

Abstract
We examined the effects of spermine binding to aspartate at site 172 on the accessibility of internal trimethylammonioethylmethane thiosulphonate (MTSET) to substituted cysteines within the pore of a Kir2.1 channel. Spermine prevented MTSET modification in Q164C and G168C mutants, indicating that sites 164 and 168 are located externally to the spermine binding site. The rates of MTSET modification were significantly reduced by spermine in I176C mutants, indicating that site 176 is located internally to D172 and that the bound spermine hinders the reaction of MTSET with cysteine at site 176. Spermidine, putrescine and Mg2+ also decreased MTSET modification at site 176. The order of effect is putrescine > spermidine ≈ spermine ≈ Mg2+. To account for the electrostatic and physical repulsion between MTSET and polyamines, possible locations of polyamines in the pore are discussed. In D172C mutants, the spermine that bound to sites 224 and 299 completely inhibited channels at +40 mV, yet MTSET remained accessible to site 172. In addition, in the D172C mutant, spermine did not affect the exit rate of Ba2+ bound to the threonine at the site 141. These results indicate that spermine bound at the cytoplasmic pore induces channel closure at positions 141–172. The effects of spermine on the accessibility of amino acids in the pore may shed light on the structural and functional relationships of the Kir2.1 channels during inward rectification.
Inward rectifier K+ channels (Kir) conduct inward currents at potentials more negative than the reversal potential of K+, but permit much smaller currents at potentials positive than the reversal potential (Hille, 2001). Under physiological conditions, the mechanism underlying this inward rectification is the voltage (membrane potential, Vm)-dependent blockade of outward K+ currents by both intracellular Mg2+ (Matsuda et al. 1987; Vandenberg, 1987) and polyamines binding to the aspartate at site 172 (D172; Lu & MacKinnon, 1994; Stanfield et al. 1994; Wible et al. 1994), the glutamate at site 224 (E224; Taglialatela et al. 1995; Yang et al. 1995) and the glutamate at site 299 (E299; Kubo & Murata, 2001). D172 has been shown to provide a strong energetic contribution to spermine and Mg2+ binding in the pore of the Kir2.1 channel (Lopatin et al. 1994; Stanfield et al. 1994; Wible et al. 1994) and thus it is generally considered to be a binding site, or to be located close to the binding site for these internal blockers. However, it is unclear to what extent a large molecule such as spermine occupies space within the pore.
Kir channels are integral membrane proteins that consist of two transmembrane segments (M1 and M2) flanking a pore-forming loop (P), and N- and C-terminal cytoplasmic domains (Fig. 1A). Recently, it has been shown that the M2 segment of a prokaryotic inward rectifier K+ channel, KirBac1.1, is a helix, as determined by X-ray crystallography (Kuo et al. 2003). Site-directed mutagenesis and cysteine scanning studies, have also shown that the M2 segments of several eukaryotic Kir channels are also helical (Choe et al. 1995; Minor et al. 1999; Loussouarn et al. 2000). However, other studies show that the sequences in the M2 segment do not have obvious periodicity corresponding to α helices or β strands (Collins et al. 1997; Lu et al. 1999). Scanning cysteine accessibility studies have revealed that several amino acids in the M2 segment of the cloned Kir2.1 channel line the inner pore of the channel. According to the secondary structure (Kubo et al. 1993), the amino end of the M2 segment is located externally to the carboxyl end. Yet, the relative positions of the amino acids of the M2 segment during a functional state remain unknown.
Figure 1. Schematic plot of a Kir2.1 subunit.

A, secondary structure of a Kir2.1 subunit. B, amino acid sequence of the M2 segment of the Kir2.1 subunit. Asterisks mark the residues that were mutated into cysteines and were accessible to MTSET.
In this study we employed the spermine bound at D172 to determine the relative positions of the pore-lining residues of the M2 segment (Fig. 1B). We rationalized that if amino acids are located externally to the spermine bound to D172, then their accessibility to internally applied trimethylammonioethylmethane thiosulphonate (MTSET) will be abolished by spermine. On the other hand, for those amino acids positioned internally to the spermine bound to D172 and located far enough from it, their accessibility to MTSET will remain unaffected by spermine. In addition, since spermine occupies a range of spaces and carries charges, the accessibility of MTSET to the amino acids adjacent to the spermine binding site will be somewhat reduced, through physical and/or electrostatic repulsion. In addition to D172, spermine also induces inward rectification through interaction with E224 and E299. Therefore, we also examined the effects of spermine on the accessibility of cysteine residues introduced in the channels with a neutral residue (asparagine or cysteine) replacing the aspartate at site 172. We provide evidence that the inward rectification induced by the spermine bound to E224 and E299 is not due to a direct occlusion of the cytoplasmic pore in the Kir2.1 channel. Possible mechanisms underlying the involvement of E224 and E299 in inward rectification are discussed.
METHODS
Molecular biology and preparation of Xenopus oocytes
Site-directed mutations were generated in a methanethiosulphonate-insensitive channel, IRK1J (C54V, C76V, C89I, C101L, C149F and C169V; Lu et al. 1999) using ‘Altered Sites II: in vitro Mutagenesis Systems’ (Promega, Madison, WI, USA). cRNA was obtained by in vitro T7 transcription (mmessage mMachine, Ambion, Dallas, TX, USA). Xenopus oocytes were isolated by partial ovariectomy from frogs anaesthetized with 0.1 % tricaine (3-aminobenzoic acid ethyl ester). The incision was sutured and the animals were monitored during the recovery period before being returned to their tank. Following the last oocyte collection, frogs were anaesthetized as described and sacrificed by decapitation. All surgical and anaesthetic procedures were reviewed and approved by institutional animal use committees. Oocytes were maintained at 18 °C in Barth's solution containing (mm): NaCl (88), KCl (1), NaHCO3 (2.4), CaN2O6 (0.3), CaCl2 (0.41), MgSO4 (0.82) and Hepes (15), pH 7.6, with gentamicin (20 µg ml−1). Oocytes were used 1-3 days after cRNA injection.
