

Abstract
Activation of both small-conductance (SKCa) and intermediate-conductance (IKCa) Ca2+-activated K+ channels in endothelial cells leads to vascular smooth muscle hyperpolarization and relaxation in rat mesenteric arteries. The contribution that each endothelial K+ channel type makes to the smooth muscle hyperpolarization is unknown. In the presence of a nitric oxide (NO) synthase inhibitor, ACh evoked endothelium and concentration-dependent smooth muscle hyperpolarization, increasing the resting potential (approx. −53 mV) by around 20 mV at 3 μm. Similar hyperpolarization was evoked with cyclopiazonic acid (10 μm, an inhibitor of sarcoplasmic endoplasmic reticulum calcium ATPase (SERCA)) while 1-EBIO (300 μm, an IKCa activator) only increased the potential by a few millivolts. Hyperpolarization in response to either ACh or CPA was abolished with apamin (50 nm, an SKCa blocker) but was unaltered by 1-[(2-chlorophenyl) diphenylmethyl]-1H-pyrazole (1 μm TRAM-34, an IKCa blocker). During depolarization and contraction in response to phenylephrine (PE), ACh still increased the membrane potential to around −70 mV, but with apamin present the membrane potential only increased just beyond the original resting potential (circa −58 mV). TRAM-34 alone did not affect hyperpolarization to ACh but, in combination with apamin, ACh-evoked hyperpolarization was completely abolished. These data suggest that true endothelium-dependent hyperpolarization of smooth muscle cells in response to ACh is attributable to SKCa channels, whereas IKCa channels play an important role during the ACh-mediated repolarization phase only observed following depolarization.
Endothelium-derived hyperpolarizing factor (EDHF) is a nitric oxide (NO) and prostanoid-independent mechanism, which relaxes vascular smooth muscle cells by evoking hyperpolarization. For a number of reasons, providing an identity for EDHF has proved both problematic and controversial. In part, this is because there may be a number of EDHFs of variable importance throughout the vasculature, and in part because in some vessels smooth muscle hyperpolarization might result from the passive spread of endothelial cell hyperpolarization. The latter is thought to reflect myoendothelial coupling and to operate in parallel or in place of a diffusible EDHF. However, from a physiological standpoint what is clear is that EDHF has a major functional influence on vascular smooth muscle tone in small resistance arteries. Furthermore, it is equally clear that whatever the precise factor or structures responsible for EDHF-evoked vasodilatation, a crucial early step involves potassium channel activation leading to endothelial cell hyperpolarization. A characteristic feature of this step is susceptibility to block with a combination of the K+ channel blockers apamin and charybdotoxin, but not with apamin and iberiotoxin (see Edwards & Weston, 2001; Busse et al. 2002). This reflects a pivotal role for small- and intermediate-conductance calcium-activated potassium channels (SKCa and IKCa channels) in the EDHF response. Both of these K+ channel types have now been shown to be localized on arterial endothelial cells, but are not present on the smooth muscle (Edwards et al. 1998; Doughty et al. 1999; Walker et al. 2001). It is the activation of these K+ channels which causes endothelial cell hyperpolarization leading to smooth muscle hyperpolarization and relaxation.
In spite of this pivotal role for SKCa and IKCa channels, the possibility that each might provide an independent input to the EDHF pathway has received little attention. As recent evidence indicates that quite significant changes in arterial SKCa levels and activity occur, for example, in association with liver cirrohosis and oestrogen deficiency (Barriere et al. 2001; Liu et al. 2001), the importance of defining the individual K+ channel inputs to EDHF-evoked hyperpolarization and relaxation is clear.
