplemented with 10 % v/v fetal bovine serum (GibcoBRL), 80 U l−1 insulin (Sigma), 0.6 % (w/v) glucose, 2 mml-glutamine and 1 % penicillin-streptomycin (Gibco). Electrophysiological recordings were made 2-4 days after cells were plated in the case of separate cultures, and after 3-5 days in co-cultures.
Current-clamp recordings
Methods for recording chemosensory responses from co-cultured petrosal neurones and type I cell clusters have been described elsewhere (Zhong et al. 1997). These recordings were made using the perforated-patch technique to preserve cytoplasmic integrity, and patch pipettes contained intracellular solution plus 300 µg ml−1 nystatin. Recordings were carried out at ≈35 °C in bicarbonate/CO2-buffered extracellular fluid of the following composition (mm): NaCl, 115; NaHCO3, 24; KCl, 5; CaCl2, 2; MgCl2, 1; glucose, 10; and sucrose, 12; pH 7.4 maintained by bubbling 95 % air-5 % CO2. The intracellular solution contained (mm): potassium glutamate or gluconate, 115; KCl, 25; NaCl, 5; CaCl2, 1; and Hepes, 10; pH 7.2. Hypoxia (PO2 ≈5 mmHg) was generated by bubbling 95 % N2-5 % CO2 into the extracellular fluid. PO2 was measured using a commercial O2 electrode (Diamond Electro-Tech Inc., MI, USA). Drug solutions were applied by gravity perfusion in the recording solution for at least 1 min before examining their effects, and recordings of membrane potential were only made after this period of application. Hypoxic (PO2 ≈5 mmHg) solutions were applied via a fast perfusion pipette placed within 300 µm of the cells under investigation. A piezoelectric switch allowed rapid changes between a barrel carrying normoxic perfusate and one containing hypoxic perfusate. With this technique, changes in PO2 local to the cells of interest were reached almost instantaneously (< 3 s; Zhong et al. 1997) following switching. In petrosal neurones, resting membrane potential was usually more negative than -50 mV; type I cell resting potential was within the range of - 38 to - 58 mV.
Voltage-clamp recordings
Cultures were transferred to a perfused recording chamber mounted on the stage of a Zeiss Axiovert S100 microscope. Patch-clamp recordings were made using patch electrodes of resistance 4-7 MΩ when filled with intracellular solution. Patch electrodes were fabricated from 1.5/0.75 mm o.d./i.d. borosilicate glass (WPI) using a P-97 Brown-Flaming horizontal electrode puller (Sutter) and fire-polished. All voltage-clamp experiments were performed at room temperature (21-24 °C). Cells were voltage-clamped at -60 mV (−80 mV for Ca2+ currents), and currents evoked by step depolarisations to various test potentials for 100 ms at a frequency of 0.1 Hz. In some experiments, membrane potential was ramped between -60 mV and +50 mV over 1 s. Current traces were filtered at 5 kHz, digitised at 10 kHz and stored on a PC for later analysis. Capacitative transients were minimised by analog means. All analyses and voltage protocols were performed using an EPC 9 amplifier with integrated AD/DA converter and ITC-16 interface, and Pulse software (HEKA). Steady-state outward currents were measured as the average current between 90 and 99 ms of the voltage step. Inward (Ca2+) currents were measured at their peak amplitude. Voltage-clamp data were analysed using Pulsefit software (HEKA). Results are expressed as means ± s.e.m., and statistical comparisons made using Student's t test, ANOVA or the Mann-Whitney test, as appropriate.
Solutions
K+ currents
These currents were recorded using the perforated-patch configuration. Electrodes were filled with a solution containing (mm): NaCl, 5; KCl, 35; potassium gluconate, 95; Hepes, 10; CaCl2, 2; and nystatin, 300 µg ml−1. Cells were perfused with a solution composed of (mm): NaCl, 135; KCl, 5; MgCl2, 1.2; Hepes, 5; CaCl2, 2.5; and d-glucose, 10 (pH 7.4). In experiments using symmetrical K+ solutions, cells were perfused with a similar solution containing 135 mm KCl and 5 mm NaCl.
Ca2+ currents
Ca2+ channel currents were recorded using the conventional whole-cell configuration, and isolated by perfusing with an extracellular solution consisting of (mm): NaCl, 125; CsCl, 5; MgCl2, 0.6; CaCl2 5; Hepes, 5; d-glucose, 10; and TEA-Cl, 20 (pH 7.4 with NaOH). The pipette solution contained (mm): CsCl, 120; TEA-Cl, 20; MgC12, 2; EGTA, 10; Hepes, 10; and ATP, 2 (pH 7.2 with CsOH).
Drug solutions
Baclofen, 5-aminovaleric acid, hydroxysaclofen, H-7, H-89, chelerythrine hydrochloride and anandamide were obtained from Sigma. CGP 55845 was obtained from Tocris Cookson.
RT-PCR
Type I clusters and petrosal neurones were harvested from their culture dishes by suction into a broken glass microelectrode. Isolation of total RNA from type I clusters and subsequent DNase treatment and reverse transcription were as described for petrosal neurones (Prasad et al. 2001). DNA was amplified in a single PCR reaction consisting of the following (mm unless stated): Tris-HCl, 20; KCl, 50; MgCl2, 1.5; each dNTP, 0.2; each primer, 0.2 µm; 5 µl template; and Platinum Taq polymerase (Gibco), 2.5 U µl−1; total reaction volume 25 µl. Gene-specific primers used were (listed as sequence amplified, forward and reverse primers and expected size of product):
 |
GABAB(1) primers were designed against a common region and identify multiple isoforms of this subunit. The reaction was held at 94 °C for 2 min and subsequently cycled 35 times. Each cycle consisted of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 1 min, followed by a 10 min final extension at 72 °C. The specificity of the PCR primers was demonstrated by automated fluorescence sequencing (Mobix, McMaster University) of PCR products to verify their identity.