Electrophysiology
Currents were recorded at room temperature (23-25 °C) using the giant patch-clamp technique (Hamill et al. 1981; Hilgemann, 1995) with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA). The resistance of the electrode pipette ranged from 0.15 to 0.25 MΩ when filled with the internal solution. The internal and external solutions contained (mm): KCl + KOH (100), EDTA (5) and Hepes (5), pH 7.4. In the Mg2+ experiments, the internal solution contained (mm): KCl + KOH (100), EDTA (5), MgCl2 (6) and Hepes (5), pH 7.4. In the Ba2+ experiments the external solution contained (mm): KCl + KOH (100), BaCl2 (0.01) and Hepes (5), pH 7.4. MTSET (Toronto Research Chemicals, North York, Ontario, Canada) was stored at -20 °C and dissolved immediately before application. The rundown of channel activity was delayed by treating inside-out patches with l-α-phosphatidylinositol-4,5-bisphosphate (Sigma, St. Louis, MO, USA; Huang et al. 1998; Shieh et al. 1998).
The command Vm and data-acquisition functions were processed using a Pentium-based personal computer, a DigiData board and pCLAMP6 software (Axon Instruments). In MTSET-modification experiments, currents were recorded with a 20 ms voltage pulse to -140 mV from a holding potential of + 40 mV at 0.2-2 Hz. Using such a protocol, the channels were held at +40 mV (to facilitate the interaction of the channel with spermine) for 96-99.6 % of the total recording time. In Ba2+-recovery experiments, a two-pulse protocol was elicited. The fraction of channel blocked by 10 µm Ba2+ at -120 mV was tested by the first pulse, whereas the fraction of channels that recovered from blockade after a given time interval at Vm = +40 mV was recorded by the second pulse. Data were sampled at 10 kHz and filtered at 2 kHz with an eight-pole, low-pass filter (Frequency Devices, Rochester, NY, USA). Capacitive currents were corrected using the capacitance neutralization function of the Axopatch 200A amplifier. The series resistance was about 0.3 MΩ, as evaluated by the ratio of input capacitance to the time constant of current relaxation in the current transient generated by a voltage step (Marty & Neher, 1995). The voltage-clamp error was 6 mV at its maximum (20 nA × 0.3 MΩ) and was mostly less than 3 mV. This uncompensated clamp error did not affect the conclusions of this study.
Data analysis
In MTSET experiments, current amplitude was measured at the end of the voltage step to -140 mV. The time courses of current inhibition by MTSET followed a single-exponential decay, except for the V169C mutant, for which the time course followed a double-exponential decay. Time constants for MTSET modification were obtained by fitting the time courses of current inhibition. The apparent second-order rate constants for MTSET modification were then calculated as the reciprocal of the respective time constant divided by the concentration of MTSET. In Ba2+ experiments, the instantaneous current measured at -120 mV was obtained by fitting the inward current with an exponential function and then extrapolating to the beginning of the pulse.
Results are presented as mean ± s.e.m. Statistical significance was assessed using Student's independent t test.
RESULTS
The effects of spermine on MTSET modification of substituted cysteines at the internal vestibule of Kir2.1 channels
We first examined the effect of channel block by spermine (1 µm) on the rate of MTSET modification in cysteine mutants. Membrane patches were held at +40 mV and stepped to -140 mV for a very short period (20 ms) to monitor channel activity. Using this protocol, the channels were held at +40 mV (to facilitate the entry of spermine into the pore) for > 96 % of the total recording time. Figure 2A shows that in control conditions, MTSET (2 mm) progressively inhibited D172C mutants. Figure 2B shows that 1 µm spermine inhibited the outward current recorded at the holding potential, presumably due to the channel closure effected by spermine binding to E224 and E299 (Yang et al. 1995; Kubo & Murata, 2001). However, MTSET remained capable of irreversibly blocking the D172C mutant. In addition, the rate of MTSET modification was significantly faster as compared to the control (Table 1). The effects of MTSET could not be reversed by washout of MTSET, but was completely reversed by 50 mm 1,4-dithiothreitol, a reducing agent that breaks disulphide bonds (data not shown; Lu et al. 1999). Furthermore, MTSET could only slightly block the pseudo-wild-type channel, IRK1J (Lu et al. 1999). These results suggest that the inhibition of channel activity by MTSET is due to the reaction of MTSET with the cysteine at site 172 (172C).
Figure 2. Effects of spermine on MTSET modification in the D172C mutant.
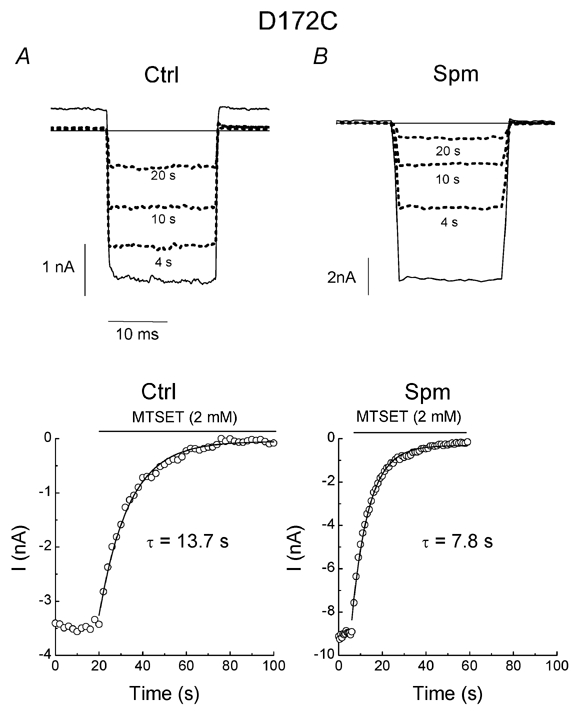
A and B, current traces in the absence and (solid lines) and presence (dotted lines) of MTSET (2 mm) mutant in the control (Ctrl) and 1 µm spermine (Spm), respectively. The number of seconds below each dotted line indicates the time after MTSET treatment. The time courses of MTSET modification in the control and spermine are shown in the lower panels. The horizontal lines indicate the zero current levels.
