

Abstract
We tested the hypothesis that nitric oxide (NO) is responsible for blunting sympathetic α-adrenergic vasoconstriction in the active muscles of humans (functional sympatholysis). We measured forearm blood flow (Doppler ultrasound) and calculated the reductions in forearm vascular conductance (FVC) in response to α-adrenergic receptor stimulation during rhythmic handgrip exercise and during a control non-exercise vasodilator condition (intra-arterial adenosine), before and after local NO synthase (NOS) inhibition in healthy men. The forearm vasoconstrictor responses to endogenous noradrenaline release (intra-arterial tyramine) were significantly blunted during moderate exercise compared with adenosine, and these vasoconstrictor responses were not restored by NOS inhibition with NG-monomethyl-l-arginine (l-NMMA; n = 6) or NG-nitro-l-arginine methyl ester (l-NAME; n = 8). Similarly, l-NAME did not restore the vasoconstrictor responses to tyramine in contracting muscle during heavy rhythmic handgrip exercise (n = 4). In four additional subjects, we also found that the vasoconstrictor responses evoked by tyramine during exercise or adenosine were repeatable in the absence of NOS inhibition (i.e. time control). Finally, in five subjects the forearm vasoconstrictor responses to direct α1-adrenergic (phenylephrine) and α2-adrenergic (clonidine) receptor stimulation were blunted during moderate exercise compared with adenosine; these responses were also unaffected by l-NAME. Taken together, our results demonstrate that NO is not obligatory for functional sympatholysis in contracting skeletal muscles of healthy men.
During dynamic exercise, there is competition between local metabolic vasodilatation and sympathetic vasoconstriction as determinants of skeletal muscle vascular tone and blood flow. Although sympathetic vasoconstriction persists in active skeletal muscle and is important for appropriate blood pressure regulation during exercise (Marshall et al. 1961), the vascular responses to a variety of sympathetic α-adrenergic vasoconstrictor stimuli are blunted in active compared with resting muscle, a phenomenon known as ‘functional sympatholysis’ (Remensnyder et al. 1962; Anderson & Faber, 1991; Thomas et al. 1994; Buckwalter et al. 2001; Ruble et al. 2002; Tschakovsky et al. 2002). This observed reduction in vascular responsiveness to sympathetic stimulation might optimize blood flow distribution and oxygen delivery under conditions in which there are potential mismatches in oxygen supply and demand (Strandell & Shepherd, 1967; Van Teeffelen & Segal, 2003).
Skeletal muscle contractions evoke the release of a number of substances from both the active muscle and vascular endothelium that can potentially modulate sympathetic α-adrenergic vasoconstriction. Adenosine, prostaglandins, and more recently, nitric oxide (NO) have been postulated as putative ‘sympatholytic’ factors that are involved in this integrative regulation of muscle blood flow during exercise (Hansen et al. 2000). Importantly, under a variety of experimental conditions, all of these substances can reduce the vasoconstrictor responses to sympathetic α-adrenergic stimulation (Lippton et al. 1981; Faber et al. 1982; Nishigaki et al. 1991; Ohyanagi et al. 1992). In this context, data derived from a series of studies in experimental animals suggest that NO is involved, at least in part, in the blunted α-adrenergic vasoconstrictor responses in active muscle (Thomas & Victor, 1998). The source of NO during muscle contractions appears to be derived from neuronal NO synthase (nNOS), and the postulate is that calcium released from contracting skeletal muscle stimulates nNOS activity (a calcium-calmodulin-dependent enzyme) which subsequently increases NO synthesis and release (Hansen et al. 2000; Grange et al. 2001). Under these conditions, NO appears to blunt α-adrenergic vasoconstriction by limiting smooth muscle regulatory light chain phosphorylation (Grange et al. 2001). Collectively, the available evidence from these studies in animals indicates that this elevation in NO interferes with α-adrenergic vasoconstriction via activation of ATP-sensitive potassium (KATP) channels, but does not completely ‘restore’ this vasoconstriction (Thomas et al. 1997; Thomas & Victor, 1998).
To date, only two studies in humans have addressed whether NO is responsible for blunting sympathetic vasoconstriction in active skeletal muscle and have provided discrepant findings. Wilson & Kapoor (1993) used venous occlusion plethysmography to estimate forearm blood flow during wrist flexion exercise with and without concomitant intra-brachial noradrenaline infusion, and found that local NOS inhibition via NG-monomethyl-l-arginine (l-NMMA) did not affect the blood flow responses. However, these data are difficult to interpret because blood flow measures obtained with this technique reflect post-exercise rather than active muscle blood flow, an issue that has received recent attention (Shoemaker et al. 1997; Frandsen et al. 2001). In contrast, Chavoshan et al. (2002) demonstrated that the blunted vasoconstrictor responses in contracting forearm muscle during reflex increases in sympathetic activity (estimated via reductions in muscle oxygenation with near infrared spectroscopy) were completely reversed after systemic NOS inhibition with NG-nitro-l-arginine methyl ester (l-NAME). From this study it is unclear what effects NOS inhibition had on the forearm hemodynamic responses during exercise, which would seem of importance with respect to both muscle blood flow and systemic blood pressure regulation during exercise.
In addition to these potential limitations of each respective study, the discrepant findings might be related to the use of l-NMMA compared with l-NAME to inhibit NOS. Specifically, l-NAME is believed to be a more potent NOS inhibitor and might inhibit nNOS to a greater degree than l-NMMA and therefore, might be the more appropriate NOS inhibitor to use for studies involving muscle contractions (Frandsen et al. 2001).
With this information as a background, the purpose of the present investigation was to test the hypothesis that NO is responsible for the blunted α-adrenergic vasoconstrictor responses in the vascular beds of contracting skeletal muscle. To do so, we measured forearm hemodynamics (Doppler ultrasound) during rhythmic handgrip exercise and intra-arterial adenosine (‘control’ vasodilator), and determined the vasoconstrictor responses to α-adrenergic stimulation before and after local NOS inhibition. Utilizing several experimental approaches, our findings indicate that acute NOS inhibition does not restore α-adrenergic vasoconstrictor responses in contracting muscle, thus suggesting that NO is not obligatory to observe functional sympatholysis in healthy humans.
METHODS
Subjects
With Institutional Review Board approval and after giving written informed consent, a total of 28 young healthy men (age 25 ± 1 years; weight 77.6 ± 2.3 kg; height 180 ± 1 cm; body mass index 23.6 ± 0.5 kg m−2; means ± s.e.m.) participated in the present study. All were non-smokers, non-obese, normotensive, and not taking any medications. Studies were performed after a 4 h fast with the subjects in the supine position. All studies were performed according to the Declaration of Helsinki.
Arterial catheterization
A 20 gauge, 5 cm catheter was placed in the brachial artery of the non-dominant arm under aseptic conditions after local anaesthesia (1 % lidocaine (lignocaine)) for local administration of study drugs. The catheter was connected to a pressure transducer for mean arterial pressure (MAP) measurement and continuously flushed at 3 ml h−1 with heparinized saline (Dietz et al. 1994)
Forearm blood flow and vascular conductance
A 4 MHz pulsed Doppler probe (Model 500V, Multigon Industries, Mt Vernon, NY, USA) was used to measure brachial artery mean blood velocity (MBV) with the probe securely fixed to the skin over the brachial artery proximal to the catheter insertion site as previously described by our laboratory (Tschakovsky et al. 2002). The probe insonation angle was 60 deg. A linear 7.0 MHz echo Doppler ultrasound probe (Acuson 128XP, Mountain View, CA, USA) was placed in a holder securely fixed to the skin immediately proximal to the velocity probe to measure brachial artery diameter. Forearm blood flow was calculated as:
where the FBF is in millilitres per minute, the MBV is in centimetres per second, the brachial diameter is in centimetres, and 60 is used to convert from millilitres per second to millilitres per minute. Forearm vascular conductance (FVC) was calculated as (FBF/MAP) × 100, and expressed as millilitres per minute per 100 mmHg.
Rhythmic handgrip exercise
Rhythmic forearm handgrip exercise was performed using either a 6.4 kg weight that was ≈10-15 % of maximal voluntary contraction (MVC), or a 12.1 kg weight that was approximately ≈20-25 % MVC. The weight was lifted 4-5 cm over a pulley at a duty cycle of 1 s contraction-2 s relaxation (20 contractions per minute) using a signal light to insure the correct timing.