Immunofluorescence
Cryostat sections of the carotid bifurcation and cultures of type 1 cells, obtained from 7- to14-day-old rat pups, were processed for immunofluorescence. In preparation for tissue sections, animals were first anaesthetized by intraperitoneal administration of Somnotol (65 mg kg−1), before perfusion via the aorta with phosphate-buffered saline (PBS) followed by PBS containing 4 % paraformaldehyde-0.5 % glutaraldehyde. The carotid bifurcation was then excised and post-fixed for 1 h at room temperature in 4 % paraformaldehyde-0.5 % glutaraldehyde. The tissue was then washed in PBS (3 × 5 min each) and incubated overnight in 30 % sucrose at 4 °C. Sections (thickness, 10-12 µm) of the bifurcation containing the carotid body were cut in a cryostat and collected on glass slides coated with 2 % silane (Sigma). After air drying, sections were stored at -20 °C until ready for immunostaining. Following rehydration in PBS, sections were incubated overnight at 4 °C with primary antisera diluted in 1 % BSA in PBS, 0.5 % Triton X-100. Primary antibodies were guinea-pig polyclonal antisera raised against (i) GABAB(1) receptor N-terminal region (1:500 dilution); (ii) GABAB(2) receptor C-terminal region (1:500 dilution); and (iii) glutaraldehyde-conjugated GABA (1:500 dilution; all Chemicon). After rinsing in PBS (3 × 10 min each), the sections were incubated in the dark for 1 h at room temperature with the secondary antibodies diluted in blocking solution (1 % BSA/PBS, 0.5 % Triton X-100). The secondary antibody was an FITC-conjugated goat anti-guinea-pig (1:20 dilution; Jackson Immunoresearch Laboratories). Samples were washed in PBS (3 × 5 min each) and covered with Vectashield Mounting Medium (Vector Laboratories, Burlington, Ontario) before viewing under a Bio-Rad Microradiance 2000 confocal microscope, equipped with argon (two lines, 488 and 514 nm) and helium-neon (543 nm). Lasersharp software (Bio-Rad, Mississauga, ON, Canada) was used for image acquisition. In control experiments, sections were either processed as described above except that the primary antibody incubation step was omitted or, in the case of GABA immunoreactivity, the primary antibody was pre-adsorbed with excess antigen.
RESULTS
GABAB receptor blockers enhance the receptor potential in type I cells and modulate chemosensory transmission
Since GABA is localised to type I cells of the mouse carotid body (Oomori et al. 1994) and G-protein-coupled GABAB receptors participate in autoreceptor feedback in other neurosecretory cell types (Castro et al. 1989), we initially examined whether selective GABAB receptor blockade modulates receptor potential in type I cells of the rat carotid body via an autocrine-paracrine feedback mechanism. When recording type I cell membrane potential under current-clamp conditions, application of either of the GABAB receptor antagonists hydroxysaclofen (OHS; 50 µm), 5-aminovaleric acid (5-AVA; 100 µm) or CGP 55845 (100 nm) caused membrane depolarisation, consistent with the tonic activation of GABAB receptors. Membrane potential was shifted by 1.2 ± 0.6 mV (n = 6) in the presence of OHS, by 1.7 ± 0.8 mV (n = 6) in the presence of 5-AVA and by 2.7 ± 0.5 mV (n = 5) in the presence of CGP 55845. During application of these blockers, the hypoxic response was reversibly potentiated (Fig. 1A-C). Hypoxia depolarised cells by 1.6 ± 0.2 mV under control conditions, and by 3.5 ± 0.4 mV in the presence of OHS (n = 11; P < 0.001, ANOVA; Fig. 2A). Similarly, hypoxic depolarisation was increased by 5-AVA (from 2.4 ± 0.6 mV to 4.7 ± 1.2 mV, n = 7; P < 0.001, ANOVA; Fig. 1B) and by CGP 55845 (from 2.1 ± 0.5 mV to 3.3 ± 0.8 mV, n = 6; P < 0.01, ANOVA; Fig. 1C). In all cases, resting membrane potential was between -38 and -55 mV.
Figure 1. GABAB receptor blockade enhances receptor potential in chemoreceptor cells.
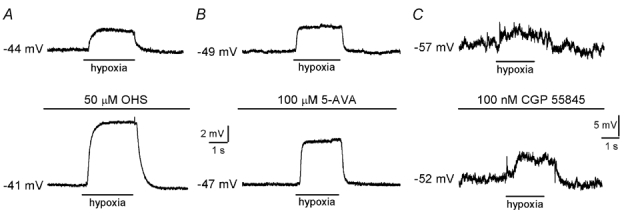
A, typical current-clamp recordings made from a type I cell cluster. Application of hypoxia (PO2, 5 mmHg) is indicated by the horizontal bar below each trace. Under control conditions (upper), hypoxia induced a depolarisation which was increased in the presence of 50 µm hydroxysaclofen (OHS; lower). B and C, as in A, except the effects of further specific blockers of GABAB receptors, 5-aminovaleric acid (5-AVA; 100 µm) and CGP 55845 (100 nm), were examined. Note the different scale bars in C. In all cases, effects of GABAB receptor blockers were fully reversed on washout of the drug (not shown).
Figure 2. Postsynaptic depolarisation due to hypoxia is modulated by GABAB receptor inhibition.
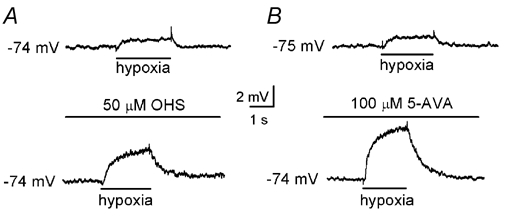
A, typical current-clamp recordings made from a petrosal neurone juxtaposed to a type I cluster in co-culture. Application of hypoxia (PO2, 5 mmHg) is indicated by the horizontal bar below each trace. Under control conditions (upper), hypoxia induced a postsynaptic depolarisation which was increased in the presence of the GABAB receptor antagonist, OHS (50 µm; lower). B, as in A, except the effect of a further specific GABAB receptor antagonist, 5-aminovaleric acid (5-AVA; 100 µm), was examined.
These data implicate GABA as a presynaptic modulator of chemoreceptor function via slow-acting G-protein-coupled GABAB autoreceptors. We further investigated the effect of GABAB receptor blockade on the postsynaptic response of juxtaposed petrosal neurones that functionally innervated type I clusters in co-culture. Postsynaptic depolarisation due to hypoxia was significantly and reversibly enhanced by 50 µm OHS (Fig. 2A). Under control conditions, hypoxia depolarised the neurones by 2.2 ± 0.3 mV; in the presence of OHS, the depolarisation due to hypoxia was increased to 6.3 ± 0.5 mV (n = 14; P < 0.001, ANOVA). Similarly, in the presence of 100 µm 5-AVA, hypoxia evoked a postsynaptic depolarisation of 6.5 ± 0.3 mV, a value significantly different from that recorded in the absence of the drug (2.4 ± 0.1 mV; n = 6; P < 0.005, ANOVA; Fig. 2B). Responses to both OHS and 5-AVA were fully reversed on removal of the GABAB receptor blocker. These data support the hypothesis that presynaptic GABAB receptors are involved in regulating hypoxic chemotransmission in the rat carotid body.