Electrophysiological studies generally use apamin and charybdotoxin together to block EDHF hyperpolarization in uncontracted arteries. Interestingly though, in both rabbit and rat mesenteric arteries, apamin alone has been reported to block hyperpolarization to ACh (Murphy & Brayden, 1995; Chen & Cheung, 1997). However, during contraction, maintaining smooth muscle impalements is very difficult, so the effect of blocking either SKCa or IKCa channels individually, on both hyperpolarization and relaxation, has not been clearly demonstrated. A further complication has been the fact that charybdotoxin blocks smooth muscle large-conductance calcium-activated K+ channels (BKCa) and also voltage-dependent K+ channels (KV), in addition to the endothelial IKCa channels. The recent development of selective blocking agents for IKCa channels (Wulff et al. 2000) has enabled assessment of the input of each channel type to EDHF-evoked relaxation, and shown that, individually, SKCa or IKCa appear to be able to support relaxation to a similar extent (Hinton & Langton, 2003). However, it is clearly important to know if the underlying smooth muscle hyperpolarization has been modified in any way, particularly with submaximal relaxation and in light of recent evidence that the intensity of constrictor stimulation modifies the mechanisms responsible for EDHF-relaxation (Dora & Garland, 2001; Richards et al. 2001; Dora et al. 2002).
We have used the recently developed, selective IKCa blocker TRAM-34 (Wulff et al. 2000) and the selective SKCa blocker apamin in both uncontracted and phenylephrine- (PE) stimulated mesenteric arteries. Our aim was to investigate the relative functional importance of SKCa and IKCa channels for smooth muscle repolarization and relaxation in precontracted arteries.
METHODS
Male Wistar rats (200-250 g) were killed by cervical dislocation and exsanguination (schedule 1 procedure, UK Animals (Scientific Procedures) Act 1986). A third order branch of the superior mesenteric artery was cleared of adherent tissue and mounted in Krebs buffer at 37° C in a small vessel myograph (Danish Myotechnology, Denmark), at a tension equivalent to that generated at 0.9 times the diameter of the vessel at 100 mmHg (Garland & McPherson, 1992). Endothelium viability was assessed by the ability of 1 µm ACh to induce > 95 % relaxation of a submaximal contraction to phenylephrine (3 µm). The artery was superfused (3-4 ml min−1) with oxygenated Krebs buffer at 37° C, and flow stopped during experimental recording, when the buffer was maintained at 37° C in the myograph and drugs mixed by continuous gassing with 5 % CO2-95 % O2. The NO synthase inhibitor, Nω-nitro-l-arginine methyl ester (l-NAME, 100 µm) was present throughout all experiments. Smooth muscle cells were impaled with sharp glass electrodes (filled with 2m KCl, tip resistances approximately 80-100 MΩ) and membrane potential and tension were measured simultaneously (Garland & McPherson, 1992)
Solutions and drugs
All experiments used Krebs buffer of the following composition (mm): NaCl 118.0, NaHCO3 25.0, KCl 3.6, MgSO4.7H2O 1.2, KH2PO4 1.2, glucose 11.0, CaCl2 2.5. Drugs were all from Sigma except for apamin (Latoxan), 1-ethyl-2-benzimidazolinone (1-EBIO, Aldrich) and 1-[(2-chlorophenyl) diphenylmethyl]-1H-pyrazole (TRAM-34, an IKCa blocker, a gift from Dr H. Wulff, University of California Irvine, USA). 1-EBIO, cyclopiazonic acid (CPA) and TRAM-34 were each dissolved in DMSO and then diluted. Preliminary experiments indicated that the DMSO vehicle control had no effect. All other stock solutions were prepared using distilled water.
Data analysis
Results are summarized as means ± s.e.m. of n replicates. Raw data were compared using one-way analysis of variance, and changes in membrane potential and percentage relaxation were compared using the Mann-Whitney test. P < 0.05 was considered statistically significant.
RESULTS
Membrane potential changes in uncontracted arteries
In the presence of l-NAME, ACh evoked reproducible, concentration- and endothelium-dependent smooth muscle hyperpolarization in arteries of 200-300 µm (o.d.). From a threshold concentration of ≈30 nm, ACh increased resting potential from -51.7 ± 0.6 mV to a maximum of -74.6 ± 0.9 mV (with 3 µm ACh), equivalent to an increase of 22.7 ± 1.1 mV (n = 25, Fig. 1).
Figure 1. Effect of apamin or TRAM-34 on smooth muscle hyperpolarization in response to ACh.