Sympathetic α-adrenergic vasoconstrictor drugs
The following drugs were infused via the brachial artery catheter: Tyramine was infused at 4 and 8 µg (per decilitre forearm volume per minute). These doses were based on the effects of forearm exercise on the tyramine vasoconstrictor dose-response curves reported by Tschakovsky et al. (2002). Tyramine evokes endogenous noradrenaline release from sympathetic nerve endings (Frewin & Whelan, 1968) and subsequent post-junctional α1- and α2-adrenergic vasoconstriction (Jie et al. 1987). Importantly, tyramine does not have any direct vasoconstrictor effects (Frewin & Whelan, 1968), and the vascular responses to tyramine are abolished by non-selective α-adrenergic blockade (Dinenno et al. 2002a,b). Phenylephrine (a selective α1 agonist) was infused at 0.0312 µg (dl forearm volume)−1 min−1 and clonidine (an α2 agonist) was infused at 0.15 µg (dl forearm volume)−1 min−1. The doses of phenylephrine and clonidine were based on our experience at rest (Dinenno et al. 2002a) and during handgrip exercise (Rosenmeier et al. 2003). All vasoconstrictor drug infusions were adjusted for the hyperaemic conditions (see below).
Given that exercise increases forearm blood flow, adenosine was infused to elevate resting forearm blood flow to similar levels observed during exercise. We have previously demonstrated that exercise blunts the vasoconstrictor responses to tyramine, phenylephrine and clonidine, whereas these vasoconstrictor responses are maintained when blood flow is elevated with adenosine and hence it was used to create a ‘high flow’ control state (Tschakovsky et al. 2002; Rosenmeier et al. 2003). In an effort to normalize the concentration of each vasoconstricting drug in the blood perfusing the forearm, the infusions were adjusted on the basis of forearm blood flow and forearm volume (measured via water displacement). Care was taken so that various concentrations of each compound were available and the absolute infusion rate was less than 3 ml min−1 in every trial.
Experimental protocols
General experimental protocol
Figure 1 is an example of a time-line for the specific trials. The subjects performed either a bout of forearm exercise or they received intra-arterial adenosine; the total time for each trial was 9 min. After 2 min of baseline measurements, exercise or adenosine infusion was initiated and steady-state FBF was reached within 3 min. Between 3 and 4 min of hyperaemia (minutes 5 and 6 of Fig. 1) the dose of the vasoconstricting agent was calculated on the basis of forearm volume and blood flow. The vasoconstrictor infusion began at the 6-minute mark and lasted for 3 min.
Figure 1. Experimental trial.
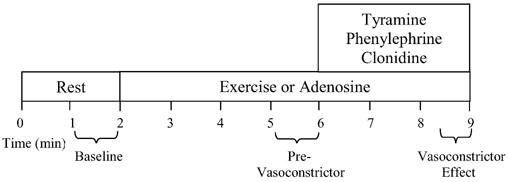
Each trial consisted of a 2-min rest (baseline) period. After this time period, subjects either began rhythmic forearm exercise or received intra-arterial adenosine to elevate resting forearm blood flow to similar levels observed during exercise (control non-exercise vasodilator). During minutes 5 and 6 (pre-vasoconstrictor), the dose of the α-adrenergic agonist was calculated on the basis of steady state hyperaemic forearm blood flow and forearm volume. Subsequently, the α-agonist (tyramine, phenylephrine or clonidine) was infused at minute 6 and lasted for 3 min. An average of the forearm blood flow and mean arterial blood pressure during the last 30 s of α-agonist infusion was used to calculate the vasoconstrictor effect during both hyperaemic conditions.
Protocol 1. Effects of NOS inhibition with l-NMMA on sympathetic vasoconstrictor responses during handgrip exercise
In the first six subjects, the vasoconstrictor responses to tyramine (4 and 8 µg (dl forearm volume)−1 min−1) were assessed during control vasodilator infusion of adenosine (6.25 µg (dl forearm volume)−1 min−1) and during moderate rhythmic handgrip exercise (6.4 kg). The order of tyramine dose and exercise or adenosine was randomized, and the subjects rested for 15 min between each trial. After these four initial trials (both doses of tyramine during adenosine and exercise), l-NMMA was administered at 5 mg for 10 min (total dose = 50 mg) to inhibit NOS (Dietz et al. 1994). We have previously documented that this dose reduces basal FBF as well as the vasodilator responses to acetylcholine, consistent with effective NOS blockade (Dietz et al. 1994; Eisenach et al. 2002). A maintenance dose of l-NMMA (1 mg min−1) was continued throughout the rest of the experimental protocol (Eisenach et al. 2002). Subsequently, the vasoconstrictor responses to tyramine were assessed again during adenosine and handgrip exercise in randomized order. In this protocol and those that follow, NOS inhibition was performed after the first set of adenosine and exercise trials due to the long half-life of NOS inhibition.
Protocol 2. Effects of NOS inhibition with l-NAME on sympathetic vasoconstrictor responses during handgrip exercise
l-NAME has been suggested to inhibit nNOS more effectively than l-NMMA, and thus might be the more appropriate NOS inhibitor for studies involving muscle contractions (Frandsen et al. 2001). We therefore repeated protocol 1 in eight additional subjects using l-NAME as the NOS inhibitor. In these subjects, the vasoconstrictor responses to tyramine (4 and 8 µg (dl forearm volume)−1 min−1) were assessed during control vasodilator infusion of adenosine (6.25 µg (dl forearm volume)−1 min−1) and during moderate rhythmic handgrip exercise (6.4 kg). The order of tyramine dose and exercise or adenosine was randomized, and the subjects rested for 15 min between each trial. After these initial trials, l-NAME was administered at 5 mg for 10 min (total dose = 50 mg) to inhibit NOS. A maintenance dose of l-NAME (1 mg min−1) was continued throughout the rest of the experimental protocol. The efficacy of this dose of l-NAME to inhibit NOS was tested in the subjects who participated in protocol 4 (see below). Subsequently, the vasoconstrictor responses to tyramine were assessed during adenosine and handgrip exercise in randomized order.
Protocol 3. Repeatability of vasoconstrictor responses to tyramine
The purpose of this protocol was to determine whether the forearm vasoconstrictor responses to tyramine were consistent over time throughout the length of the experimental protocol in the absence of NOS inhibition (time control). In four additional subjects, the vasoconstrictor responses to tyramine (4 and 8 µg (dl forearm volume)−1 min−1) were assessed during control vasodilator infusion of adenosine (6.25 µg (dl forearm volume)−1 min−1) and during moderate rhythmic handgrip exercise (6.4 kg). The order of tyramine dose and exercise or adenosine was randomized, and the subjects rested for 15 min between each trial. In these subjects we did not administer a NOS inhibitor. The vasoconstrictor responses to tyramine were then repeated during adenosine and handgrip exercise in randomized order.
Protocol 4. Effects of l-NAME on sympathetic vasoconstrictor responses during heavy handgrip exercise
It is well known that the blunted sympathetic vasoconstrictor responses during exercise are graded with the level of exercise intensity such that a greater reduction in vasoconstriction (greater sympatholysis) is observed during higher workloads (Anderson & Faber, 1991; Buckwalter et al. 2001; Tschakovsky et al. 2002). Additionally, in rats it appears that nNOS might be preferentially located in fast-twitch muscle fibres (Kobzik et al. 1994), although this might not be the case in humans (Frandsen et al. 1996). Thus, it is possible that nNOS might be activated to a greater extent during heavier workloads when a greater proportion of fast-twitch muscle fibres are recruited during contractions. Therefore, the purpose of this protocol was to determine whether NO blunts sympathetic vasoconstriction during heavy exercise. In four additional subjects, the vasoconstrictor responses to tyramine (8 µg (dl forearm volume)−1 min−1) were assessed during control vasodilator infusion of adenosine (12.50 µg (dl forearm volume)−1 min−1) and during heavy rhythmic handgrip exercise (12.1 kg). Only one dose of tyramine was administered in these subjects to reduce the number of exercise trials in an effort to minimise any potential effects of muscle fatigue associated with this workload. l-NAME was then administered at 5 mg min−1 for 10 min (total dose = 50 mg) and a maintenance dose of l-NAME (1 mg min−1) was continued throughout the rest of the experimental protocol. In previous studies in humans utilizing l-NAME to inhibit NOS during exercise, a systemic dose was administered intravenously over 1 h and NOS activity was reduced by ≈70 % (Frandsen et al. 2001). Therefore, in this protocol, the vasoconstrictor responses to tyramine were assessed during adenosine and heavy handgrip exercise after 1 h of the termination of the 10 min loading dose of l-NAME to ensure maximal reduction in NOS activity.