GABAB receptors regulate presynaptic excitability and release of fast excitatory neurotransmitters from type I cells
In the CNS the efficacy of synaptic transmission mediated by fast-acting neurotransmitters can be modulated by the actions of slow-acting transmitters (Greengard, 2001). Since spontaneous firing sometimes occurred in type I cells that were members of a large cluster (Zhang & Nurse, 2000) we tested whether constitutive activation of GABAB receptors may control presynaptic excitability. In the presence of 50 µm OHS, spontaneous spike activity observed under current-clamp conditions was enhanced (Fig. 3A). Since ‘slow’ transmitter receptors can alter synaptic transmission by regulating presynaptic efficacy (Greengard, 2001), it is plausible that the major presynaptic effect of GABA is to inhibit release of the fast transmitters ACh and ATP from type I cells during chemosensory stimulation (Nurse & Zhang, 1999; Zhang et al. 2000; Prasad et al. 2001). In functional chemosensory units in co-culture, perfusion of OHS (50 µm) induced spike activity in neurones juxtaposed to type I clusters (n = 6; Fig. 3B). This activity was reversibly reduced in the presence of either 1 µm mecamylamine or 25 µm suramin, blockers of nicotinic and purinergic receptors, respectively, and abolished in the presence of both blockers (Fig. 3B). This demonstrates a ‘tonic’ release of GABA from type I cells which reduces excitability, and that removal of this inhibition enhances release of the excitatory neurotransmitters ACh and ATP from type I cells onto postsynaptic neurones.
Figure 3. GABAB receptor blockers enhance excitability and release of ACh and ATP from type I cells.
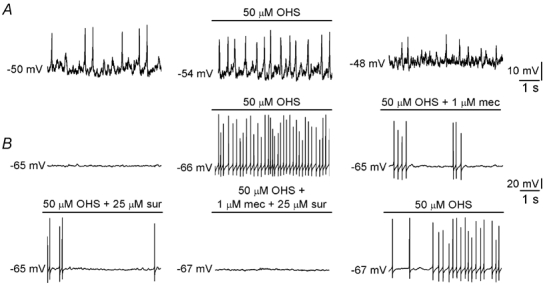
A, effect of 50 µm hydroxysaclofen (OHS) on membrane potential in a presynaptic type I cell. This cell exhibited spontaneous activity, which was reversibly enhanced by OHS. B, effect of 50 µm OHS on membrane potential in a petrosal neurone juxtaposed to a type I cluster in co-culture. Application of this GABAB receptor antagonist induced spiking in this cell, representative of 6 cells that behaved in this way. This effect is due to disinhibition of ACh and ATP release since responses were reduced in the presence of 1 µm mecamylamine (mec) or 25 µm suramin (sur), blockers of nicotinic and purinergic receptors, respectively. In the presence of both blockers OHS-induced spike activity in the neurone was abolished. Scale bars apply to all traces. Together with the data in A, this demonstrates a GABAB receptor-mediated ‘braking’ mechanism which is constitutively active and reduces presynaptic excitability and the release of fast excitatory neurotransmitters onto postsynaptic neurones.
GABAB receptor-mediated modulation of neurotransmission occurs via G-protein coupled regulation of PKA
During current-clamp recordings of membrane potential from type I cells in clusters, enhancement of hypoxia-induced depolarisation by OHS persisted in the presence of the specific PKC blocker chelerythrine chloride (100 µm; Fig. 4A and B). In 12 experiments, hypoxia caused a depolarisation of 3.7 ± 0.9 mV under control conditions; this depolarisation increased to 7.3 ± 1.8 mV in the presence of 50 µm OHS, a value not significantly different from that seen in the presence of both OHS and chelerythrine (7.1 ± 3.2 mV; P > 0.05, ANOVA). Similar effects were observed using 5-AVA (not shown), and together these data suggest that modulation of PKC is not involved in the enhanced hypoxic depolarisation due to GABAB receptor blockade. In contrast, in the presence of the specific PKA blocker, H-89 (50 µm; Fig. 4C), or following pre-treatment for 24 h with PTX (500 ng ml−1; Fig. 4D), the increase in the hypoxic depolarisation seen in the presence of OHS was abolished. In 11 PTX-treated cells examined, mean hypoxic depolarisation was 2.3 ± 0.4 mV under control conditions and 2.1 ± 0.5 mV in the presence of 50 µm OHS (P > 0.05, ANOVA). These data demonstrate that the modulation of presynaptic hypoxic depolarisation due to GABAB receptor antagonists involves inhibition of PKA coupled to the inhibitory G-protein, Gi.
Figure 4. Mechanism of GABA-mediated regulation of synaptic transmission.
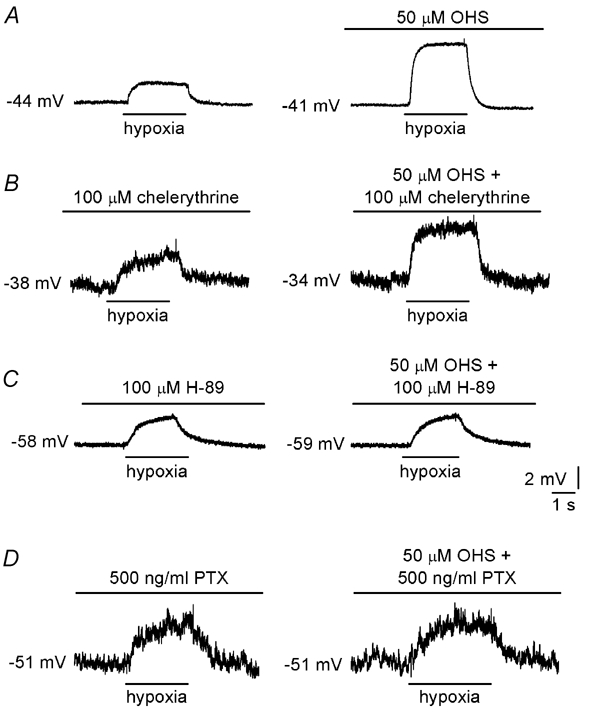
A, current-clamp recordings made from a type I cell in a cluster. Application of hypoxia (PO2, 5 mmHg) is indicated by the horizontal bars below each trace. Under control conditions (left), hypoxia induced a depolarisation which was increased in the presence of 50 µm OHS (right). B, enhanced depolarisation still occurred in the presence of the selective PKC blocker chelerythrine chloride (100 µm, right). In contrast, enhancement was abolished in the presence of the selective PKA blocker H-89 (50 µm; C) or following pretreatment for 24 h with PTX (500 ng ml−1; D). In B and C, the effects of chelerythrine and H-89 were fully reversible on washout of the kinase inhibitors (not shown).
Baclofen enhances K+ current in type I cells by activating a TASK-1-like conductance
During perforated-patch recordings from type I cells in clusters using asymmetrical K+ solutions, the GABAB receptor agonist baclofen (50 µm) enhanced outward current at more positive potentials (Fig. 5A). This enhancement was 14.0 ± 2.8 % relative to control at a test potential of +30 mV (n = 5). The I-V relationship for the baclofen-sensitive difference current was outwardly rectifying and reversed at -78.7 ± 2.5 mV (n = 5), close to the predicted K+ equilibrium potential (EK = -83 mV). Under both physiological and symmetrical K+ conditions, 50 µm baclofen reversibly enhanced currents even at negative potentials, where voltage-dependent K+ currents are inactive. At a test potential of -60 mV, enhancement was 8.8 ± 0.4 % (n = 5) under physiological conditions, and 12.1 ± 0.3 % (n = 7) under symmetrical K+ conditions. In symmetrical K+ solutions, the baclofen-sensitive (difference) current (see Fig. 5B) was linear and reversed at -4.8 ± 1.9 mV (n = 7), close to the Nernst equilibrium potential for K+ (EK = 0 mV under symmetrical K+ conditions), suggesting that baclofen activates a K+-selective, voltage-independent conductance. Enhancement of this current by baclofen was virtually abolished in the presence of 5 µm anandamide (Fig. 5C), a selective blocker of TASK-1 (Maingret et al. 2001) which appears to be expressed in rat type I cells (Buckler et al. 2000). Potentiation due to baclofen was reduced significantly from 15.0 ± 3.3 % in control conditions to 1.5 ± 1.3 % in the presence of anandamide (n = 9 and 7, respectively; P < 0.01, Mann-Whitney test). Similarly, a non-specific inhibitor of TASK-1, 10 mm Ba2+, also reduced the degree of potentiation due to baclofen from, 12.4 ± 3.4 % to 3.0 ± 1.8 % (n = 4; P < 0.05, Mann-Whitney test).