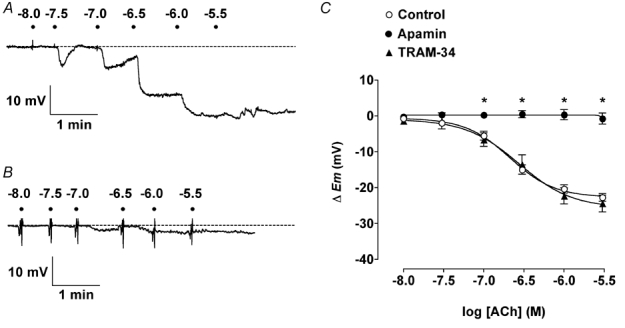
A and B, representative original traces show ACh cumulative concentration-increases in smooth muscle cell membrane potential in rat isolated mesenteric arteries. Each dot represents the addition of ACh (log molar concentration). A, in the presence of 100 µml-NAME (Control in C) the impaled cell hyperpolarized from a resting membrane potential (rmp) of − 53.5 mV (dashed line) to a maximum of − 78.1 mV. In the presence of 50 nm apamin (B) the rmp (−52.6 mV, dashed line) was not significantly increased. C, summarized data showing the average change in membrane potential (▵Em) to cumulative increases in [ACh] under control conditions (rmp = −51.7 ± 0.6 mV, n = 17–25), and in the presence of 50 nm apamin (rmp = −52.8 ± 1.0 mV, n = 5) or 1 µm TRAM-34 (rmp = −51.1 ± 1.0 mV, n = 6–7). Apamin alone was fully able to abolish hyperpolarization to ACh, whereas TRAM-34 had no effect. * P < 0.05vs. control.
Hyperpolarization of a similar maximum amplitude was evoked with 10 µm CPA (−53.7 ± 0.8 mV to -70.6 ± 1.7 mV, an increase of 16.9 ± 1.7 mV, n = 9, Fig. 2), but only a very small hyperpolarization was recorded in response to 300 µm 1-EBIO, an activator of IKCa channels (4.3 ± 1.2 mV increase to -55.3 ± 1.3 mV, n = 7).
Figure 2. Effect of apamin or TRAM-34 on smooth muscle hyperpolarization in response to CPA.
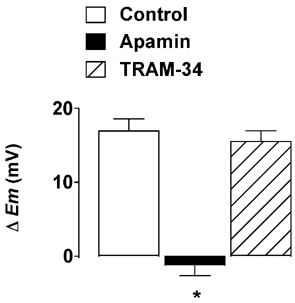
Under control conditions, cyclopiazonic acid (CPA, 10 µm) hyperpolarized smooth muscle cells by an average of 16.9 ± 1.7 mV (n = 9). This hyperpolarization was abolished in the presence of 50 nm apamin (n = 5), but unaffected by 1 µm TRAM-34 (n = 3). * P < 0.05vs. control. ▵Em, change in membrane potential.
Neither apamin nor TRAM-34 alone altered the smooth muscle resting potential (apamin: -52.8 ± 1.0 mV, n = 5; TRAM-34: -51.1 ± 1.0 mV, n = 7). However, hyperpolarization in response to either ACh or CPA was abolished in the presence of apamin (50 nm), but unaltered with 1 µm TRAM-34 (ACh maximum increase: 24.4 ± 2.4 mV, n = 7; CPA maximum increase: 15.5 ± 1.5 mV, n = 3; Fig. 1 and Fig. 2) or 10 µm ryanodine (ACh maximum increase: 20.8 ± 1.7 mV, n = 3).