Because we are unaware of any studies in humans that have administered intra-arterial l-NAME, we tested the efficacy of NOS inhibition in these subjects by determining the vasodilator responses to brachial artery infusions of acetylcholine (which is partly NO mediated) before and after l-NAME. Therefore, 20 min after the initial adenosine and exercise trials, acetylcholine (1, 2 and 4 µg (dl forearm volume)−1 min−1) was infused for 2 min at each dose, and these infusions were subsequently repeated 10 min after l-NAME administration.
Protocol 5. Effects of l-NAME on post-junctional α-adrenergic vasoconstrictor responses during handgrip exercise
The purpose of this protocol was to determine whether NOS inhibition restores post-junctional α-adrenergic vasoconstriction during exercise. Because the tyramine-induced release of noradrenaline during adenosine infusions and exercise (both before and after NOS inhibition) is difficult to assess, in five additional subjects we determined the vasoconstrictor responses to direct α-adrenergic stimulation via phenylephrine (selective α1 agonist) and clonidine (α2 agonist). Four subjects received both phenylephrine and clonidine, and one subject received clonidine only. The vasoconstrictor responses to phenylephrine and clonidine were assessed during control vasodilator infusion of adenosine (6.25 µg (dl forearm volume)−1 min−1) and during moderate rhythmic handgrip exercise (6.4 kg). The order of α-agonist administration and exercise or adenosine was randomized, and the subjects rested for 15 min between each trial. After these initial trials, our standard l-NAME loading dose was administered and a maintenance dose was continued throughout the rest of the experimental protocol. Subsequently, the vasoconstrictor responses to phenylephrine and clonidine were assessed during adenosine and handgrip exercise, but the exercise trials were performed last to make sure that these were performed 1 h post l-NAME administration.
Data acquisition and analysis
Data were collected and stored on computer at 250 Hz and analysed off-line with signal-processing software (WinDaq, DATAQ Instruments, Akron, OH, USA). Mean arterial pressure (MAP) was determined from the arterial pressure waveform. Baseline FBF and MAP represent an average of the last minute of the resting time period, the hyperaemic values represent an average of minutes 3-4 (minutes 5-6 of Fig. 1; pre-vasoconstrictor) during adenosine or exercise, and the effects of the α-agonists represent an average of the final 30 s of drug infusion (post-vasoconstrictor).
The percentage reduction in FVC during vasoconstrictor administration was calculated as:
We used percentage reduction in FVC as our standard index to compare vasoconstriction across conditions when forearm blood flow and vascular conductance might differ. After much discussion this index has emerged as the most appropriate way to compare interventions that cause vasodilatation or vasoconstriction under conditions where there might be marked difference in baseline blood flow (Lautt, 1989; O'Leary, 1991; Thomas et al. 1994; Tschakovsky et al. 2002).
Statistics
All values are reported as means ± s.e.m. Specific hypothesis testing within each of the exercise or adenosine trials with the three different drug infusions was performed using repeated measures ANOVA. Comparison of the haemodynamic values at specific time points between the exercise and adenosine conditions were made with unpaired t tests, and the values within each hyperaemic condition (exercise or adenosine) before and after NOS inhibition were made with paired t tests. Significance was set at P < 0.05.
RESULTS
Protocol 1. Effects of NOS inhibition with l-NMMA on sympathetic vasoconstrictor responses during handgrip exercise
Forearm haemodynamics and MAP are presented in Table 1. Adenosine increased FBF and FVC significantly, but the steady-state forearm haemodynamics were slightly less than achieved during exercise. The vasoconstrictor responses to the low and high doses of tyramine were significantly blunted during exercise (▵FVC = -21 ± 4 and -20 ± 4 %, respectively) compared with the responses during adenosine (−44 ± 6 and -64 ± 5 %; P < 0.001vs. exercise; Fig. 2). NOS inhibition via l-NMMA reduced baseline FVC ≈40 %, and the steady-state FVC during adenosine (≈15-30 %) and handgrip exercise (≈10 %). However, the low and high doses of tyramine evoked similar vasoconstrictor responses during adenosine (▵FVC = -45 ± 6 and -62 ± 6 %, respectively), and this reduction was still significantly blunted during exercise (−20 ± 2 and -22 ± 2 %; Fig. 2). Importantly, the tyramine-induced vasoconstriction was similar during exercise before and after l-NMMA (P > 0.05).
Table 1.
Forearm and systemic haemodynamics for moderate exercise before and after NG-monomethyl-L-arginine (l-NMMA) (protocol 1: n = 6)
| Tyramine | Adenosine | Exercise | ||||
|---|---|---|---|---|---|---|
| Variable | dose | Time | Pre l-NMMA | Post l-NMMA | Pre l-NMMAPost | l-NMMA |
| FBF (ml min−1) | Low | Baseline | 75 ± 11 | 50 ± 6* | 61 ± 9 | 44 ± 6* |
| Pre-tyramine | 232 ± 47 | 187 ± 33 | 297 ± 27 | 282 ± 32 | ||
| Post-tyramine | 123 ± 20 | 96 ± 9 | 240 ± 27† | 231 ± 25† | ||
| High | Baseline | 73 ± 11 | 43 ± 6* | 65 ± 13 | 45 ± 8 | |
| Pre-tyramine | 224 ± 46 | 217 ± 45 | 296 ± 21 | 284 ± 31 | ||
| Post-tyramine | 76 ± 11 | 75 ± 11 | 245 ± 25† | 232 ± 29† | ||
| FVC (ml min−1 (100 mmHg)−1) | Low | Baseline | 85 ± 13 | 51 ± 6* | 68 ± 11 | 45 ± 6* |
| Pre-tyramine | 263 ± 54 | 193 ± 35 | 315 ± 27 | 283 ± 30* | ||
| Post-tyramine | 133 ± 21 | 98 ± 10 | 248 ± 25† | 225 ± 23† | ||
| High | Baseline | 79 ± 13 | 43 ± 6* | 71 ± 15 | 45 ± 8* | |
| Pre-tyramine | 253 ± 57 | 213 ± 43 | 307 ± 19 | 280 ± 34 | ||
| Post-tyramine | 81 ± 11‡ | 72 ± 9 | 247 ± 24† | 221 ± 29† | ||
| MAP (mmHg) | Low | Baseline | 88 ± 1 | 98 ± 4* | 89 ± 2 | 98 ± 3 |
| Pre-tyramine | 89 ± 1 | 97 ± 3* | 94 ± 3 | 99 ± 4 | ||
| Post-tyramine | 92 ± 2 | 98 ± 4 | 97 ± 3 | 103 ± 3 | ||
| High | Baseline | 93 ± 4 | 100 ± 4* | 92 ± 3 | 99 ± 3 | |
| Pre-tyramine | 91 ± 4 | 101 ± 3* | 96 ± 3 | 103 ± 3 | ||
| Post-tyramine | 94 ± 3 | 103 ± 5* | 99 ± 3 | 106 ± 2 | ||
FBF, forearm blood flow; FVC, forearm vascular conductance; MAP, mean arterial pressure.
P < 0.05 post vs. pre l-NMMA within same hyperaemic condition
P < 0.05 exercise vs. adenosine within same l-NMMA condition
P < 0.05vs. tyramine low during adenosine and within same l-NMMA condition.
Figure 2. Effects of l-NMMA on forearm vasoconstrictor responses to tyramine.