Figure 5. Baclofen activates a TASK-1-like conductance in type I cells.
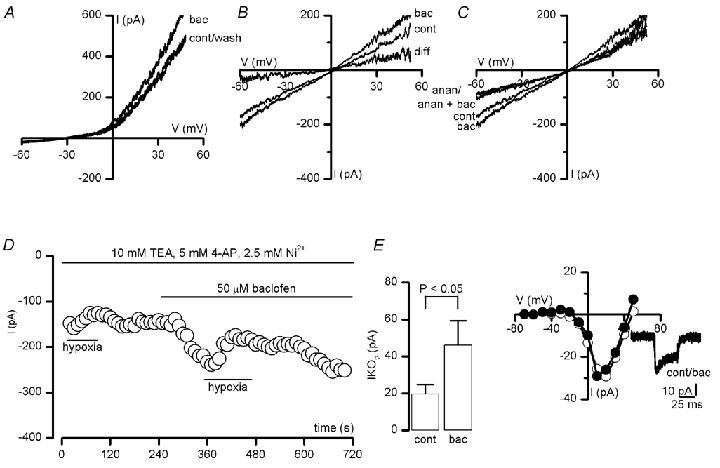
A, voltage-clamp recordings obtained from a type I cell in a cluster. Currents were evoked by ramp depolarisations between -60 and +50 mV under asymmetrical K+ conditions, in the absence (cont) and presence of 50 µm baclofen (bac) and following washout (wash), as indicated. B, as in A, except currents were recorded under symmetrical K+ conditions. Subtraction of the current obtained in the presence of baclofen from that seen under control conditions (to give the indicated difference current) shows that baclofen activates a linear K+ conductance. C, the selective TASK-1 blocker anandamide (anan; 5 µm) ablated the response to baclofen. D, time-series recording demonstrating the enhancement of the oxygen-sensitive background K+ current (IKO2) by 50 µm baclofen. Typical of 4 such recordings, which were made under Ca2+-free, symmetrical K+ conditions and in the presence of 2.5 mm Ni2+, 10 mm TEA and 5 mm 4-AP to block voltage- and Ca2+-dependent K+ channels (Buckler et al. 2000). Periods of application of baclofen and hypoxia (PO2, 5 mmHg) are indicated by the horizontal bars. Currents were measured at a test potential of -60 mV. Inset shows the magnitude of the hypoxic response, obtained by subtracting the current evoked at -60 mV under normoxic conditions from that obtained in hypoxia. Plotted are mean (± s.e.m.) data obtained in 4 cells, under control conditions (cont) and in the presence of 50 µm baclofen (bac), as indicated. E, Ca2+ channel current-voltage relationships obtained from a type I cell under control conditions (○) and in the presence of 50 µm baclofen (•). Each point shows the peak current amplitude evoked by a 50 ms step depolarisation to the indicated test potential, from a holding potential of -80 mV. Ca2+ (5 mm) was used as charge carrier. Inset, individual current traces obtained by step-depolarising to +10 mV for 50 ms, under control conditions and in the presence of 50 µm baclofen (bac).
The above data suggest that baclofen activates a K+ current with characteristics of the TASK-1-like O2-sensitive background K+ channel (Buckler et al. 2000). To test this possibility further, whole-cell recordings were made under Ca2+-free, symmetrical K+ conditions and in the presence of 10 mm TEA, 5 mm 4-AP and 2.5 mm Ni2+ to block voltage- and Ca2+-dependent K+ and Ca2+ channels and to isolate background K+ current (Buckler et al. 2000). Under these conditions, the magnitude of the hypoxia-sensitive background current (IKO2), obtained by subtracting the current obtained during hypoxia from that obtained under control conditions, was enhanced by baclofen (e.g. Fig. 5D). At a test potential of -60 mV, IKO2 was increased from 19.5 ± 5.2 pA under control conditions to 46.0 ± 13.2 pA in the presence of 50 µm baclofen (n = 4; P < 0.05, Student's paired t test; see Fig. 5D, inset). Taken together, these voltage-clamp data demonstrate that type I cell GABAB receptor activation augments current through TASK-1-like channels, which underlie part of the O2 sensitivity of these cells (Buckler et al. 2000).
In previous studies, GABAB receptor activation caused inhibition of presynaptic neuronal voltage-gated Ca2+ channels (Bowery & Enna, 2000). Under voltage-clamp, 50 µm baclofen was without effect on Ca2+ currents in 10 type I cells examined (Fig. 5D). Mean Ca2+ current induced by step depolarising to +10 mV from a holding potential of -80 mV was -38.2 ± 6.8 pA under control conditions and -41.8 ± 8.5 pA in the presence of 50 µm baclofen (n = 10; P = 0.37, Student's paired t test).
GABA and GABAB receptor subunit expression in type I cells
To verify the presence of GABA in the rat carotid body, sections of the intact carotid body and cultured type I clusters were immunostained with a GABA-specific polyclonal antibody. Positive immunoreactivity was confined to type I clusters in sections (Fig. 6A) and cultures (Fig. 6B). To test for expression of GABAB receptor subunits, RT-PCR and immunohistochemical techniques were used on isolated type I clusters and carotid body sections, respectively. Using gene-specific primers, PCR products corresponding to target sequences of both the GABAB(1) and GABAB(2) receptor subunits were amplified from mRNA extracted from type I cell clusters (Fig. 6E). Sequencing demonstrated the correct identity of PCR products compared to published sequences. When examining the localisation of GABAB(1) and GABAB(2) receptor subunits in sections of the carotid body by immunofluorescence, positive immunoreactivity for both receptor subunits was prominent in type I clusters (Fig. 6C and D, respectively). In control experiments, no staining was observed when sections were exposed to the secondary antibody without prior exposure to the primary antibody or, in the case of GABA immunoreactivity, following pre-adsorption of the primary antibody with an excess of GABA (not shown).
Figure 6. Presence of GABA and GABAB receptors in type I cells.