Membrane potential and tension changes in the presence of phenylephrine
PE (0.6 µm) provoked sustained smooth muscle depolarization (to -40.0 ± 1.1 mV) and contraction (to 15.9 ± 0.9 mN, n = 15), often with the generation of spontaneous, depolarizing spike discharges. ACh (3 µm) hyperpolarized the membrane potential to -70.7 ± 0.8 mV and caused a simultaneous relaxation of 96.7 ± 0.5 % (n = 15). In the presence of 50 nm apamin, the membrane potential and contraction with PE were -40.8 ± 1.8 mV and 11.7 ± 0.5 mN (n = 5), and the hyperpolarization to ACh was significantly suppressed, such that the membrane potential now only attained -58.8 ± 1.5 mV. This increase in potential was associated with a maximum relaxation of 89.3 ± 2.4 % (n = 5; Fig. 3A and Fig. 3C). In contrast, 1 µm TRAM-34, did not block the ability of ACh-hyperpolarization to overshoot the original membrane potential. The membrane potential and contraction with PE were -35.4 ± 1.8 mV and 13.0 ± 1.0 mN (n = 5), respectively, in this series. ACh increased the potential to -68.7 ± 1.0 mV associated with 96.4 ± 1.5 % relaxation (n = 5, Fig. 3C). In contrast, when apamin and TRAM-34 were present in combination (membrane potential and contraction to PE of -37.5 ± 2.1 mV and 14.2 ± 1.3 mN, n = 3), both hyperpolarization and relaxation was abolished (Fig. 3B and Fig. 3C), and a slight depolarization and contraction recorded (to -36.9 ± 1.9 mV from -37.4 ± 2.1 mV and -13.6 ± 12.7 % of maximum relaxation, n = 3, respectively) However, marked hyperpolarization (to -78.6 ± 1.4 mV) and relaxation (93.5 ± 0.6 % of maximum, n = 3) could then be stimulated by the addition of the KATP channel activator, 3 µm levcromakalim (Fig. 3B). 300 µm 1-EBIO, which did not hyperpolarize uncontracted arteries by more than a few millivolts, now reversed the depolarization to PE, increasing the membrane potential from -37.2 ± 3.9 mV to -56.9 ± 0.7 mV (original resting membrane potential (rmp) -51.2 ± 1.7 mV), associated with 97.5 ± 0.7 % relaxation (n = 3).
Figure 3. Effect of apamin and TRAM-34 on smooth muscle hyperpolarization in response to ACh in the presence of phenylephrine.
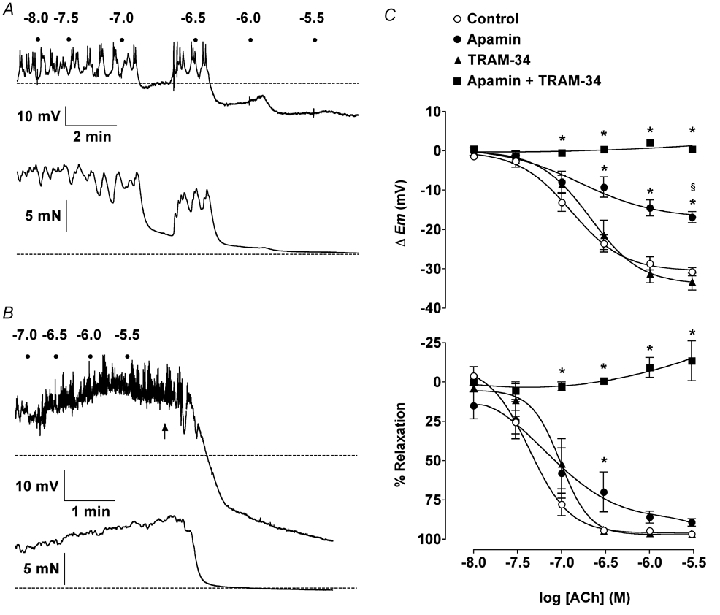
A and B, representative records show simultaneous smooth muscle cell hyperpolarization and relaxation in response to ACh in rat isolated mesenteric arteries contracted with phenylephrine. Dots represent addition of ACh (log molar concentrations). A, in the presence of 100 µml-NAME (Control) 0.6 µm phenylephrine caused depolarization from −52.5 mV (rmp, dashed line) to − 44.6 mV and contraction from 2.3 mN (resting tension, dashed line) to 16.0 mN. ACh stimulated hyperpolarization to levels beyond rmp (maximum −70.1 mV), associated with almost complete relaxation. B, in the combined presence of 50 nm apamin and 1 µm TRAM-34, 1.3 µm phenylephrine stimulated depolarization from −51.5 mV to −39.3 mV and contraction from 3.2 mN to 15.4 mN. Under these conditions, ACh was unable to stimulate hyperpolarization or relaxation, but the subsequent addition of 3 µm levcromakalim (indicated by arrow) was able to stimulate hyperpolarization (to − 81.3 mV) and relaxation. C, summarized data showing the average change in smooth muscle membrane potential (▵Em, top panel) and tension (% Relaxation, bottom panel) to cumulative increases in [ACh]. TRAM-34 had no effect on ACh responses, whereas apamin prevented hyperpolarization beyond rmp with slightly reduced relaxation. The combination of apamin plus TRAM-34 abolished hyperpolarization or relaxation to ACh.