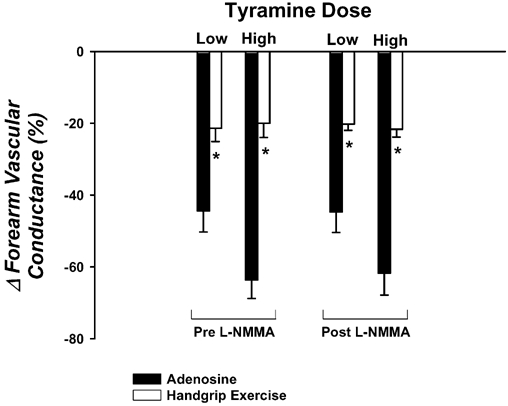
The vasoconstrictor responses to tyramine are significantly blunted during rhythmic handgrip exercise (open bars) compared with a control vasodilator condition (adenosine; filled bars). Intra-arterial administration of l-NMMA to inhibit nitric oxide synthase (NOS) does not augment the vasoconstrictor responses during adenosine or (more importantly) during handgrip exercise. * P < 0.05vs. adenosine for given tyramine dose within same l-NMMA condition.
Protocol 2. Effects of NOS inhibition with l-NAME on sympathetic vasoconstrictor responses during handgrip exercise
Forearm haemodynamics and MAP are presented in Table 2. Steady-state FBF and FVC during adenosine and exercise were similar. The vasoconstrictor responses to the low and high doses of tyramine were significantly blunted during exercise (▵FVC = -16 ± 3 and -24 ± 2 %, respectively) compared with the responses during adenosine (−54 ± 6 and -74 ± 5 %; P < 0.001vs. exercise; Fig. 3). NOS inhibition via l-NAME reduced baseline FVC ≈50 %, and the steady-state FVC during adenosine (≈55 %) and handgrip exercise (≈20 %). However, the low and high doses of tyramine evoked similar vasoconstrictor responses during adenosine (▵FVC = -47 ± 4 and -67 ± 5 %, respectively), and this reduction was still significantly blunted during exercise (−16 ± 3 and -26 ± 2 %; Fig. 3). Importantly, the tyramine-induced vasoconstriction was similar during exercise before and after l-NAME (P > 0.05).
Table 2.
Forearm and systemic haemodynamics for moderate exercise before and after l-NAME (protocol 2: n = 8)
| Tyramine | Adenosine | Exercise | ||||
|---|---|---|---|---|---|---|
| Variable | dose | Time | Pre l-NAME | Post l-NAME | Pre l-NAME | Post l-NAME |
| FBF (ml min−1) | Low | Baseline | 69 ± 9 | 43 ± 4* | 72 ± 13 | 36 ± 4* |
| Pre-tyramine | 267 ± 26 | 134 ± 16* | 277 ± 21 | 252 ± 25† | ||
| Post-tyramine | 119 ± 13 | 69 ± 5 * | 238 ± 16† | 215 ± 16⋆† | ||
| High | Baseline | 69 ± 10 | 46 ± 5 * | 64 ± 10 | 39 ± 3* | |
| Pre-tyramine | 272 ± 38 | 149 ± 18* | 282 ± 31 | 266 ± 22† | ||
| Post-tyramine | 68 ± 9‡ | 48 ± 5*‡ | 219 ± 24† | 204 ± 19† | ||
| FVC (ml min−1(100 mmHg)−1) | Low | Baseline | 75 ± 9 | 40 ± 6* | 74 ± 12 | 33 ± 3* |
| Pre-tyramine | 287 ± 22 | 125 ± 14* | 283 ± 16 | 227 ± 18*† | ||
| Post-tyramine | 126 ± 14 | 63 ± 4* | 236 ± 12† | 187 ± 10*† | ||
| High | Baseline | 75 ± 10 | 42 ± 4* | 69 ± 10 | 36 ± 3 * | |
| Pre-tyramine | 298 ± 41 | 137 ± 14* | 286 ± 25 | 241 ± 15*† | ||
| Post-tyramine | 72 ± 11‡ | 44 ± 4*‡ | 217 ± 18† | 179 ± 14*† | ||
| MAP (mmHg) | Low | Baseline | 92 ± 2 | 107 ± 4* | 95 ± 3 | 107 ± 2* |
| Pre-tyramine | 92 ± 3 | 107 ± 3* | 98 ± 4 | 110 ± 3* | ||
| Post-tyramine | 95 ± 3 | 109 ± 2* | 101 ± 4 | 114 ± 4* | ||
| High | Baseline | 91 ± 2 | 108 ± 3* | 93 ± 3 | 107 ± 3* | |
| Pre-tyramine | 91 ± 2 | 107 ± 3* | 98 ± 4 | 110 ± 3* | ||
| Post-Tyramine | 96 ± 3 | 109 ± 3* | 100 ± 4 | 113 ± 4* | ||
P < 0.05 post vs. pre l-NAME within same hyperaemic condition
P < 0.05 exercise vs. adenosine within same l-NAME condition
P < 0.05 vs. tyramine low during denosine and within same l-NAME condition.
Figure 3. Effects of l-NAME on forearm vasoconstrictor responses to tyramine.
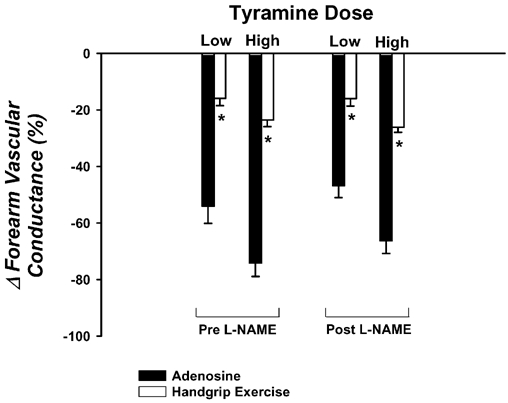
The vasoconstrictor responses to tyramine are significantly blunted during rhythmic handgrip exercise (open bars) compared with a control vasodilator condition (adenosine; filled bars). Intra-arterial administration of l-NAME to inhibit NOS does not augment the vasoconstrictor responses during adenosine or (more importantly) during handgrip exercise. * P < 0.05vs. adenosine for given tyramine dose within same l-NAME condition.
Protocol 3. Repeatability of vasoconstrictor responses to tyramine
Forearm haemodynamics and MAP are presented in Table 3. In trial 1, adenosine increased FBF and FVC significantly, but the steady-state forearm haemodynamics were slightly less than achieved during exercise. The vasoconstrictor responses to the low and high doses of tyramine were significantly blunted during exercise (▵FVC = -14 ± 2 and -18 ± 5 %, respectively) compared with the responses during adenosine (−47 ± 5 and -61 ± 4 %; P < 0.001vs. exercise). In trial 2, the steady-state forearm haemodynamics were similar to trial 1. Additionally, the reductions in FVC to the low and high dose of tyramine during adenosine (−46 ± 6 and -61 ± 4 %, respectively) as well as during exercise (−14 ± 3 and -19 ± 4 %, respectively) were similar during both trials. These data indicate that the vasoconstrictor responses to tyramine are consistent throughout the experimental protocol, and any lack of effect of NOS inhibition in protocols 1 and 2 cannot be explained by tachyphylaxis to tyramine.
Table 3.