Confocal images showing carotid body sections which were immunostained with a specific antibodies raised against GABA (A), GABAB(1) (B) and GABAB(2) (C) receptor subunits and visualised by secondary FITC fluorescence. Positive immunostaining of type I clusters is seen in each case. Scale bars represent 20 µm. In all cases, staining was abolished either when sections were exposed to the secondary antibody without prior exposure to the primary antibody, or in the case of GABA the primary antibody was pre-adsorbed with excess antigen (not shown). D, micrograph of a 2 % agarose gel stained with ethidium bromide and viewed under UV illumination. RT-PCR was carried out on isolated type I clusters following extraction of mRNA, and using gene-specific primers for the GABAB(1) and GABAB(2) subunits, and β-actin. Marker lane (M) shows bands at 100 bp increments with the 600 bp fragment at increased intensity. In negative control reactions without RT (−) no PCR products were observed.
DISCUSSION
A role for a TASK-1-like conductance in GABAergic modulation of fast synaptic transmission
Background (leak) K+ currents control neuronal excitability and shape action potentials by controlling the resting membrane potential (Goldstein et al. 2001). Enhancement of leak conductances stabilises cells at hyperpolarised potentials, while inhibition leads to membrane depolarisation and excitation. Background currents are carried by members of the K2P family of tandem-pore-domain K+ channels, of which at least 14 members have been cloned (Goldstein et al. 2001, 2003). K2P channel activity is tightly controlled by intracellular factors such as cyclic nucleotide levels and metabolic status, and by extracellular factors such as neurotransmitters, including 5-HT, noradrenaline, substance P, glutamate, TRH and ACh (Millar et al. 2000; Talley et al. 2000; Goldstein et al. 2001). In hypoglossal motoneurones, where TASK-1 is abundantly expressed, native TASK-1-like currents were inhibited by agonists at several G-protein-coupled neurotransmitter receptors (Talley et al. 2000). This provides a mechanism whereby neurotransmitters can regulate neuronal excitability and provide slow regulation of fast neurotransmission via intracellular effectors. Here, we demonstrate that a selective agonist at metabotropic receptors for the inhibitory neurotransmitter GABA activates a K+-selective background conductance in presynaptic chemoreceptor cells of the rat carotid body. Block of this conductance by either anandamide (Maingret et al. 2001) or Ba2+ ions, alongside data demonstrating that baclofen activates a linear K+ conductance under conditions where voltage- and Ca2+-dependent K+ channels are inhibited, suggests that this effect is attributable to activation of a tandem-pore-domain K+ channel with characteristics of TASK-1. This link between GABAB receptors and this background K+ channel provides a novel mechanism for regulating neuronal excitability and synaptic signalling.
In the present study, we examined the effects of several GABAB receptor antagonists on the depolarising responses to hypoxia in both isolated type I clusters and in petrosal neurons juxtaposed to type I clusters in co-culture. The responses to hypoxia obtained in these studies were modest, particularly when compared to those obtained previously by others when recording hypoxic responses in single type I cells (e.g. Buckler & Vaughan Jones, 1994). The reasons for this are not fully understood, but one possibility is the operation of autoreceptor feedback mechanisms within clusters, such as that reported here, that serve to limit the degree of depolarisation during hypoxia. Such mechanisms are expected to be most effective when recording from clusters as opposed to single cells. In general, the receptor potential in a chemoreceptor cluster reflects a balance between inhibitory (e.g. GABA) and excitatory (e.g. 5-HT; Zhang et al. 2003) feedback influences, and this may vary in culture from one cluster to another.
A recent study demonstrated the activation by baclofen of a background K+ conductance in mouse cerebellar purkinje neurones with pharmacological characteristics of the background channel THIK-1 (Rajan et al. 2001; Bushell et al. 2002). Furthermore, we recently demonstrated the O2 sensitivity of a similar background K+ current in glossopharyngeal (GPN) neurones (Campanucci et al. 2003). However, the THIK-1-like, O2-sensitive K+ current in GPN neurones is anandamide-insensitive (Campanucci et al. 2003), making it unlikely that these channels mediate O2 and/or baclofen sensitivity in type I cells.
GABAB receptors linked to TASK-1 provide presynaptic autoregulatory feedback during hypoxia
GABA is a well characterised inhibitory CNS neurotransmitter and its effects at presynaptic metabotropic GABAB receptors are thought to underlie, amongst other processes, autoregulation of neurotransmitter release. In many cases regulation involves receptors coupled by G-proteins to plasmalemmal Ca2+ and K+ channels (Misgeld et al. 1995; Bowery & Enna, 2000) or to the exocytotic machinery itself (Wu & Saggau, 1997). Here, we present immunohistochemical evidence for the presence of GABA in presynaptic type I cells of the rat carotid body. Examination of type I cell clusters in co-culture with their postsynaptic (petrosal neurone) partners revealed that selective inhibitors of GABAB receptor function markedly enhanced synaptic transmission in response to hypoxia. This is attributable, at least in part, to a presynaptic mechanism since GABAB receptor blockade also enhanced the hypoxia-induced depolarisation or receptor potential in type I cells cultured alone.
The mechanism by which GABAB receptor inhibition enhances the efficacy of hypoxic chemotransmission involves modulation of the activity of PKA, since blockers of this kinase (and not of PKC) inhibited the enhancing effect of the GABAB receptor blocker hydroxysaclofen on presynaptic depolarisation. Evidence from this study further points to a mechanism involving GABA-mediated activation of a background K+ channel, with properties similar to those of TASK-1, during hypoxia. Inhibition of this TASK-1-like K+ channel (Buckler et al. 2000), is thought to be at least partly responsible for the hypoxic depolarisation or receptor potential in type I cells. The rat isoform of TASK-1 possesses two C-terminal consensus sites for phosphorylation by PKA and furthermore, current through TASK-1 is inhibited by stimulation of this kinase (Leonoudakis et al. 1998; Lopes et al. 2000). Neuronal GABAB receptors couple to the inhibitory G protein, Gi (Isaacson, 1998; Leaney & Tinker, 2000), leading to K+ channel activation (Misgeld et al. 1995). Moreover, Gi has also been shown to couple functionally to a GABAB receptor-adenylyl cyclase system (Nishikawa et al. 1997). In our system, inhibition of Gi by pretreatment with PTX abolished presynaptic sensitivity to GABAB receptor blockade. We suggest therefore that in the rat carotid body, presynaptic GABAB receptor activation by GABA released from type I cells couples to Gi, resulting in inhibition of PKA activity and activation of background K+ channels. This pathway is enhanced during hypoxic stimulation, due to depolarisation-evoked GABA release which leads to activation of presynaptic GABAB autoreceptors and subsequently activation of the TASK-1-like conductance via Gi-mediated inhibition of PKA. This cascade gives rise to a hyperpolarisation which effectively blunts the depolarising receptor potential due to hypoxia. The end result is an autoregulatory feedback mechanism (see Fig. 7 for schematic representation of this cascade) that modulates the release of neurotransmitters from the receptor cells during hypoxia via convergence of two separate signalling pathways onto the same K+ channels. We further suggest that the basal activation of GABAB receptors exerts control over the excitability of presynaptic type I cells, since blockade of this constitutive activity with hydroxysaclofen enhanced the excitability of presynaptic type I cells in which spontaneous activity was observed. Taken with our data, this suggests that basal GABAB receptor activation stimulates Gi, causing inhibition of adenylate cyclase and a reduction in cAMP levels, which would reduce type I cell excitability via the activation of the TASK-1-like conductance.