Table 1.
The rates of MTSET modification in various cysteine mutants (s−1 M−1)
| Q164 | Q164C(−40mV) | Q164C/D172N | G168C | V169C | I171C | D172C | I176C | |
|---|---|---|---|---|---|---|---|---|
| Ctrl | 221 ± 46 (fast) | |||||||
| 22.0 ± 3.3 | 5.2 ± 0.6 | 63.5 ± 5.8 | 18.9 ± 2.4 | 15.7 ± 1.2 (slow) | 5.5 ± 0.5 | 42.4 ± 6.0 | 115.4 ± 1.8 | |
| (n = 12) | (n = 11) | (n = 7) | (n = 13) | (n = 8) | (n = 6) | (n = 5) | (n = 3) | |
| Spm | n.d | 7.7 ± 0.5* | 55.6 ± 6.8 | n.d | 12.8 ± 1.1 | 3.0 ± 0.5* | 65.6 ± 4.9* | 67.0 ± 5.3*** |
| (n = 5) | (n = 5) | (n = 6) | (n = 4) | (n = 4) | (n = 6) | (n =10) | (n = 3) |
Fast indicates the fast component and slow denotes the slow component of MTSET modification with two phases. Ctrl, experiment with control solution, Spin, experiment with spermine solution.
P < 0.05
P < 0.001, n.d. denotes not determined.
Figure 3 illustrates the representative time courses of MTSET (2 mm) modification of the indicated cysteine mutants exposed to the control and 1 µm spermine-added solutions. In Q164C mutants, MTSET progressively inhibited channel activity in the control (Fig. 3A). However, in the presence of 1 µm spermine, MTSET could no longer inhibit the channel. To further confirm that the effect of spermine on MTSET modification in the Q164C mutant is attributed to channel block, we next examined MTSET modification in the Q164C mutant at -40 mV, where spermine does not block the channel. Figure 3B shows the time course of MTSET modification in the Q164C mutant at -40 mV. In the control, the averaged modification rate at -40 mV was slower than that at +40 mV (Table 1), suggesting that MTSET modification is Vm dependent. In 1 µm spermine, MTSET was still able to inhibit the Q164C mutant held at -40 mV. We also carried out experiments using the Q164C/D172N mutant to verify that the effect of spermine is associated with D172. Figure 3C illustrates that in both the control and 1 µm spermine, MTSET progressively inhibited the Q164C/D172N mutant. The rate of MTSET modification in spermine appeared to be slightly slower than that in the control solution (Table 1). These results suggest that when spermine is bound to D172, MTSET can no longer access the cysteine at site 164.
Figure 3. Effects of spermine on MTSET modification in various cysteine mutants.
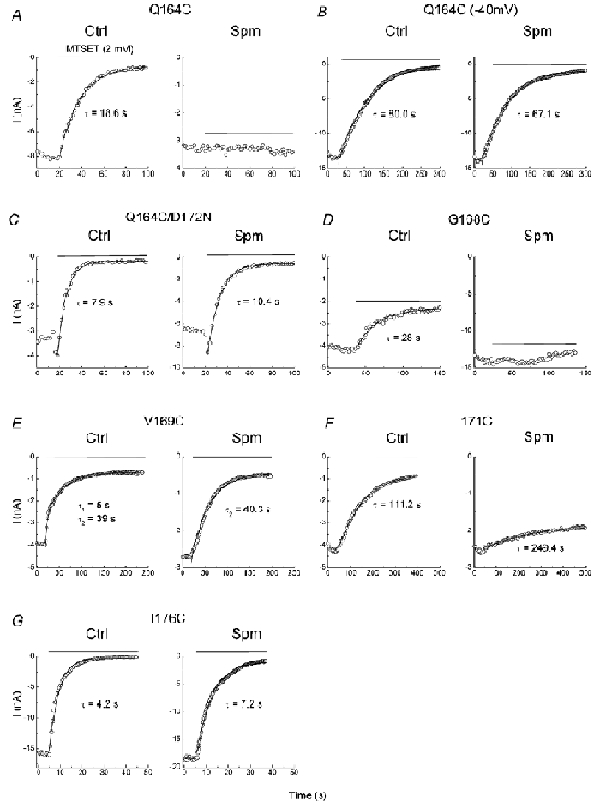
A–G, the time courses of MTSET modification in the indicated cysteine mutants in the control and spermine (1 µm). The horizontal lines indicate the time span of MTSET (2 mm) treatment.
Figure 3D shows that spermine also prevented MTSET from modifying G168C mutants. In V169C mutants, the time course of MTSET modification was best fitted by two rates in the control (Fig. 3E). The fast and slow component made up 33 and 67 %, respectively, of the total modified channel activities. In the presence of 1 µm spermine, MTSET modification was described by one rate, which was similar to the predominantly slow component in the control. Figure 3F demonstrates that spermine decreased both the rate and degree of MTSET modification in I171C mutants. MTSET inhibited I176C mutants at a slower rate in spermine than in the control (Fig. 3G).
Table 1 lists the averaged rates of MTSET modification in various cysteine mutants in the control and with 1 µm spermine. The major findings are summarized as follows. First, after the channels were blocked by spermine, MTSET could no longer access the cysteines at positions 164 and 168, indicating that these sites are located externally to the spermine bound to D172, as predicted by the secondary structure. Second, in the presence of spermine, MTSET could still block the V169C and I171C mutants. Third, in the D172C mutant, in which spermine can no longer bind to the site at 172, internal spermine speeded up the rate of MTSET modification. Fourth, spermine slowed down but did not completely abolish MTSET modification in I176C, suggesting that the spermine bound within the pore hinders the accessibility of cysteine at 176 (176C) to MTSET.