Forearm and systemic haemodynamics for time control trials (protocol 3: n = 4)
| Tyramine | Adenosine | Exercise | ||||
|---|---|---|---|---|---|---|
| Variable | dose | Time | Trial 1 | Trial 2 | Trial 1 | Trial 2 |
| FBF (ml min−1) | Low | Baseline | 37 ± 5 | 41 ± 6 | 31 ± 3 | 40 ± 6 |
| Pre-tyramine | 118 ± 18 | 166 ± 26* | 180 ± 23 | 192 ± 19 | ||
| Post-tyramine | 62 ± 7 | 92 ± 17 | 163 ± 22 | 175 ± 16 | ||
| High | Baseline | 41 ± 6 | 38 ± 3 | 33 ± 5 | 36 ± 5 | |
| Pre-tyramine | 142 ± 19 | 144 ± 33 | 183 ± 24 | 193 ± 23 | ||
| Post-tyramine | 54 ± 6 | 54 ± 8 | 157 ± 22 | 171 ± 29 | ||
| FVC (ml min−1(100 mmHg)−1) | Low | Baseline | 41 ± 5 | 45 ± 6 | 35 ± 4 | 42 ± 6 |
| Pre-tyramine | 133 ± 19 | 179 ± 24 | 190 ± 17 | 198 ± 11 | ||
| Post-tyramine | 70 ± 7 | 96 ± 14 | 164 ± 5 | 171 ± 14 | ||
| High | Baseline | 46 ± 8 | 41 ± 4 | 38 ± 7 | 39 ± 5 | |
| Pre-tyramine | 157 ± 19 | 156 ± 36 | 195 ± 19 | 198 ± 13 | ||
| Post-tyramine | 58 ± 6 | 58 ± 8 | 160 ± 19 | 162 ± 15 | ||
| MAP (mmHg) | Low | Baseline | 88 ± 2 | 91 ± 3 | 89 ± 2 | 94 ± 2 |
| Pre-tyramine | 88 ± 3 | 91 ± 3 | 94 ± 5 | 97 ± 4 | ||
| Post-tyramine | 89 ± 4 | 94 ± 4 | 99 ± 6 | 101 ± 7 | ||
| High | Baseline | 90 ± 2 | 93 ± 1 | 90 ± 3 | 94 ± 2 | |
| Pre-tyramine | 90 ± 3 | 92 ± 1 | 93 ± 4 | 97 ± 5 | ||
| Post-tyramine | 93 ± 5 | 94 ± 1 | 98 ± 8 | 104 ± 9 | ||
P <0.05 trial 2 vs. trial 1 within same hyperaemic condition.
Protocol 4. Effects of l-NAME on sympathetic vasoconstrictor responses during heavy handgrip exercise
Forearm haemodynamics and MAP are presented in Table 4. Adenosine increased FBF and FVC significantly, but the steady-state forearm haemodynamics were less than achieved during heavy exercise. The vasoconstrictor responses to the high dose of tyramine were significantly blunted during exercise (▵FVC = -18 ± 5 %) compared with the responses during adenosine (−51 ± 4 %; P < 0.001vs. exercise; Fig. 4). NOS inhibition via l-NAME reduced baseline FVC ≈35 %, and the steady-state FVC during adenosine (≈36 %) and handgrip exercise (≈10 %). However, the high dose of tyramine evoked similar reductions in FVC during adenosine as before l-NAME (−56 ± 4 %), and this reduction was still significantly blunted during exercise (−14 ± 7 %; Fig. 4). Importantly, the tyramine-induced vasoconstriction was blunted to a similar extent during heavy exercise before and after l-NAME (P > 0.05).
Table 4.
Forearm and systemic haemodynamics for heavy exercise before and after l-NAME (protocol 4: n = 4)
| Tyramine | Adenosine | Exercise | ||||
|---|---|---|---|---|---|---|
| Variable | dose | Time | Pre l-NAME | Post l-NAME | Pre l-NAME | Post l-NAME |
| FBF (ml min−1) | High | Baseline | 53 ± 11 | 41 ± 7 | 54 ± 10 | 38 ± 7* |
| Pre-tyramine | 238 ± 59 | 171 ± 40 | 353 ± 62 | 370 ± 66† | ||
| Post-tyramine | 115 ± 27 | 72 ± 15 | 317 ± 74† | 338 ± 80† | ||
| FVC (ml min−1(100 mmHg)−1) | High | Baseline | 59 ± 11 | 40 ± 6* | 59 ± 10 | 36 ± 7* |
| Pre-tyramine | 267 ± 62 | 170 ± 38 | 348 ± 61 | 326 ± 54† | ||
| Post-tyramine | 125 ± 25 | 71 ± 15* | 289 ± 62 | 289 ± 68† | ||
| MAP (mmHg) | High | Baseline | 88 ± 5 | 101 ± 4* | 90 ± 3 | 104 ± 1* |
| Pre-tyramine | 87 ± 4 | 100 ± 2* | 102 ± 4* | 113 ± 3*† | ||
| Post-tyramine | 89 ± 5 | 101 ± 2 | 109 ± 6* | 116 ± 5*† | ||
P < 0.05 post vs. pre l-NAME within same hyperaemic condition
P < 0.05 exercise vs. adenosine with in same l-NAME condition.
Figure 4. Effects of l-NAME on forearm vasoconstrictor responses to tyramine during heavy rhythmic handgrip exercise.
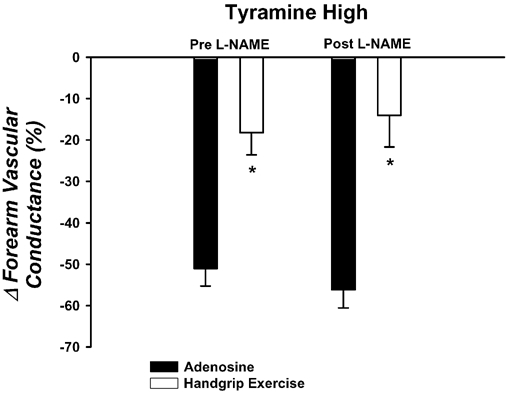
The vasoconstrictor responses to tyramine are significantly blunted during heavy handgrip exercise (open bars) compared with a control vasodilator condition (adenosine; filled bars). NOS inhibition with l-NAME does not augment the vasoconstrictor responses during either adenosine or exercise. * P < 0.05vs. adenosine within same l-NAME condition.
In these subjects, l-NAME reduced baseline FVC by ≈37 % before acetylcholine infusion. Additionally, the vasodilator responses to all three doses of acetylcholine were significantly reduced after l-NAME (▵FVC before l-NAME = 194 ± 39, 222 ± 40 and 291 ± 41 ml min−1 (100 mmHg)−1; ▵FVC after l-NAME = 62 ± 16, 71 ± 18 and 116 ± 22 ml min−1 (100 mmHg)−1; P < 0.05; Fig. 5). Collectively, these data indicate that the dose of l-NAME used in our studies effectively inhibits NOS.
Figure 5. Effects of intra-arterial l-NAME on forearm vasodilator responses to acetylcholine.
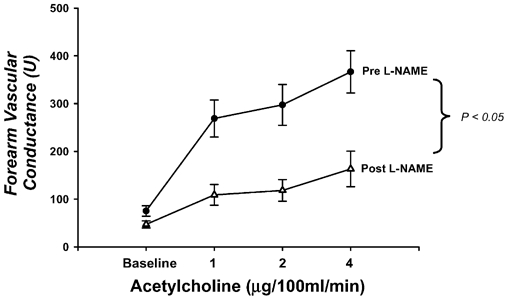
Baseline forearm vascular conductance (FVC) is reduced ≈40 % and the increases in FVC to acetylcholine are significantly blunted after NOS inhibition with l-NAME. Taken together, these data are consistent with NOS inhibition in the human forearm.
Protocol 5. Effects of l-NAME on post-junctional α-adrenergic vasoconstrictor responses during handgrip exercise
Forearm haemodynamics and MAP are presented in Table 5. Adenosine increased FBF and FVC significantly, but the steady-state forearm haemodynamics were slightly less than achieved during exercise. Phenylephrine (−36 ± 4 %) and clonidine (−46 ± 6 %) evoked significant reductions in FVC during adenosine, and this reduction was significantly blunted during exercise (−14 ± 3 and -15 ± 5 %, respectively; P < 0.05; Fig. 6). NOS inhibition via l-NAME reduced baseline FVC ≈40 %, and the steady-state FVC during adenosine (≈40 %) and handgrip exercise (≈15 %). However, phenylephrine and clonidine evoked similar reductions in FVC during adenosine as before l-NAME (−37 ± 4 and -47 ± 9 %, respectively), and this reduction was still significantly blunted during exercise (−12 ± 5 and -12 ± 6 %; Fig. 6). Importantly, the vasoconstrictor responses to phenylephrine and clonidine were similar during exercise before and after l-NAME (P > 0.05).
Table 5.