Figure 7. Schematic representation of the autoregulatory pathways involved in the GABA-mediated regulation of neurotransmitter release from type I cells during hypoxia.
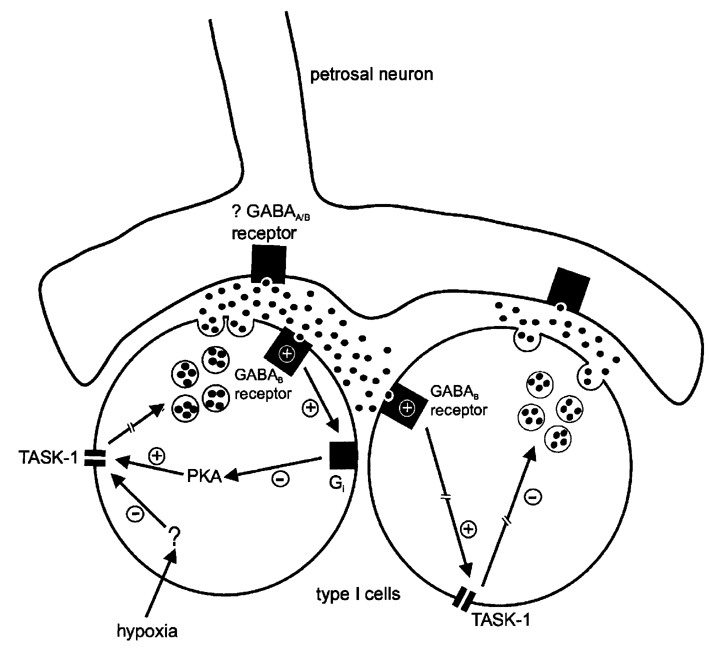
Via an as yet uncharacterised intracellular pathway, hypoxia inhibits TASK-1-like background channels in type I cells, leading to membrane depolarisation and ultimately (broken arrow) neurotransmitter release. In this process GABA (black circles) is released from type I cells, and acts at presynaptic GABAB receptors on either the same type I cell (autocrine) or on an adjacent type I cell (paracrine) in the cluster. This causes stimulation of the pertussis toxin-sensitive inhibitory G protein Gi, causing inhibition of protein kinase A (PKA) and subsequently activation of TASK-1. This would serve to hyperpolarise the type I cell and limit the degree of depolarisation during exposure to hypoxia, regulating the further release of transmitters. GABA may also act at postsynaptic ionotropic or metabotropic GABA receptors to modulate chemoreceptor output. For clarity, the involvement of other K+ channels and neurotransmitters in chemotransmission, and the intracellular events leading to transmitter release, have been omitted.
In the rabbit carotid body dopamine, acting at presynaptic D2 receptors, exerts autoregulatory feedback to inhibit the further release of this transmitter (Bairam et al. 2000), presumably via the inhibition of voltage-dependent Ca2+ currents (Benot & Lopez-Barneo, 1990). The presence of dopamine and presynaptic D2 receptors (Gauda et al. 1996; Donnelly, 2000) suggests that similar autoreceptor feedback loops may control neurotransmitter output in the rat carotid body, although this has not been directly tested. Since dopamine D2 receptors are linked to inhibition of adenylate cyclase and reduce cellular cAMP levels upon activation, it is possible that dopamine acting via presynaptic D2 receptors elicits a similar autoinhibitory feedback loop as that evoked by GABAB receptor activation. To support this possibility, D2 receptor activation has been shown in many cases to activate neuronal K+ conductances (Lacey et al. 1987; Freedman & Weight, 1988; Casteletti et al. 1989). On the other hand, recent studies from this laboratory indicate that paracrine release of 5-HT from clustered type I cells produces the opposite effect and augments the receptor potential via PKC-mediated inhibition of K+ channels (Zhang et al. 2003).
Autoreceptor regulation of neurotransmitter release in the CNS is mediated in part by activation of presynaptic GABAB receptors which modulate voltage-gated Ca2+ channels (Misgeld et al. 1995; Bowery & Enna, 2000). However, in the present studies GABAB receptor activation was without effect on Ca2+ channel activity in presynaptic type I cells. In the neonatal rat carotid body, Ca2+ current is carried almost exclusively by L-type channels (Stea et al. 1995; Peers et al. 1996). Although the inhibitory effects of GABAB receptor activation on neuronal N- (e.g. Lambert & Wilson, 1996) and P/Q-type (e.g. Chen & van den Pol, 1998) Ca2+ channels are well documented, there is little evidence to suggest that L-type Ca2+ channels are modulated by GABAB receptors. Moreover in some neurones which express multiple Ca2+ channel subtypes, effects of GABAB receptor activation on L-type Ca2+ currents are negligible (Harayama et al. 1998) or non-existent (Doze et al. 1995) compared to effects on N- and P/Q-type channels. Also consistent with our data, there is no evidence of regulation by PKA of L-type Ca2+ channels in type I cells from other species (Summers et al. 2000). Thus, autoregulation of neurotransmitter release from type I cells is mediated by presynaptic activation of a K+ current rather than Ca2+ channel inhibition. Activation of K+ conductances by GABAB receptor agonists has mostly been described in postsynaptic CNS neurones (Misgeld et al. 1995), while other studies have demonstrated no effect of GABAB receptor activation on presynaptic K+ conductances (Isaacson, 1998). However, Wagner & Dekin (1993, 1997) demonstrated the regulation by cAMP and activation by baclofen of K+ channels in presynaptic respiratory neurones with pharmacological and biophysical characteristics similar (although not identical) to those described for background K+ channels. Since TASK-1 is expressed in respiratory neurones (Bayliss et al. 2001), it is possible that GABAB receptors mediate presynaptic regulation of TASK-1 in these cells. Furthermore, background K+ channels and GABAB receptors are co-expressed in a variety of neuronal cell types and these findings open the possibility that GABAB autoreceptors linked to background channels may be a general mechanism for controlling CNS excitability.
Acknowledgments
This work was supported by a Wellcome Trust International Prize Traveling Research Fellowship (Ref. 06154) to I.M.F. and by a grant from the Canadian Institutes for Health Research (MOP 12037) to C.A.N.