The fast component of MTSET modification in V169C mutant is related to D172
Figure 3E shows that after exposure to spermine, the fast component of MTSET modification disappeared in the V169C mutant. To determine whether D172 is involved in the fast component of the modification, we next examined MTSET modification in the V169C/D172N mutant. Figure 4 shows that regardless of whether spermine was present, MTSET modification was described only by the slow rate. The rate of MTSET modification increased twofold in spermine as compared to the control (Table 2). When spermine is bound to D172 (Fig. 3E) or when D172 is replaced by an asparagine residue (Fig. 4), the fast component of MTSET modification in V169C is abolished, suggesting that the fast component of MTSET modification is related to the positive charge at site 172. It is possible that the fast component is due to MTSET binding to D172, thereby blocking the channel in a reversible way (similar to the action of polyamines). We next examined whether MTSET modification could be washed out. We found that MTSET modification could not be reversed by washout of MTSET, but was reversed by 1,4-dithiothreitol (data not shown), indicating that the block of channels is due to the covalent modification of channels by MTSET. All other cysteine mutants used in this study showed the same reversibility.
Figure 4. Effects of spermine on MTSET modification in the V169C/D172N mutant.
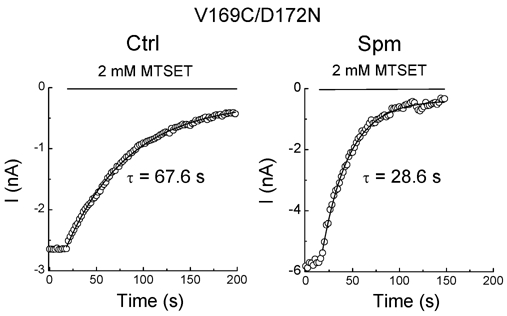
Each panel illustrates the time course of MTSET modification in the control (left panel) and in spermine (right panel).
Table 2.
The rates of MTSET modification in various cysteine mutants (s−1 m−1)
| V169C/D172N | I176C/D172N | D172C/E224G/E299S | I176C/D172N/E224G/E299S | |
|---|---|---|---|---|
| Ctrl | 8.2 ± 0.9 | 22.7 ± 2.3 | 50.5 ± 8.2 | 23.6 ± 1.5 |
| (n = 4) | (n = 3) | (n = 4) | (n = 7) | |
| Spm | 14.7 ± 1.4** | 33.8 ± 3.9* | 62.6 ± 8.3 | 24.1 ± 2.9 |
| (n = 6) | (n = 5) | (n = 5) | (n = 5) |
P < 0.05
P < 0.005.
Spermine bound to D172 hinders the accessibility of MTSET to 176C
Figure 3G shows that the rate of MTSET modification in the I176C mutant is slower in spermine than in the control solution, suggesting that the bound spermine interacts with the accessibility of MTSET to 176C. To further confirm this possibility we examined the rate of MTSET modification in I176C/D172N mutants. Figure 5 demonstrates that the rate of the MTSET modification was increased in the presence of spermine. The average rate was increased by 1.5-fold when the mutant was exposed to spermine as compared to the control (Table 2). These results suggest that the spermine bound to D172 does indeed hinder MTSET modification at 176C. This possibility is further explored in Fig. 7.
Figure 5. Effects of spermine on MTSET modification in the I176C/D172N mutant.
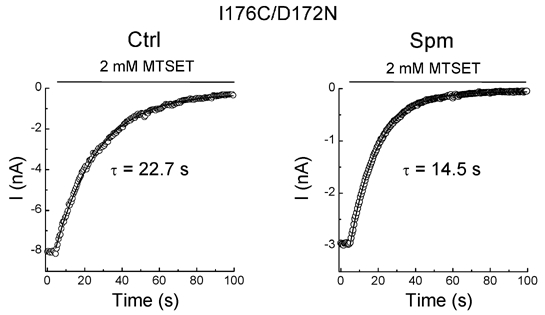
Each panel illustrates the time courses of MTSET modification in the control (left panel) and in spermine (right panel).
Figure 7. Effects of polyamines and Mg2+ on MTSET modification in the I176C/E224G/E299S mutant.
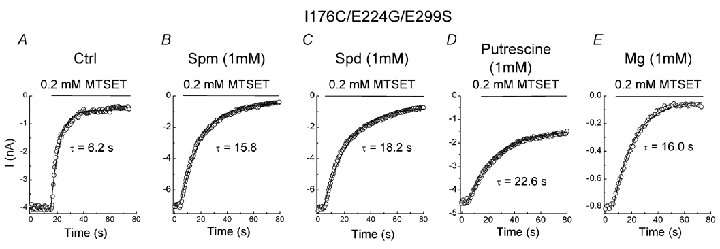
A–E, the time course of MTSET modification in the control solution and the indicated blocker.
E224 and E299 are involved in the effects of spermine accelerating MTSET modification
The results from mutants D172C (Fig. 2), V169C/D172N (Fig. 4) and I176C/D172N (Fig. 5) share the following major common feature. When the site at position 172 could no longer bind to spermine, application of internal spermine enhanced the rates of MTSET modification in these cysteine mutants held at +40 mV. In addition to D172, spermine can also bind to E224 and E299. Therefore, it is likely that E224 and E299 are involved in the accelerating effect of spermine on MTSET modification. We next measured the rates of MTSET modification in mutants D172C/E224G/E299S and I176C/D172N/E224G/E299S. Figure 6 shows that outward currents were recorded in mutants D172C/E224G/E299S (Fig. 6A) and I176C/D172N/E224G/E299S (Fig. 6B) in control solution and in spermine (1 µm). MTSET (2 mm) progressively inhibited the channel activities in both mutants. The corresponding time course of MTSET modification in the control and spermine are shown in the lower panels of Fig. 6A and B. The rates of modification were not statistically different (P > 0.3) between the control and spermine in either mutants D172C/E224G/E299S or I176C/D172N/E224G/E299S (Table 2). The results shown in Figs 2, 4, 5 and 6 suggest that E224 and E299 are indeed involved in the accelerating effect of spermine on MTSET modification.