Forearm and systemic haemodynamics for moderate exercise before and after l-NAME (protocol 5)
| Adenosine | Exercise | |||||
|---|---|---|---|---|---|---|
| Variable | Vasoconstrictor | Time | Pre l-NAME | Post l-NAME | Pre l-NAME | Post l-NAME |
| FBF (ml min−1) | Phenylephrine | Baseline | 28 ± 2 | 20 ± 4* | 31 ± 5 | 18 ± 7 |
| (n = 4) | Pre-phenylephrine | 143 ± 46 | 88 ± 24 | 185 ± 16 | 164 ± 15† | |
| Post-phenylephrine | 96 ± 35 | 57 ± 19 | 163 ± 8 | 151 ± 22† | ||
| Clonidine | Baseline | 36 ± 7 | 26 ± 4 | 38 ± 7 | 23 ± 3 * | |
| (n = 5) | Pre-clonidine | 148 ± 31 | 92 ± 29* | 205 ± 21 | 188 ± 19† | |
| Post-clonidine | 78 ± 15 | 45 ± 12* | 185 ± 25† | 171 ± 25† | ||
| FVC (ml min−1 (100 mm Hg)−1) | Phenylephrine | Baseline | 31 ± 2 | 21 ± 4* | 35 ± 5 | 18 ± 7* |
| Pre-phenylephrine | 166 ± 52 | 94 ± 28 | 194 ± 14 | 163 ± 17* | ||
| Post-phenylephrine | 107 ± 37 | 59 ± 20 | 167 ± 15 | 146 ± 22† | ||
| Clonidine | Baseline | 42 ± 8 | 27 ± 5* | 44 ± 9 | 24 ± 4 * | |
| Pre-clonidine | 172 ± 35 | 101 ± 33* | 226 ± 27 | 188 ± 21* | ||
| Post-clonidine | 89 ± 17 | 47 ± 12* | 198 ± 32† | 168 ± 27*† | ||
| MAP (mmHg) | Phenylephrine | Baseline | 90 ± 2 | 95 ± 2 | 90 ± 1 | 99 ± 3* |
| Pre-phenylephrine | 86 ± 1 | 95 ± 2 | 95 ± 3 | 101 ± 2 | ||
| Post-phenylephrine | 89 ± 2 | 96 ± 2* | 97 ± 4 | 104 ± 1† | ||
| Clonidine | Baseline | 89 ± 2 | 94 ± 1 | 88 ± 1 | 99 ± 2* | |
| Pre-clonidine | 86 ± 2 | 92 ± 1 | 91 ± 3 | 100 ± 3 *† | ||
| Post-clonidine | 89 ± 3 | 96 ± 1* | 96 ± 4 | 103 ± 2† | ||
P < 0.05 post vs. pre l-NAME within same hyperaemic condition
P < 0.05 exercise vs. adenosine within same l-NAME condition.
Figure 6. Effects of l-NAME on forearm vasoconstrictor responses to phenlyephrine and clonidine.
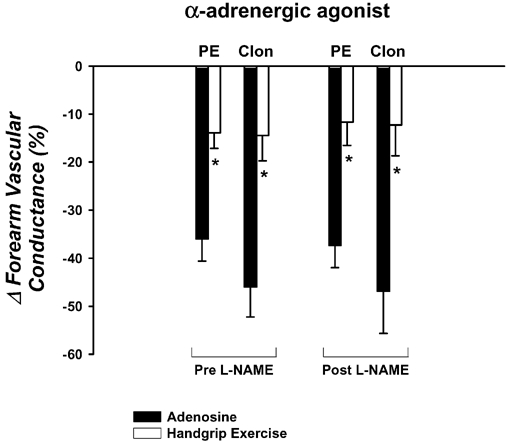
The vasoconstrictor responses to the direct α1-agonist (phenylephrine; PE) and α2-agonist (clonidine; Clon) are significantly blunted during exercise compared with adenosine. Local NOS inhibition with l-NAME does not augment the vasoconstrictor responses to either α-agonist during adenosine or rhythmic handgrip exercise. * P < 0.05vs. adenosine for specific α-agonist within same l-NAME condition.
DISCUSSION
Recent studies in both experimental animals and conscious humans have clearly demonstrated that the vasoconstrictor responses to sympathetic α-adrenergic stimulation are blunted in contracting compared with resting (quiescent) skeletal muscle (Thomas et al. 1994; Buckwalter et al. 2001; Tschakovsky et al. 2002). What is less clear is the mechanism(s) by which skeletal muscle contractions can interfere with or blunt sympathetic vasoconstriction. Despite evidence that NO is involved in functional sympatholysis in experimental animals (Thomas & Victor, 1998), the role of NO in blunting sympathetic vasoconstriction in contracting muscle of healthy humans has been more difficult to determine. The primary new finding from the present investigation is that local inhibition of NOS via both l-NMMA or l-NAME does not restore the vasoconstrictor responses to local endogenous noradrenaline release (via tyramine) during moderate or heavy rhythmic handgrip exercise. Importantly, these observations cannot be explained by any potential differences in neurotransmitter release because NOS inhibition did not restore the vasoconstrictor responses to direct α1- or α2-adrenergic receptor stimulation during exercise. Taken together, our findings demonstrate that NO is not obligatory to observe blunted post-junctional α-adrenergic vasoconstriction (i.e. functional sympatholysis) during muscle contractions in healthy humans.
Early studies in the rat hindlimb demonstrated that sympathetic vasoconstrictor responses were blunted during muscle contractions, and that these responses could be (in part) ‘normalized’ by administration of the KATP channel antagonist glibenclamide (Thomas et al. 1997). These data indicated that muscle contractions activated these metabolically sensitive channels and inhibited the vasoconstrictor responses to sympathetic nerve stimulation, and that this might be specific for post-junctional α2-adrenergic receptors. Additional studies by Thomas and colleagues (Thomas & Victor, 1998) also implicated a role for NO, but not adenosine or prostaglandins, in the blunted vasoconstrictor responses during exercise. Interestingly, in this latter study, blockade of KATP channels after NOS inhibition did not further augment the hindlimb sympathetic vasoconstrictor responses during contractions, strongly suggesting that the actions of NO interact with KATP channels to evoke functional sympatholysis.
Other studies performed in a mouse model of Duchenne muscular dystrophy (in which nNOS levels are substantially low), as well as in an nNOS knockout mouse model, have provided evidence that NO is involved in this phenomenon in animals (Thomas et al. 1998). A lack of functional sympatholysis has also been suggested (via near-infrared spectroscopy; see below) in children with Duchenne muscular dystrophy (in whom nNOS levels are greatly decreased), providing indirect evidence that NO derived from nNOS is an important modulator of sympathetic vasoconstriction in contracting muscle (Sander et al. 2000).
In healthy humans, a role for NO in functional sympatholysis has been more difficult to demonstrate. Wilson and Kapoor (1993) documented that intra-arterial infusions of noradrenaline during exercise evoked similar forearm vasoconstrictor responses before and after local NOS inhibition via l-NMMA. In this study, the investigators used venous occlusion plethysmography to estimate muscle blood flow, a technique that necessitates the interruption of muscle contractions during the time required for blood flow measurements. The major problem with this approach is that the regulatory mechanisms of muscle blood flow control studied using this technique reflect the control of post-exercise hyperaemia, and not active muscle blood flow regulation (Dyke et al. 1995; Shoemaker et al. 1997; Radegran & Saltin, 1999; Frandsen et al. 2001).
In contrast, Chavoshan et al. (2002) demonstrated that the blunted vasoconstrictor responses (estimated via near-infrared spectroscopy) normally observed during reflex increases in muscle sympathetic nerve activity (via lower body negative pressure) in contracting skeletal muscle were completely reversed after systemic NOS inhibition via l-NAME. These data not only support a role for NO in functional sympatholysis in humans but also indicate that NO is the putative sympatholytic factor and contrasts the findings in experimental animals indicating that NO is only in part responsible for blunting sympathetic vasoconstriction. Unfortunately, the use of near-infrared spectroscopy does not provide measures of active muscle blood flow regulation or the whole-limb haemodynamic responses (flow and pressure) during exercise and subsequently during sympathetic vasoconstriction. This latter point is significant because much of the discussion regarding functional sympatholysis concerns how the sympathetic nerves and metabolic vasodilators interact to regulate blood pressure during exercise (Rowell, 1997).