REFERENCES
- Bairam A, Neji H, De Grandpre P, Carroll JL. Autoreceptor mechanism regulating carotid body dopamine release from adult and 10-day-old rabbits. Resp Physiol. 2000;120:27–34. doi: 10.1016/s0034-5687(00)00092-x. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Talley EM, Sirois JE, Lei Q. TASK-1 is a highly modulated pH-sensitive ‘leak’ K+ channel expressed in brainstem respiratory neurones. Resp Physiol. 2001;129:159–174. doi: 10.1016/s0034-5687(01)00288-2. [DOI] [PubMed] [Google Scholar]
- Benot AR, Lopez-Barneo J. Feedback inhibition of Ca2+ currents by dopamine in glomus cells of the carotid body. Eur J Neurosci. 1990;2:809–812. doi: 10.1111/j.1460-9568.1990.tb00473.x. [DOI] [PubMed] [Google Scholar]
- Bowery N, Enna SJ. γ-Aminobutyric acidB receptors: first of the functional metabotropic heterodimers. J Pharmacol Exp Ther. 2000;2:2–7. [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell T, Clarke C, Mathie A, Robertson B. Pharmacological characterization of a non-inactivating outward current observed in mouse cerebellar Purkinje neurones. Br J Pharmacol. 2002;135:705–712. doi: 10.1038/sj.bjp.0704518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanucci VA, Fearon IM, Nurse CA. A novel O2-sensing mechanism in glossopharyngeal neurones mediated by a halothane-inhibitable background K+ conductance. J Physiol. 2002;548:731–743. doi: 10.1113/jphysiol.2002.035998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelletti L, Memo M, Missale C, Spano PF, Valerio A. Potassium channels involved in the transduction mechanism of dopamine D2 receptors in rat lactotrophs. J Physiol. 1989;410:251–265. doi: 10.1113/jphysiol.1989.sp017531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro E, Oset-Gasque MJ, Gonzalez MP. GABAA and GABAB receptors are functionally active in the regulation of catecholamine secretion by bovine chromaffin cells. J Neurosci Res. 1989;23:290–296. doi: 10.1002/jnr.490230307. [DOI] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Presynaptic GABAB autoreceptor modulation of P/Q-type calcium channels and GABA release in rat suprachiasmatic nucleus neurones. J Neurosci. 1998;18:1913–1922. doi: 10.1523/JNEUROSCI.18-05-01913.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couve A, Moss SJ, Pangalos MN. GABAB receptors: a new paradigm in G protein signaling. Mol Cell Neurosci. 2000;1:296–312. doi: 10.1006/mcne.2000.0908. [DOI] [PubMed] [Google Scholar]
- Donnelly DF. Are oxygen dependent K+ channels essential for carotid body chemo-transduction? Respir Physiol. 1997;110:211–218. doi: 10.1016/s0034-5687(97)00085-6. [DOI] [PubMed] [Google Scholar]
- Donnelly DF. Developmental aspects of oxygen sensing by the carotid body. J Appl Physiol. 2000;88:2296–2301. doi: 10.1152/jappl.2000.88.6.2296. [DOI] [PubMed] [Google Scholar]
- Doze VA, Cohen GA, Madison DV. Calcium channel involvement in GABAB receptor-mediated inhibition of GABA release in area CA1 of the rat hippocampus. J Neurophysiol. 1995;74:42–53. doi: 10.1152/jn.1995.74.1.43. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Weight FF. Single K+ channels activated by D2 dopamine receptors in acutely dissociated neurones from rat corpus striatum. Proc Natl Acad Sci USA. 1988;85:3618–3622. doi: 10.1073/pnas.85.10.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauda EB, Bamford O, Gerfen CR. Developmental expression of tyrosine hydroxylase, D2-dopamine receptor and substance P genes in the carotid body of the rat. Neuroscience. 1996;75:969–977. doi: 10.1016/0306-4522(96)00312-0. [DOI] [PubMed] [Google Scholar]
- Goldstein SAN, Bockenhauer D, O'Kelly I, Zilderberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- Goldstein SAN, Bayliss DA, Desir GV, Kim D, Lazdunski M, Lesage F, O'Kelly I. NC-IUPHAR subcommittee on potassium channels: K2P group. In: Catterall WA, Chandy KG, Gutman GA, editors. The IUPHAR Compendium of Voltage-Gated Ion Channels. Leeds: IUPHAR Media; 2003. pp. 174–189. [Google Scholar]
- Gonzalez C, Almarez L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Harayama N, Shinuya I, Tanaka K, Kabashima N, Ueta Y, Yamashita H. Inhibition of N- and P/Q-type calcium channels by postsynaptic GABAB receptor activation in rat supraoptic neurones. J Physiol. 1998;509:371–383. doi: 10.1111/j.1469-7793.1998.371bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS. GABAB receptor-mediated modulation of presynaptic currents and excitatory transmission at a fast central synapse. J Neurophysiol. 1998;80:1571–1576. doi: 10.1152/jn.1998.80.3.1571. [DOI] [PubMed] [Google Scholar]
- Jackson A, Nurse C. Dopaminergic properties of cultured rat carotid body chemoreceptors grown in normoxic and hypoxic environments. J Neurochem. 1997;69:645–654. doi: 10.1046/j.1471-4159.1997.69020645.x. [DOI] [PubMed] [Google Scholar]
- Kerr DI, Ong J. GABAB receptors. Pharmacol Ther. 1995;67:187–246. doi: 10.1016/0163-7258(95)00016-a. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Roy A, Rozanov C, Mokashi A. K+-current modulated by PO2 in type I cells in rat carotid body is not a chemosensor. Brain Res. 1998;94:162–165. doi: 10.1016/s0006-8993(98)00276-5. [DOI] [PubMed] [Google Scholar]
- Lambert NA, Wilson WA. High-threshold Ca2+ currents in rat hippocampal interneurones and their selective inhibition by activation of GABAB receptors. J Physiol. 1996;492:115–127. doi: 10.1113/jphysiol.1996.sp021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaney JL, Tinker A. The role of members of the pertussis toxin-sensitive family of G proteins in coupling receptors to the activation of the G protein-gated inwardly rectifying potassium channel. Proc Natl Acad Sci USA. 2000;97:5651–5656. doi: 10.1073/pnas.080572297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Gray AT, Winegar BD, Kindler CH, Harada M, Taylor DM, Chavez RA, Forsayeth JR, Yost CS. An open rectifier potassium channel with two pore domains in tandem cloned from rat cerebellum. J Neurosci. 1998;18:868–877. doi: 10.1523/JNEUROSCI.18-03-00868.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CMB, Gallagher PG, Buck ME, Butler MH, Goldstein SAN. Proton block and voltage gating are potassium-dependent in the cardiac leak channel KCNK3. J Biol Chem. 2000;275:16969–16978. doi: 10.1074/jbc.M001948200. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen-sensing by ion channels and the regulation of cellular functions. Trends Neurosci. 1996;19:435–440. doi: 10.1016/0166-2236(96)10050-3. [DOI] [PubMed] [Google Scholar]