Figure 6. Effects of spermine on MTSET modification in the D172C/E224G/E299S and I176C/D172N/E224G/E299S mutants.
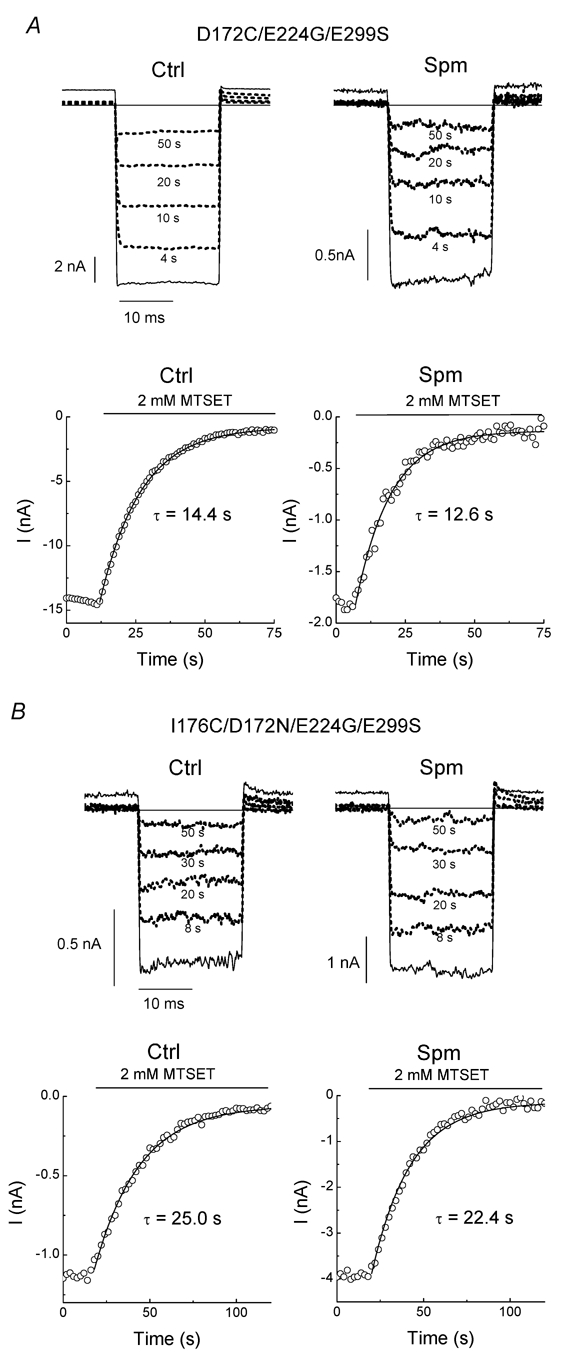
A and B, current traces in the absence and (solid lines) and presence (dotted lines) of MTSET (2 mm) in the D172C/E224G/E299S and I176C/D172N/E224G/E299S mutants, respectively, exposed to the control solution (left panel) and spermine-added solution (right panel). The corresponding time course of MTSET modification is shown below the traces.
Effects of smaller pore blockers on MTSET modification at 176C
We showed that the accelerating effect of spermine involves E224 and E299. To further justify that the spermine bound to D172 interferes with MTSET modification at 176C, we studied MTSET modification in I176C/E224G/E299S mutants. Figure 7A and B shows that spermine slowed down the modification rate of MTSET (0.2 mm) in the I176C/E224G/E299S mutant as compared to the control (Table 3). Since the affinity for spermine in the mutant containing the mutation of E224G and E299S is much reduced (Kubo & Murata, 2001), 1 mm spermine was used to completely block the outward current (presumably by binding to D172). Spermine (1 µm), which could no longer block the outward current, did not significantly (P = 0.19) change the rate of MTSET modification (Table 3). These results suggest that the spermine bound to D172 slowed down MTSET modification at 176C possibly via physical and electrostatic interaction with MTSET.
Table 3.
The effects of various blockers on the rates of MTSET modification (s−1 M−1) in the I176C/E224G/E299S mutant
| Ctrl | Spm (1μM) | Spm (1mM) | Spd (1mM) | Put (1mM) | Mg2+(1mM) |
|---|---|---|---|---|---|
| 714.5 ± 67.8 | 595.6 ± 32.5 | 322.7 ± 20.4** | 281.1 ± 6.3*** | 220.4 ± 15.6*** | 307.9 ± 9.6** |
| (n = 5) | (n = 4) | (n = 4) | (n = 4) | (n = 5) | (n = 4) |
Spd, spermidine; Put, putrescine. *P < 0.05
P < 0.005
P < 0.001.
We next examined the effects of blockers of smaller size and fewer charges on MTSET modification in the I176C/E224G/E299S mutant. Figure 7C shows that 1 mm spermidine decreased MTSET modification to a rate similar to spermine. However, the rate of MTSET modification is slower in putrescine (1 mm) than in spermine and spermidine (Fig. 7D). Finally, 1 mm Mg2+ also decreased MTSET modification (Fig. 7E). These results further support the idea that electrostatic and physical interactions between the blocker and MTSET in the pore are involved in the reducing MTSET modification at 176C.
Probing the closure site during inward rectification induced by spermine bound to E224 and E299
Figure 2B shows that spermine inhibited the D172C mutant at +40 mV. In addition, Fig. 6A shows that spermine could no longer inhibit the current at +40 mV in the D172C/E224G/E299S mutant. These results support the hypothesis that E224 and E299 are involved in the inhibition of the D172C mutant at +40 mV. However, 172C remained accessible to MTSET when the mutants were inhibited by spermine (Fig. 2). Taken together, these results suggest that the closure of the D172C mutant induced by spermine has to occur at a position more external than site 172. It may be that the closure occurs at the narrow pore region. Ba2+ blocks the Kir2.1 channel in the narrow pore (Alagem et al. 2001). If the closure happens in the pore near the selectivity filter, Ba2+ blockade may be affected by spermine. To test this possibility, we next examined the effect of spermine on the exit rate of Ba2+ bound in the D172C mutant.