Thus, ACh increased the membrane potential to a similar value whether or not the arteries were contracted to PE, while apamin abolished any significant increase beyond the level of the normal resting potential (Fig. 4).
Figure 4. Effect of apamin on smooth muscle hyperpolarization in response to ACh.
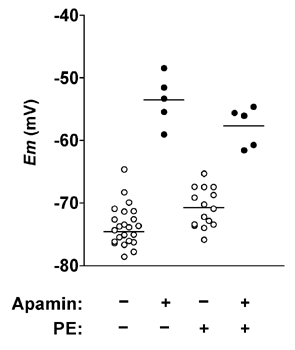
Scatter of individual impalements, showing the maximum steady-state membrane potential in response to 3 µm ACh with or without 50 nm apamin present, and in both uncontracted arteries or arteries contracted with phenylephrine (PE). Each horizontal line represents the mean for that data set. Membrane potentials achieved with ACh were independent of resting potential in the absence but not the presence of apamin.
DISCUSSION
The activation of both SKCa and IKCa channels in the arterial endothelium is a key step leading to the smooth muscle hyperpolarization and relaxation ascribed to EDHF (Bolz et al. 1999; Busse et al. 2002). Our data indicate that EDHF hyperpolarization in non-contracting arteries (true hyperpolarization) can be explained solely by the activation of SKCa channels, while during smooth muscle depolarization and contraction with PE, EDHF hyperpolarization can be separated into two components, reflecting separate input from SKCa and IKCa channels. In the presence of TRAM-34 to block IKCa channels, EDHF hyperpolarization increases the membrane potential to the same level as it does in the absence of PE. The depolarization is reversed and membrane potential overshoots resting levels to approach the equilibrium potential for potassium, associated with 100 % relaxation. With apamin present to block SKCa channels, EDHF hyperpolarization only increases the membrane potential back to the original resting potential (repolarization), but this change is still sufficient to evoke complete relaxation. So the ability of SKCa channels to drive membrane potential beyond resting values may indicate a distinct physiological role for this component of the EDHF response, perhaps analogous to the after-hyperpolarization in neurones.
CPA evoked endothelium-dependent hyperpolarization of similar magnitude to the EDHF-hyperpolarization with ACh. In both cases, hyperpolarization was blocked by apamin but unaffected by TRAM-34, indicating a selective activation of SKCa channels. Interestingly, neither apamin nor TRAM-34 alone caused depolarization or contraction in the wire-mounted mesenteric arteries, indicating that the basal levels of Ca2+ are not sufficient to activate either SKCa or IKCa in these vessels. The concentration of TRAM-34 used in the present study has been shown selectively to block IKCa currents in dispersed rat carotid endothelial cells, with higher concentrations not affecting either BKCa, Kv or ATP-sensitive (KATP) currents (Eichler et al. 2003). The selectivities of both TRAM-34 and apamin, together with the inability of the IKCa activator 1-EBIO to stimulate more than a few millivolts of hyperpolarization, provide data which are consistent with a predominant role for SKCa channel activity in the EDHF response in uncontracted arteries. Of course, 1-EBIO does have other actions apart from activating IKCa, so for example in the mesenteric artery it stimulates smooth muscle relaxation by an undefined mechanism in far lower concentrations than those required for hyperpolarization. However, the endothelium-dependent hyperpolarization that is evoked with 1-EBIO (>100 µm) in this artery appears only to reflect activation of IKCa (Walker et al. 2001).
Overall, these data are supported by previous reports that apamin alone can block ACh evoked hyperpolarization in rabbit and rat mesenteric arteries (Murphy & Brayden, 1995; Chen & Cheung, 1997). In the latter study, the rat superior mesenteric artery was used and an attempt was made to unravel the relative importance of apamin- and charybdotoxin-sensitive channels in contracted arteries. However, the overall magnitude of hyperpolarization to ACh only increased by a couple of millivolts during contraction, probably because the EDHF response in the superior mesenteric artery is much less than in the smaller resistance size arteries we used (Hwa et al. 1994). This, together with the use of charybdotoxin, meant that no clear conclusion could be made with regard to the relative importance of the different types of K+ channel (Chen & Cheung, 1997). In fact, sensitivity to charybdotoxin was interpreted to indicate a role for BKCa channels, even though by that time EDHF responses were known to be resistant to block with iberiotoxin (Zygmunt & Högestatt, 1996).