- Madden TE, Johnson SW. γ-Hydroxybutyrate is a GABAB receptor agonist that increases a potassium conductance in rat ventral tegmental dopamine neurones. J Pharmacol Exp Ther. 1998;287:261–265. [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lazdunski M, Honore E. The endocannabinoid anandamide is a direct and selective blocker of the background K+ channel TASK-1. EMBO J. 2001;20:47–54. doi: 10.1093/emboj/20.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JA, Barratt L, Southan AP, Page KM, Fyffe RE, Robertson B, Mathie A. A functional role for the two-pore domain potassium TASK-1 in cerebellar granule neurones. Proc Natl Acad Sci USA. 2000;97:3614–3618. doi: 10.1073/pnas.050012597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Bijak M, Jarolimek W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian CNS. Prog Neurobiol. 1995;46:423–462. doi: 10.1016/0301-0082(95)00012-k. [DOI] [PubMed] [Google Scholar]
- Mott DD, Lewis DV. The pharmacology and function of central GABAB receptors. Int Rev Neurobiol. 1994;36:97–223. doi: 10.1016/s0074-7742(08)60304-9. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Hirouchi M, Kuriyama K. Functional coupling of Gi subtype with GABAB receptor/adenylyl cyclase system: analysis using a reconstituted system with purified GTP-binding protein from bovine cerebral cortex. Neurochem Int. 1997;31:21–25. doi: 10.1016/s0197-0186(96)00138-6. [DOI] [PubMed] [Google Scholar]
- Nurse CA, Zhang M. Acetylcholine contributes to hypoxic chemotransmission in co-cultures of rat type 1 cells and petrosal neurones. Resp Physiol. 1999;115:189–199. doi: 10.1016/s0034-5687(99)00017-1. [DOI] [PubMed] [Google Scholar]
- Ohtomo K, Fukuhara K, Yoshizaki K. Immunohistochemical study of the carotid body during hibernation. Adv Exp Med Biol. 2000;475:815–821. doi: 10.1007/0-306-46825-5_82. [DOI] [PubMed] [Google Scholar]
- Oomori Y, Iuchi H, Nakaya K, Tanaka H, Ishikawa K, Satoh Y, Ono K. Gamma-aminobutyric acid (GABA) immunoreactivity in the mouse adrenal gland. Histochemistry. 1993;100:203–213. doi: 10.1007/BF00269093. [DOI] [PubMed] [Google Scholar]
- Oomori Y, Nakaya K, Tanaka H, Iuchi H, Ishikawa K, Sato Y, Ono K. Immunohistochemical and histochemical evidence for the presence of noradrenaline, serotonin and γ-aminobutyric acid in chief cells of the mouse carotid body. Cell Tiss Res. 1994;278:249–254. doi: 10.1007/BF00414167. [DOI] [PubMed] [Google Scholar]
- Osanai S, Buerk DG, Mokashi A, Chugh DK, Lahiri S. Cat carotid body chemosensory discharge (in vitro) is insensitive to charybdotoxin. Brain Res. 1997;747:324–327. doi: 10.1016/s0006-8993(96)01313-3. [DOI] [PubMed] [Google Scholar]
- Pardal R, Ludewig U, Garcia-Hirschfeld J, Lopez-Barneo J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci USA. 2000;97:2361–2366. doi: 10.1073/pnas.030522297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C. Hypoxic suppression of K+ currents in type I carotid body cells: selective effect on the Ca2+-activated K+ current. Neurosci Lett. 1990;119:253–256. doi: 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- Peers C, Carpenter E, Hatton CJ, Wyatt CN, Bee D. Ca2+ channel currents in type I carotid body cells of normoxic and chronically hypoxic neonatal rats. Brain Res. 1996;739:251–257. doi: 10.1016/s0006-8993(96)00832-3. [DOI] [PubMed] [Google Scholar]
- Prasad M, Fearon IM, Zhang M, Laing M, Vollmer C, Nurse CA. Expression of P2X2 and P2X3 receptor subunits in rat carotid body afferent neurones: role in chemosensory signalling. J Physiol. 2001;537:667–677. doi: 10.1111/j.1469-7793.2001.00667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, Karschin A, Derst C. THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem. 2001;276:7302–7311. doi: 10.1074/jbc.M008985200. [DOI] [PubMed] [Google Scholar]
- Stea A, Jackson A, Macintyre L, Nurse CA. Long-term modulation of inward currents in O2 chemoreceptors by chronic hypoxia and cyclic AMP in vitro. J Neurosci. 1995;15:2192–2202. doi: 10.1523/JNEUROSCI.15-03-02192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers BA, Overholt JL, Prabhakar NR. Augmentation of L-type calcium current by hypoxia in rabbit carotid body glomus cells: evidence for a PKC-sensitive pathway. J Neurophysiol. 2000;84:1636–1644. doi: 10.1152/jn.2000.84.3.1636. [DOI] [PubMed] [Google Scholar]
- Talley EM, Lei Q, Sirois JE, Bayliss DA. TASK-1, a two-pore-domain K+ channel, is modulated by multiple neurotransmitters in motoneurones. Neuron. 2000;25:399–410. doi: 10.1016/s0896-6273(00)80903-4. [DOI] [PubMed] [Google Scholar]
- Urena J, Fernandez-Chacon R, Benot AR, Alvarez de Toledo GA, Lopez-Barneo J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci USA. 1994;91:10208–10211. doi: 10.1073/pnas.91.21.10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PG, Dekin MS. GABAB receptors are coupled to a barium-insensitive outward rectifying potassium conductance in premotor respiratory neurones. J Neurophysiol. 1993;69:286–289. doi: 10.1152/jn.1993.69.1.286. [DOI] [PubMed] [Google Scholar]
- Wagner PG, Dekin MS. cAMP modulates an S-type K+ channel coupled to GABAB receptors in mammalian respiratory neurones. Neuroreport. 1997;8:1667–1670. doi: 10.1097/00001756-199705060-00021. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Hori T, Tajahashi T. Presynaptic inhibition by muscimol through GABAB receptors. Eur J Neurosci. 2000;12:3433–3436. doi: 10.1046/j.1460-9568.2000.00248.x. [DOI] [PubMed] [Google Scholar]
- Zhang M, Fearon IM, Zhong H, Nurse CA. Presynaptic modulation of rat arterial chemoreceptor function by 5-HT: role of K+ channel inhibition via protein kinase C. J Physiol. 2003;551:825–842. doi: 10.1113/jphysiol.2002.038489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Nurse CA. Does endogenous 5-HT mediate spontaneous rhythmic activity in chemoreceptor clusters of rat carotid body? Brain Res. 2000;872:199–203. doi: 10.1016/s0006-8993(00)02499-9. [DOI] [PubMed] [Google Scholar]
- Zhang M, Zhong H, Vollmer C, Nurse CA. Co-release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J Physiol. 2000;525:143–158. doi: 10.1111/j.1469-7793.2000.t01-1-00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Zhang M, Nurse CA. Synapse formation and hypoxic signalling in co-cultures of rat petrosal neurones and carotid body type 1 cells. J Physiol. 1997;503:599–612. doi: 10.1111/j.1469-7793.1997.599bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]