In the present study, we sought to overcome some of the methodological concerns that have potentially contributed to these equivocal findings. First, we used Doppler ultrasound to measure active forearm blood flow during moderate and heavy dynamic handgrip exercise and determined the vasoconstrictor responses to intra-arterial infusions of tyramine to evoke local endogenous noradrenaline release and subsequent post-junctional α-adrenergic receptor stimulation. Second, we administered both l-NMMA and l-NAME to inhibit NOS in order to determine whether the discrepant findings from prior investigations in humans are related to the specific analogue for l-arginine used to inhibit NOS in these previous studies. Finally, we extended our findings utilizing tyramine to evoke local noradrenaline release by selectively stimulating post-junctional α1- and α2-adrenergic receptors (via phenylephrine and clonidine, respectively). Collectively, our data derived from the use of several approaches indicate that acute inhibition of NOS does not restore sympathetic α-adrenergic vasoconstriction in the vascular beds of contracting skeletal muscle of healthy humans. This finding is also consistent with recent data from our laboratory indicating that exogenous administration of sodium nitroprusside (NO donor) sufficient to elevate forearm blood flow to levels observed during exercise does not blunt α-adrenergic vasoconstriction (Tschakovsky et al. 2002).
Experimental considerations
The source of NO postulated to be involved in functional sympatholysis is derived primarily from skeletal muscle nNOS (Hansen et al. 2000). We have demonstrated either previously or in the present study that NOS inhibition (via l-NMMA and l-NAME) reduces resting forearm vascular conductance and the vasodilator responses to acetylcholine, consistent with effective NOS inhibition. Despite this, it could be argued that the infusions of these l-arginine analogues only effectively inhibited eNOS (and not nNOS) and therefore could possibly explain why we did not observe ‘restored’ vasoconstrictor responses in contracting muscle after NOS inhibition. However, the dose of l-NMMA or l-NAME administered directly into the forearm vasculature in the present study (total dose ≈110 mg kg−1) is substantially greater than the dose of l-NAME infused systemically (4 mg kg−1 body mass) in humans which reduced skeletal muscle NOS activity by ≈70 % (Fransden et al. 2001). Given the nature of drug administration in the present study (intra-arterial), the concentration of either l-NMMA or l-NAME at the level of the tissue should have been much greater than in this previous study (Fransden et al. 2001). Therefore, we believe that our doses of l-NMMA and l-NAME effectively inhibited both eNOS and nNOS and that this should not limit the interpretations of our findings.
Another potential experimental consideration is our choice of using adenosine as a ‘control’ vasodilator in the present study. Previous studies in experimental animals have demonstrated that KATP channels are involved in functional sympatholysis. Thus, given that adenosine can activate these channels, one could question the use of adenosine as a control vasodilator to study this phenomenon in humans. However, our rationale for using adenosine is based on previous findings from our laboratory which indicate that elevating resting forearm blood flow (via adenosine) similar to levels observed during exercise does not blunt sympathetic vasoconstriction (Tschakovsky et al. 2002; Rosenmeier et al. 2003). Further, it has recently been demonstrated that passive vasodilatation via the NO donor sodium nitroprusside also does not blunt sympathetic vasoconstriction in both animals and humans (Tschakovsky et al. 2002; Van Teeffelen & Segal, 2003). Irrespective of which vasodilator is used to elevate resting forearm blood flow, the key finding from the present investigation is that local NOS inhibition does not restore the sympathetic α-adrenergic vasoconstrictor responses in contracting skeletal muscle.
Perspectives
Skeletal muscle blood flow control during muscle contractions is a complicated and highly integrated process involving the interactions of a number of locally produced vasodilating metabolites, mechanical factors (e.g. muscle pump), as well as sympathetic nerves (Saltin et al. 1998). With respect to the possible substance(s) that could interfere with sympathetic vasoconstriction in contracting muscle, several vasodilating factors (adenosine, prostaglandins, NO) have been demonstrated to blunt sympathetic vasoconstriction in a variety of experimental conditions (Lippton et al. 1981; Faber et al. 1982; Nishigaki et al. 1991; Ohyanagi et al. 1992). Therefore, the findings from the present study suggest that there are redundant pathways that can mediate functional sympatholysis during exercise, as is probably the case for the metabolic vasodilating substances involved in exercise hyperaemia (Joyner & Proctor, 1999). For example, prostaglandins have been demonstrated to inhibit noradrenaline release (Malik & Sehic, 1990), as well as reduce post-junctional α-adrenergic vasoconstriction (Lippton et al. 1981; Malik & Sehic, 1990). Given that acute NOS inhibition can result in greater prostacyclin synthesis and release (Osanai et al. 2000), it appears reasonable to speculate that elevated levels of vasodilating prostaglandins might compensate for the impaired NO synthesis under these conditions and continue to blunt the sympathetic vasoconstrictor responses during exercise.
In this context, a recent study (Boushel et al. 2002) demonstrated that combined inhibition of NOS and prostaglandins reduces active muscle blood flow during dynamic knee extensor exercise, a finding that contrasts what has been demonstrated by only the selective inhibition of either substance alone. It is also of interest to note that the reduction in active muscle blood flow during combined prostaglandin and NO inhibition was greater during heavy compared with light exercise (Boushel et al. 2002), a condition associated with elevations in sympathetic vasoconstrictor nerve activity (Seals et al. 1988; Hansen et al. 1994). Therefore, whether or not the reduction in muscle blood flow under these conditions reflects less available vasodilator substances or an impaired ability to blunt sympathetic vasoconstriction in active muscle is unclear, but certainly deserves further study.
Conclusions
The findings from the present investigation challenge the hypothesis that NO plays an obligatory role in the blunted α-adrenergic vasoconstrictor responses observed in the vascular beds of contracting skeletal muscles of humans. Similar to exercise hyperaemia, it is likely that there are redundant mechanisms involved in functional sympatholysis. Future studies are needed to identify and determine the potential interactions between these ‘sympatholytic’ substances in healthy humans, and how these interactions might be altered in those populations that demonstrate impaired active muscle blood flow regulation during exercise (e.g. ageing, heart failure patients).
Acknowledgments
We thank Shelly Roberts, Karen Krucker, Niki Dietz, John Eisenach, Branton Walker and Chris Johnson for their technical assistance, and the subjects who volunteered for this study. This research was supported by NIH grants HL-46493 and NS-32352 (M.J.J.), NIH General Research Center Grant RR-00585 (to the Mayo Clinic, Rochester, MN USA) and an Individual National Research Service Award AG-05912 (F.A.D.).