When mesenteric arteries were contracted with PE, separate and selective block of IKCa (with TRAM-34) or SKCa (with apamin) did not alter the ability of EDHF fully to relax the arteries (Hinton & Langton, 2003). However, we now show that smooth muscle hyperpolarization is altered in this artery by a similar pharmacological manipulation. Although TRAM-34 did not modify hyperpolarization or relaxation, the former was attenuated by around 20 mV with apamin. These observations raise the possibility that each channel type may serve a separate physiological role, particularly if they are activated individually. In neurones, SKCa channel activation underlies the overshoot of resting potential, or medium and slow after-hyperpolarization, which slows the rate of action potential firing (Kohler et al. 1996; Hosseini et al. 2001). This raises the possibility that SKCa channels may perform an equivalent role in the mesenteric artery, by ‘buffering’ excitation. An ability to operate around and in excess of the resting membrane potential will mean that hyperpolarization can be sustained as long as the intracellular calcium concentration ([Ca]i) is sufficiently high. Smooth muscle membrane potentials in pressurized mesenteric arteries are between 5-8 mV less then in wire-mounted vessels of similar size (Wesselman et al. 1997), allowing for more hyperpolarization. In smaller mesenteric arteries, which develop myogenic tone, this mechanism may then provide a key negative control on decreases in artery diameter. This suggestion is supported by very recent data from a transgenic mouse (SK3T/T), which has elevated SK3 in the arterial endothelium (three subtypes of SKCa, SK1, SK2 and SK3, have been identified; Kohler et al. 1996). In isolated pressurized mesenteric arteries, the SK3 channels were responsible for a sustained smooth muscle hyperpolarization, such that enhanced PE or pressure-induced constriction was obtained when apamin was applied (Taylor et al. 2003). The apparent inability of IKCa to increase the membrane potential much above -55 to - 60 mV, suggests that these channels will only provide a significant input to the EDHF response when the smooth muscle cells are depolarized, for example by sympathetic nerve stimulation.
Two important questions then arise, how could endothelial cell SKCa and IKCa channels be activated independently and why are the IKCa channels unable to develop as much hyperpolarization as the SKCa channels? As neither channel type displays any voltage sensitivity, the answer in both cases is likely to relate, at least in part, to [Ca2+]i. Although the concentrations of Ca2+ required for half maximum activation of SK1 and SK2 are 456-700 nm (Kohler et al. 1996; Pedarzani et al. 2001), the channels in endothelial cells are almost certainly SK3 (Burnham et al. 2002; Eichler et al. 2003). SK3 channels are much more sensitive to [Ca2+]i, and have a similar apparent EC50 to IKCa (104.2 nm, Carignani et al. 2002). This similarity suggests that any differential activation is likely to reflect distinct subcellular localization within endothelial cells.