Figure 8A shows the double-pulse protocol used to assess the recovery from Ba2+ blockade in the D172C mutant. Figure 8B and C illustrates currents recorded in control conditions and in 1 µm spermine, respectively. In the control, outward currents were observed during recovery at Vm = +40 mV. On the other hand, the current at +40 mV was instantaneously inhibited by 1 µm spermine. Figure 8D shows the time courses of recovery. The fractional recovery was calculated as I2/I1. I1 is the instantaneous current of the first pulse minus the steady-state current and I2, that of the second pulse minus the steady-state current. The recovery time course followed a monoexponential function. Since 10 µm external Ba2+ cannot block the D172C mutant at Vm > -20 mV (data not shown), the Ba2+ exit rate (koff) can be calculated as the reciprocal of the recovery time constant. The averaged koff in the control (32.9 ± 2.7 s−1; n = 4) is not significantly different from that in 1 µm spermine (33.8 ± 1.3 s−1; n = 4, P = 0.76). In addition, the channel closure induced by 1 µm spermine at +40 mV did not require the exit of Ba2+ from the pore (see Fig 8C). These results suggest that the channel closure induced by spermine binding to E224 and E299 in the D172C mutant is unlikely to be due to an occlusion at the narrow channel pore.
Figure 8. Effects of spermine on the recovery from Ba2+ block in the D172C mutant.
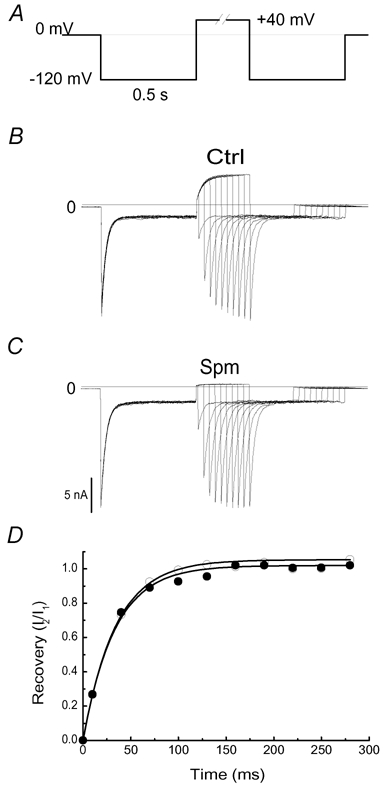
A, the two-pulse voltage protocol. The time interval between the two identical pulses was increased by 15 ms between sweeps (starting with 10 ms) to assess recovery from the block induced by 10 µm extracellular Ba2+. B and C, currents traces recorded in the control and in spermine (1 µm), respectively. D, time courses of recovery from Ba2+ block in the control (•) and 1 µm spermine (○), respectively. Continuous lines are the fit of the data to a monoexponential function.
DISCUSSION
Relative positions of amino acids in the inner pore to D172
In this study we determined the relative positions of the pore-lining residues in the M2 segment of the Kir2.1 channel. We measured the accessibility of MTSET to substituted cysteines in the presence of spermine bound to D172. We found that under these conditions, MTSET can no longer access the cysteines at positions 164 and 168, indicating that these two sites are located external to D172. MTSET is still effective in modifying the V169C and I171C mutants when spermine is bound to D172. However, MTSET modification in the V169C mutant consists of two components, suggesting that complicated processes are involved. In addition, the MTSET modification rate is very slow in the I171C mutant, even in the control, and thus the reaction may not be specific. At present we cannot conclude that sites 169 and 171 are located internally to D172. Different approaches are required to determine further the relative positions of sites 169, 171 and 172. On the other hand, the time course of MTSET modification in the I176C mutant is monoexponential and the rate of modification is relatively high. Spermine slows down, but does not prevent MTSET modification in this mutant. These results suggest that site 176 is located internally to D172 and that these two sites are close enough so that the spermine bound to D172 interferes with the accessibility of MTSET to 176C via physical and electrostatic interaction.
Estimating the binding positions of polyamines in the pore
Spermine decreases the rate of MTSET modification in the I176C and I176C/E224G/E299S mutants but not in the I176C/D172N/E224G/E299S mutant, suggesting that the spermine bound to D172 hinders the accessibility of MTSET to 176C. Spermidine, putrescine and Mg2+ also obstruct the reaction between MTSET and 176C. Although each spermine carries more charges than a Mg2+ ion, the charges are more spread apart in the former molecule. This may explain why Mg2+, a smaller blocker of fewer charges, produces a comparable effect in reducing MTSET modification at 176C. Among the polyamines tested, putrescine carries the fewest charges and is smallest in size, yet it is most effective in slowing MTSET modification at 176C. We suggest that the different effects of various polyamines are attributable to their actual binding positions. Figure 9A shows the molecular structures and dimensions of spermine, spermidine and putrescine. We speculate that all of the polyamines bind to D172 at the second amine, with the first amine located internally. It then appears that the first amine of putrescine protrudes more internally than that of spermine and spermidine, and thus it may assume a position that exerts a stronger physical and electrostatic interaction, with MTSET accessing 176C. The assumption is perhaps reasonable. According to the structure of the MthK channel (Jiang et al. 2002), which is the only K+ channel whose structure has been determined in the open configuration, the space between A58 (T141 in Kir2.1) and F87 (D172 in Kir2.1) is about 12 Å in length. This space is not long enough to fit an entire vertical spermine molecule (Lopatin et al. 1995), which is about 16 Å in length (Fig. 9A). If the second amine of spermine binds to D172, then the rest of spermine (about 11 Å) may fit in and block the channel pore in the deeper position (Fig. 9B). This may also explain why spermine is much more efficient than putrescine in blocking the channel. We only estimated the distance between the backbones instead of side chains because the residues of concern are not conserved between the Kir2.1 and MthK channels. Exactly how polyamines obstruct the accessibility of 176C to MTSET and how they fit into the cavity to produce channel blockade require further structural determination of the Kir2.1 channel.