REFERENCES
- Anderson KM, Faber JE. Differential sensitivity of arteriolar α1- and α2-adrenoceptor constriction to metabolic inhibition during rat skeletal muscle contraction. Circ Res. 1991;69:178–184. doi: 10.1161/01.res.69.1.174. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces skeletal muscle blood flow during exercise. J Physiol. 2002;543:691–698. doi: 10.1113/jphysiol.2002.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckwalter JB, Naik JS, Valic Z, Clifford PS. Exercise attenuates alpha-adrenergic-receptor responsiveness in skeletal muscle vasculature. J Appl Physiol. 2001;90:172–178. doi: 10.1152/jappl.2001.90.1.172. [DOI] [PubMed] [Google Scholar]
- Chavoshan B, Sander M, Sybert TE, Hansen J, Victor RG, Thomas GD. Nitric oxide-dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J Physiol. 2002;540:377–386. doi: 10.1113/jphysiol.2001.013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz NM, Rivera JM, Eggener ES, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol. 1994;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA, Dietz NM, Joyner MJ. Aging and forearm postjunctional α-adrenergic vasoconstriction in healthy men. Circulation. 2002a;106:1349–1354. doi: 10.1161/01.cir.0000028819.64790.be. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Eisenach JH, Dietz NM, Joyner MJ. Post-junctional α-adrenoceptors and basal limb vascular tone in healthy men. J Physiol. 2002b;540:1103–1110. doi: 10.1113/jphysiol.2001.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyke CK, Proctor DN, Dietz NM, Joyner MJ. Role of nitric oxide in exercise hyperaemia during prolonged rhythmic handgripping in humans. J Physiol. 1995;488:259–265. doi: 10.1113/jphysiol.1995.sp020964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach JH, Clark ES, Charkoudian N, Dinenno FA, Atkinson JL, Fealey RD, Dietz NM, Joyner MJ. Effects of chronic sympathectomy on vascular function in the human forearm. J Appl Physiol. 2002;92:2019–2025. doi: 10.1152/japplphysiol.01025.2001. [DOI] [PubMed] [Google Scholar]
- Faber JE, Harris PD, Joshua IG. Microvascular response to blockade of prostaglandin synthesis in rat skeletal muscle. Am J Physiol. 1982;243:H51–60. doi: 10.1152/ajpheart.1982.243.1.H51. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with N(G)-nitro-l-arginine methyl ester in humans. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen U, Lopez-Figueroa M, Hellsten Y. Localization of nitric oxide synthase in human skeletal muscle. Biochem Biophys Res Commun. 1996;227:88–93. doi: 10.1006/bbrc.1996.1472. [DOI] [PubMed] [Google Scholar]
- Frewin DB, Whelan RF. The mechanism of action of tyramine on the blood vessels of the forearm in man. Br J Pharmacol. 1968;33:105–116. doi: 10.1111/j.1476-5381.1968.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange RW, Isotani E, Lau KS, Kamm KE, Huang PL, Stull JT. Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast-twitch muscles. Physiol Genomics. 2001;5:35–44. doi: 10.1152/physiolgenomics.2001.5.1.35. [DOI] [PubMed] [Google Scholar]
- Hansen J, Sander M, Thomas GD. Metabolic modulation of sympathetic vasoconstriction in exercising skeletal muscle. Acta Physiol Scand. 2000;168:489–503. doi: 10.1046/j.1365-201x.2000.00701.x. [DOI] [PubMed] [Google Scholar]
- Hansen J, Thomas GD, Jacobsen TN, Victor RG. Muscle metaboreflex triggers parallel sympathetic activation in exercising and resting human skeletal muscle. Am J Physiol. 1994;266:H2508–2514. doi: 10.1152/ajpheart.1994.266.6.H2508. [DOI] [PubMed] [Google Scholar]
- Jie K, van Brummelen P, Vermey P, Timmermans PB, van Zwieten PA. Postsynaptic alpha1 and alpha2-adrenoceptors in human blood vessels: interactions with exogenous and endogenous catecholamines. Eur J Clin Invest. 1987;17:174–181. doi: 10.1111/j.1365-2362.1987.tb02397.x. [DOI] [PubMed] [Google Scholar]
- Kobzik L, Reid MB, Bredt DS, Stamler JS. Nitric oxide in skeletal muscle. Nature. 1994;372:546–548. doi: 10.1038/372546a0. [DOI] [PubMed] [Google Scholar]
- Lautt WW. Resistance or conductance for expression of arterial vascular tone. Microvasc Res. 1989;37:230–236. doi: 10.1016/0026-2862(89)90040-x. [DOI] [PubMed] [Google Scholar]
- Lippton HL, Chapnick BM, Kadowitz PJ. Influence of prostaglandins on vasoconstrictor responses in the hindquarters vascular bed of the cat. Prostaglandins Med. 1981;6:183–202. doi: 10.1016/0161-4630(81)90089-6. [DOI] [PubMed] [Google Scholar]
- Malik KU, Sehic E. Prostaglandins and the release of the adrenergic transmitter. Ann N Y Acad Sci. 1990;604:222–236. doi: 10.1111/j.1749-6632.1990.tb31996.x. [DOI] [PubMed] [Google Scholar]
- Marshall RJ, Schirger A, Shepherd JT. Blood pressure during supine exercise in idiopathic orthostatic hypotension. Circulation. 1961;24:76–81. doi: 10.1161/01.cir.24.1.76. [DOI] [PubMed] [Google Scholar]
- Nishigaki K, Faber JE, Ohyanagi M. Interactions between α-adrenoceptors and adenosine receptors on microvascular smooth muscle. Am J Physiol. 1991;260:H1655–1666. doi: 10.1152/ajpheart.1991.260.5.H1655. [DOI] [PubMed] [Google Scholar]
- Ohyanagi M, Nishigaki K, Faber JE. Interaction between microvascular α1- and α2-adrenoceptors and endothelium-derived relaxing factor. Circ Res. 1992;71:188–200. doi: 10.1161/01.res.71.1.188. [DOI] [PubMed] [Google Scholar]
- O'Leary DS. Regional vascular resistance vs. conductance: which index for baroreflex responses. Am J Physiol. 1991;260:H632–637. doi: 10.1152/ajpheart.1991.260.2.H632. [DOI] [PubMed] [Google Scholar]
- Osanai T, Fujita N, Fujiwara N, Noakano T, Takahashi K, Guan W, Okumura K. Cross talk of shear-induced production of prostacyclin and nitric oxide in endothelial cells. Am J Physiol Heart Circ Physiol. 2000;278:H233–238. doi: 10.1152/ajpheart.2000.278.1.H233. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Proctor DN. Muscle blood flow during exercise: the limits of reductionism. Med Sci Sports Exerc. 1999;31:1036–1040. doi: 10.1097/00005768-199907000-00017. [DOI] [PubMed] [Google Scholar]
- Radegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol. 1999;276:H1951–1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- Rosenmeier JB, Dinenno FA, Fritzlar SJ, Joyner MJ. α1- and α2-adrenergic vasoconstriction is blunted in contracting human muscle. J Physiol. 2003;547:971–976. doi: 10.1113/jphysiol.2002.037937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell LB. Neural control of muscle blood flow: importance during dynamic exercise. Clin Exp Pharmacol Physiol. 1997;24:117–125. doi: 10.1111/j.1440-1681.1997.tb01793.x. [DOI] [PubMed] [Google Scholar]
- Ruble SB, Valic Z, Buckwalter JB, Tschakovsky ME, Clifford PS. Attenuated vascular responsiveness to noradrenaline release during dynamic exercise in dogs. J Physiol. 2002;541:637–644. doi: 10.1113/jphysiol.2001.014738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltin B, Radegran G, Koskolou MD, Roach RC. Skeletal muscle blood flow in humans and its regulation during exercise. Acta Physiol Scand. 1998;162:421–436. doi: 10.1046/j.1365-201X.1998.0293e.x. [DOI] [PubMed] [Google Scholar]
- Sander M, Chavoshan B, Harris SA, Iannoccone ST, Stull JT, Thomas GD, Victor RG. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DR, Victor RG, Mark AL. Plasma norepinephrine and muscle sympathetic discharge during rhythmic exercise in humans. J Appl Physiol. 1988;65:940–944. doi: 10.1152/jappl.1988.65.2.940. [DOI] [PubMed] [Google Scholar]
- Shoemaker JK, Halliwill JR, Hughson RL, Joyner MJ. Contributions of acetylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol. 1997;273:H2388–2395. doi: 10.1152/ajpheart.1997.273.5.H2388. [DOI] [PubMed] [Google Scholar]
- Strandell T, Shepherd JT. The effect in humans of increased sympathetic activity on the blood flow to active muscles. Acta Med Scand. 1967;472:146–167. doi: 10.1111/j.0954-6820.1967.tb12622.x. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Hansen J, Victor RG. Inhibition of alpha2-adrenergic vasoconstriction during contraction of glycolytic, not oxidative, rat hindlimb muscle. Am J Physiol. 1994;266:H920–929. doi: 10.1152/ajpheart.1994.266.3.H920. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Hansen J, Victor RG. ATP-sensitive potassium channels mediate contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Clin Invest. 1997;99:2602–2609. doi: 10.1172/JCI119448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of α-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Victor RG. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol. 1998;506:817–826. doi: 10.1111/j.1469-7793.1998.817bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschakovsky ME, Sujirattanawimol K, Ruble SB, Valic Z, Joyner MJ. Is sympathetic neural vasoconstriction blunted in the vascular bed of exercising human muscle? J Physiol. 2002;541:623–635. doi: 10.1113/jphysiol.2001.014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Teeffelen JW, Segal SS. Interaction between sympathetic nerve activation and muscle fibre contraction in resistance vessels of hamster retractor muscle. J Physiol. 2003;550:563–574. doi: 10.1113/jphysiol.2003.038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JR, Kapoor S. Contribution of endothelium-derived relaxing factor to exercise-induced vasodilation in humans. J Appl Physiol. 1993;75:2740–2744. doi: 10.1152/jappl.1993.75.6.2740. [DOI] [PubMed] [Google Scholar]