As CPA can mimic the EDHF hyperpolarization, it is unlikely that a close or restricted relationship between internal calcium release sites and the SKCa channels is the explanation. It is more likely that the Ca2+ influx channel is closely associated with SKCa channels. Although the subcellular topography of SKCa and IKCa channels versus Ca2+ entry sites in endothelial cells is unknown, close coupling does exist with SKCa channels in other cells. In hippocampal neurones, Ca2+ entry through L-type voltage-dependent Ca2+ channels (VDCC) links selectively to SKCa activation, with the delay between opening of the different channels indicating a separation of 50-150 nm (Marrion & Tavalin, 1998). A similar profile was observed in urinary bladder smooth muscle cells, where VDCC were thought to associate closely with SKCa channels and global rises in [Ca2+]i, whereas Ca2+ release through ryanodine receptors selectively activated large-conductance Ca2+-activated K+ channels, and not SKCa (Herrera & Nelson, 2002). This relationship does not always exist, as in both colonic smooth muscle cells and some hippocampal neurones, the release of Ca2+ from ryanodine-sensitive stores can also activate SKCa (Torres et al. 1996; Koh et al. 1997). The lack of block with ryanodine in the present study suggests this to be an unlikely mechanism in the mesenteric artery endothelial cells. However, this does not preclude activation by InsP3-mediated Ca2+ release from the endoplasmic reticulum. Indeed, spontaneous, ryanodine-insensitive focal release of Ca2+ from internal stores can occur in vascular endothelial cells (Burdyga et al. 2003). Further, although VDCC are absent from endothelial cells, the channels responsible for Ca2+ influx associated with store depletion could clearly activate SKCa. But while either of these channels could potentially provide local communication with SKCa, thus explaining a lack of IKCa activation at rest, high concentrations of ACh have been shown to increase endothelial cell Ca2+ fairly uniformly in uncontracted, superior mesenteric arteries (Oishi et al. 2001). Interestingly, Oishi et al. (2001) found that direct smooth muscle stimulation with PE indirectly increased Ca2+ in discrete clusters of endothelial cells. Presumably, due to Ca2+ or InsP3 flow through myoendothelial gap junctions, as in arterioles (Dora et al. 1997). So it may be that IKCa channels are localized near myoendothelial gap junctions, and effective inflow of Ca2+ must accrete with endothelial cell [Ca2+]i in order to activate IKCa.
Whatever the precise explanation, endothelial cell stimulation with ACh evokes EDHF hyperpolarization of the resting membrane potential which appears to be mediated entirely by the activation of endothelial cell SKCa channels, probably SK3 channels. When the vascular smooth muscle is depolarized and contracted, both SKCa and IKCa channels can contribute to hyperpolarization and relaxation, although the IKCa channels are not able to increase the membrane potential beyond resting levels, presumably reflecting the [Ca2+] in their microdomain. These individual characteristics may reflect different cellular locations for each channel type, and may link to defined physiological roles.
Acknowledgments
This work was supported by the Wellcome Trust. We are very grateful to Dr Henkie Wulff, University of California Irvine, USA for the gift of TRAM-34.
REFERENCES
- Barriere E, Tazi KA, Pessione F, Heller J, Poirel O, Lebrec D, Moreau R. Role of small-conductance Ca2+-dependent K+ channels in in vitro nitric oxide-mediated aortic hyporeactivity to alpha-adrenergic vasoconstriction in rats with cirrhosis. J Hepatol. 2001;35:350–357. doi: 10.1016/s0168-8278(01)00141-6. [DOI] [PubMed] [Google Scholar]
- Bolz SS. Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acetylcholine-induced dilation of microvessels. Br J Pharmacol. 1999;128:124–134. doi: 10.1038/sj.bjp.0702775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga T, Shmygol A, Eisner DA, Wray S. A new technique for simultaneous and in situ measurements of Ca2+ signals in arteriolar smooth muscle and endothelial cells. Cell Calcium. 2003;34:27–33. doi: 10.1016/s0143-4160(03)00019-8. [DOI] [PubMed] [Google Scholar]
- Burnham MP, Bychkov R, Feletou M, Richards GR, Vanhoutte PM, Weston AH, Edwards G. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol. 2002;135:1133–1143. doi: 10.1038/sj.bjp.0704551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- Carignani C, Roncarati R, Rimini R, Terstappen GC. Pharmacological and molecular characterisation of SK3 channels in the TE671 human medulloblastoma cell line. Brain Res. 2002;939:11–18. doi: 10.1016/s0006-8993(02)02535-0. [DOI] [PubMed] [Google Scholar]