Figure 9. Proposed interaction of polyamines with the Kir2.1 channel.
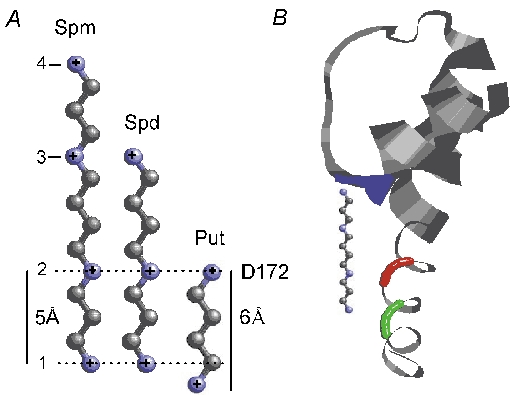
A, the molecular structures and dimensions of polyamines. Atom C is plotted in grey and atom N in blue. The dimensions of polyamines were determined by ChemIDPlus (http://chem.sis.nlm.nih.gov/chemidplus/). B, residues 49-98 of one MthK subunit. The colour codes are: blue, A58 (T141); red F87 (D172); green, V91 (I176). The structure is taken from Protein Data Bank (code 1lnq). A spermine molecule is fitted in the pore cavity with the second amine aligned horizontally with F87 (D172). Spd = spermidine, Put = putrescine
Possible mechanisms for inward rectification induced by spermine bound to E224 and E299
It has been demonstrated that E224 and E299 are involved in spermine binding, which eventually inhibits channel conductance (Yang et al. 1995; Kubo & Murata, 2001). E224 and E299 are two amino acids that appear in the cytoplasmic C terminal, which together with the N-terminal may form a cytoplasmic pore. The structures of the cytoplasmic pores in the Kir3.1 and KirBac1.1 channels have been determined (Nishida & MacKinnon, 2002; Kuo et al. 2003). These two studies have shown that the lining of the cytoplasmic pore contains polar, negatively charged residues alternated with hydrophobic amino acids. Therefore, these amino acids form a good complementary receptor for polyamines, which thus bind effectively to the cytoplasmic pore. However, there is as yet no structural evidence for a direct block of the cytoplasmic pore by polyamines and Mg2+ binding. Although KirBac1.1 crystals have been grown in the presence of Mg2+, the presence of Mg2+ in the cavity or cytoplasmic pore was not established (Kuo et al. 2003). On the other hand, functional studies suggest that the spermine bound to E224 and E299 does not directly block the channel pore. Instead, it has been suggested that these two sites are intermediate binding sites that enhance the entry and exit of spermine to and from the final channel-blocking site located deeper in the pore (Kubo & Murata, 2001; Xie et al. 2002). In this study, we provide further evidence for this possibility. Figure 2 shows that spermine inhibits the D172C mutant but does not prevent MTSET from interacting with 172C, suggesting that the spermine bound to E224 and E299 does not directly block the pore. Instead, these results suggest that the channel closure induced by spermine takes place at a location more external than site 172. We also tested whether channel closure could occur at the narrow pore. Ba2+ is a permeable blocker in the Kir2.1 channel (Shieh et al. 1998). The innermost Ba2+ binding site has been shown to be the threonine at the site 141 (T141) (Alagem et al. 2001), which is close to the Ba2+ site in the KcsA channel determined by X-ray Crystallography (Jiang & MacKinnon, 2000). We rationalize that if the closure occurs at the narrow pore, the exit of Ba2+ bound in the pore should be affected. Figure 8 shows that spermine (1 µm) had no effects on the exit rate of Ba2+ blockade in the pore of the D172C mutant. Taken together, these results suggest that the channel closure initiated by the spermine bound to E224 and E299 occurs between sites 141 and 172. The exact mechanism underlying the channel inhibition induced by the spermine associated with E224 and E299 requires further investigation. It is possible that E224 and E299 facilitate spermine binding to a site between 141 and 172 in the absence of an aspartate at site 172. Based on the structure of the MthK channel (Jiang et al. 2002), we estimated that the space between T141 and D172 in Kir2.1 (12 Å in length) would not be long enough for fitting an entire vertical spermine molecule (Fig. 9A). This argument is consistent with previous studies. It has been shown that the serine at site 165 (S165) is located between T141 and D172 and may form the narrowest part of the Kir2.1 channel (Fujiwara & Kubo, 2002). S165 has been shown to be accessible to small molecules including, Rb+, Cs+ and Mg2+ but not large molecules such as spermine and MTSET, suggesting the space at S165 is very narrow (Thompson et al. 2000; Fujiwara & Kubo, 2002). Furthermore, it is difficult to envisage how the binding of spermine between sites 141 and 172 enhances MTSET modification in the D172C, V169C/D172N, and I176C/D172N mutants.
Alternatively, the spermine bound to E224 and E299 may induce channel closure through global changes of conformation. The two possible mechanisms can be distinguished by the voltage-dependence of spermine's binding affinity, which can be evaluated by zδ (z is the valence of spermine and δ is the apparent electrical distance). If the block of spermine occurs at a site more external than D172 zδ is likely to be larger in the D172N mutant than in the wild-type. In contrast, if channel closure is due to conformational changes effected by spermine bound to E224 and E299, zδ may be smaller in the D172N mutant than in the wild-type. Since the value of zδ is 4.5 ± 0.1 in the wild-type and 3.2 ± 0.4 in the D172N mutant (Xie et al. 2002), it appears that the conformational change hypothesis may be more likely. Figures 2, 4 and 5 show that spermine enhances the rate of MTSET modification in the D172C, V169C/D172N and I176C/D172N mutants. Further neutralization of amino acids at positions 224 and 299 eliminates this effect of spermine (Fig. 6). In addition, Fig. 3C demonstrates that spermine tends to decrease the rate of MTSET modification in the Q164C/D172N mutant. These results are consistent with the conformational changes hypothesis. The binding of spermine to E224 and E299 may induce conformational changes such that the accessibilities of MTSET to the cysteines are altered.