- Chen GF, Cheung DW. Effect of K+-channel blockers on ACh-induced hyperpolarization and relaxation in mesenteric arteries. Am J Physiol Heart Circ Physiol. 1997;272:H2306–2312. doi: 10.1152/ajpheart.1997.272.5.H2306. [DOI] [PubMed] [Google Scholar]
- Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci U S A. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Garland CJ. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol. 2001;280:H2424–2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- Dora KA, Ings NT, Garland CJ. KCa channel blockers reveal hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol. 2002;283:H606–614. doi: 10.1152/ajpheart.01016.2001. [DOI] [PubMed] [Google Scholar]
- Doughty JM, Plane F, Langton PD. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am J Physiol Heart Circ Physiol. 1999;276:H1107–1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Edwards G, Weston AH. EDHF-are there gaps in the pathway? J Physiol. 2001;531:299. doi: 10.1111/j.1469-7793.2001.0299i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J, Kohler R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol. 2003;138:594–601. doi: 10.1038/sj.bjp.0705075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, McPherson GA. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br J Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera GM, Nelson MT. Differential regulation of SK and BK channels by Ca2+ signals from Ca2+ channels and ryanodine receptors in guinea-pig urinary bladder myocytes. J Physiol. 2002;541:483–492. doi: 10.1113/jphysiol.2002.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton JM, Langton PD. Inhibition of EDHF by two new combinations of K+-channel inhibitors in rat isolated mesenteric arteries. Br J Pharmacol. 2003;138:1031–1035. doi: 10.1038/sj.bjp.0705171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini R, Benton DC, Dunn PM, Jenkinson DH, Moss GW. SK3 is an important component of K+ channels mediating the afterhyperpolarization in cultured rat SCG neurones. J Physiol. 2001;535:323–334. doi: 10.1111/j.1469-7793.2001.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa JJ, Ghibaudi L, Williams P, Chatterjee M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. Am J Physiol Heart Circ Physiol. 1994;266:H952–958. doi: 10.1152/ajpheart.1994.266.3.H952. [DOI] [PubMed] [Google Scholar]
- Koh SD, Dick GM, Sanders KM. Small-conductance Ca2+-dependent K+ channels activated by ATP in murine colonic smooth muscle. Am J Physiol Cell Physiol. 1997;273:C2010–2021. doi: 10.1152/ajpcell.1997.273.6.C2010. [DOI] [PubMed] [Google Scholar]
- Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Liu MY, Hattori Y, Fukao M, Sato A, Sakuma I, Kanno M. Alterations in EDHF-mediated hyperpolarization and relaxation in mesenteric arteries of female rats in long-term deficiency of oestrogen and during oestrus cycle. Br J Pharmacol. 2001;132:1035–1046. doi: 10.1038/sj.bjp.0703899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Murphy ME, Brayden JE. Apamin-sensitive K+ channels mediate an endothelium-dependent hyperpolarization in rabbit mesenteric arteries. J Physiol. 1995;489:723–734. doi: 10.1113/jphysiol.1995.sp021086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi H, Budel S, Schuster A, Stergiopulos N, Meister J, Beny J. Cytosolic-free calcium in smooth-muscle and endothelial cells in an intact arterial wall from rat mesenteric artery in vitro. Cell Calcium. 2001;30:261–267. doi: 10.1054/ceca.2001.0233. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, Stocker M, Adelman JP, Fakler B. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J Biol Chem. 2001;276:9762–9769. doi: 10.1074/jbc.M010001200. [DOI] [PubMed] [Google Scholar]
- Richards GR, Weston AH, Burnham MP, Feletou M, Vanhoutte PM, Edwards G. Suppression of K+-induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium-derived hyperpolarizing factor. Br J Pharmacol. 2001;134:1–5. doi: 10.1038/sj.bjp.0704256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- Torres GE, Arfken CL, Andrade R. 5-Hydroxytryptamine4 receptors reduce afterhyperpolarization in hippocampus by inhibiting calcium-induced calcium release. Mol Pharmacol. 1996;50:1316–1322. [PubMed] [Google Scholar]
- Walker SD, Dora KA, Ings NT, Crane GJ, Garland CJ. Activation of endothelial cell IKCa and 1-ethyl-2-benzimidazolinone evokes smooth muscle hyperpolarization in rat isolated mesenteric artery. Br J Pharmacol. 2001;134:1548–1554. doi: 10.1038/sj.bjp.0704415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselman JPM, Schubert R, Vanbavel E, Nilsson H, Mulvany MJ. KCa-channel blockade prevents sustained pressure-induced depolarization in rat mesenteric small arteries. Am J Physiol Heart Circ Physiol. 1997;272:H2241–2249. doi: 10.1152/ajpheart.1997.272.5.H2241. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Högestatt ED. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br J Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]