Conclusions
In this study we employed the spermine bound to D172 to determine the relative positions of some pore-lining amino acids of the M2 segment in the Kir2.1 channel. The effects of different polyamines on the MTSET modification at 176C provide insights on the locations of polyamines in the pore. Finally, the effects of E224 and E299 on MTSET modification may shed light on the mechanisms underlying the inward rectification induced by the spermine binding to E224 and E299.
Acknowledgments
We thank Dr Jian Yang for kindly providing the IRK1J clone. We are grateful to Dr Cyprian V. Weaver for reading the manuscript and his suggestions, and to the reviewers for their insightful advice. This work was supported by Academia Sinica and National Science Council Grants 91-2320-B-001-036 and 91-2320-B-001-045 in Taiwan, R.O.C.
REFERENCES
- Alagem N, Dvir M, Reuveny E. Mechanism of Ba2+ block of a mouse inwardly rectifying K+ channel: differential contribution by two discrete residues. J Physiol. 2001;534:381–393. doi: 10.1111/j.1469-7793.2001.00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S, Stevens CF, Sullivan JM. Three distinct structural environments of a transmembrane domain in the inwardly rectifying potassium channel ROMK1 defined by perturbation. Proc Natl Acad Sci U S A. 1995;92:12046–12049. doi: 10.1073/pnas.92.26.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A, Chuang H, Jan YN, Jan LY. Scanning mutagenesis of the putative transmembrane segments of Kir2. 1, an inward rectifier potassium channel. Proc Natl Acad Sci U S A. 1997;94:5456–5460. doi: 10.1073/pnas.94.10.5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Kubo Y. Ser165 in the second transmembrane region of the Kir2. 1 channel determines its susceptibility to blockade by intracellular Mg2+ J Gen Physiol. 2002;120:677–693. doi: 10.1085/jgp.20028663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Heher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW. The giant membrane patch. In: Sakmann B, Neher E, editors. Single-Channel Recording. 2. New York: Plenum; 1995. pp. 307–328. [Google Scholar]
- Hille B. Potassium channels and chloride channels. In: Hille B, editor. Ionic Channels of Excitable Membranes. 3. Sunderland: Sinauer; 2001. pp. 131–167. [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- Jiang Y, MacKinnon R. The barium site in a potassium channel by x-ray crystallography. J Gen Physiol. 2000;115:269–272. doi: 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Baldwin T, Jan Y, Jan L. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Murata Y. Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2. 1 K+ channel. J Physiol. 2001;531:645–660. doi: 10.1111/j.1469-7793.2001.0645h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1. 1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. The mechanism of inward rectification of potassium channels: ‘long-pore plugging’ by cytoplasmic polyamines. J Gen Physiol. 1995;106:923–955. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G, Makhina EN, Rose T, Nichols CG. Structure and dynamics of the pore of inwardly rectifying K(ATP) channels. J Biol Chem. 2000;275:1137–1144. doi: 10.1074/jbc.275.2.1137. [DOI] [PubMed] [Google Scholar]
- Lu T, Nguyen B, Zhang X, Yang J. Architecture of a K+ channel inner pore revealed by stoichiometric covalent modification. Neuron. 1999;22:571–580. doi: 10.1016/s0896-6273(00)80711-4. [DOI] [PubMed] [Google Scholar]
- Lu Z, MacKinnon R. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 1994;371:243–246. doi: 10.1038/371243a0. [DOI] [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single-Channel Recording. 2. New York: Plenum; 1995. pp. 31–52. [Google Scholar]
- Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through the inwardly rectifying K+ channel and blocking by internal Mg2+ Nature. 1987;325:156–159. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- Minor DL, Jr, Masseling SJ, Jan YN, Jan LY. Transmembrane structure of an inwardly rectifying potassium channel. Cell. 1999;96:879–891. doi: 10.1016/s0092-8674(00)80597-8. [DOI] [PubMed] [Google Scholar]
- Nishida M, MacKinnon R. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1. 8 Å resolution. Cell. 2002;111:957–965. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- Shieh RC, Chang JC, Arreola J. Interaction of Ba2+ with the pores of the cloned inward rectifier K+ channels Kir2. 1 expressed in Xenopus oocytes. Biophys J. 1998;75:2313–2322. doi: 10.1016/S0006-3495(98)77675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, Conley EC. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J Physiol. 1994;478:1–6. doi: 10.1113/jphysiol.1994.sp020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Ficker E, Wible BA, Brown AM. C-terminus determinants for Mg2+ and polyamine block of the inward rectifier K+ channel IRK1. EMBO J. 1995;14:5532–5541. doi: 10.1002/j.1460-2075.1995.tb00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson GA, Leyland ML, Ashmole I, Sutcliffe MJ, Stanfield PR. Residues beyond the selectivity filter of the K+ channel Kir2. 1 regulate permeation and block by external Rb+ and Cs+ J Physiol. 2000;526:231–240. doi: 10.1111/j.1469-7793.2000.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg C. Inward rectification of a potassium channel in cardiac ventricular cells depends of internal magnesium ions. Proc Natl Acad Sci U S A. 1987;84:2560–2564. doi: 10.1073/pnas.84.8.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 1994;371:246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- Xie LH, John SA, Weiss JN. Spermine block of the strong inward rectifier potassium channel kir2. 1: dual roles of surface charge screening and pore block. J Gen Physiol. 2002;120:53–66. doi: 10.1085/jgp.20028576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 1995;14:1047–1054. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]