

Abstract
The apical Na+-H+ exchanger NHE3 plays an important role in fluid reabsorption in the proximal tubule. However, whether its deletion alters the salt and water transport in the distal nephron remains unknown. To answer these questions, wild-type (Nhe3+/+) and NHE3 null mice (Nhe3−/−) were placed in metabolic cages and their water balance and urine osmolality were examined. Nhe3−/− mice demonstrated a significant polydipsia (P < 0.03) and polyuria (P < 0.04), with a lower urine osmolality (P < 0.003) as compared to Nhe3+/+ mice. Northern hybridization and immunoblotting studies indicated that the mRNA expression and protein abundance of the collecting duct (CD) water channel AQP2 decreased by 52 % (P < 0.0003) and 73 % (P < 0.003) in the cortex, and by 53 % and 54 % (P < 0.002) in the inner medulla (IM) of Nhe3−/− vs. Nhe3+/+ mice. The expression of AQP2 in the outer medulla (OM) remained unchanged. Further, the mRNA expression and protein abundance of the medullary thick ascending limb (mTAL) apical Na+-K+-2Cl− cotransporter (NKCC2) decreased by 52 % (P < 0.02) and 44 % (P < 0.01), respectively, in the OM of Nhe3−/− vs. Nhe3+/+ mice. The circulating plasma levels of vasopressin as well as the mRNA expression of vasopressin prohormone were significantly increased in Nhe3−/− vs. Nhe3+/+ mice (P < 0.05). Studies in mice treated with acetazolamide indicated that increased bicarbonate and fluid delivery to distal nephron did not alter the expression of NKCC2 in mTAL and decreased AQP2 protein only in OM but not in the cortex or IM. In conclusion, mice lacking the apical NHE3 have impairment in their water balance and urine osmolality, which correlates with the downregulation of AQP2 expression. These defects occur despite increased circulating levels of vasopressin. We propose that an ADH-independent mechanism is responsible for the downregulation of AQP2 and the resulting polyuria in NHE3 null mice.
The apical Na+-H+ exchanger isoform 3 (NHE3) plays an important role in NaCl, HCO3− and fluid reabsorption in the kidney proximal tubule and intestine, and thus is essential to the regulation of extracellular fluid volume and blood pressure. The deletion of the Slc9a3 gene encoding the NHE3 protein in mouse is associated with a mild diarrhoea and a significant defect in HCO3− and fluid reabsorption by the proximal tubule (Schultheis et al. 1998). The decreased HCO3−reclamation in the proximal tubule is compensated in the collecting duct through an enhanced rate of bicarbonate absorption that is mediated via an adaptive increase in the expression of gastric H+-K+-ATPase and Cl−-HCO3− exchanger (AE1) as well as increased H+-ATPase activity (Schultheis et al. 1998; Nakamura et al. 1999). This adaptation has limited the perturbation of acid-base status of NHE3 null mice to a mild metabolic acidosis as shown by a small decrease in serum HCO3− concentration and blood pH (Schultheis et al. 1998). In addition, a decrease in glomerular filtration rate (GFR) and the upregulation of proximal tubule Na+-Pi cotransporter (NaPi2) and collecting duct γ-subunit of the epithelium Na+ channel (ENAC) has been thought to compensate for decreased Na+ reabsorption in the proximal tubule of NHE3 knockout mice (Brooks et al. 2001). The absorptive defect in the intestine of homozygous mutant mice is also compensated to a certain degree in the distal colon via an adaptive increase in the activity and expression of the epithelial Na+ channel and colonic H+-K+-ATPase (Schultheis et al. 1998). In addition, mice lacking NHE3 exhibited significant volume depletion as shown by decreased blood pressure, increased kidney renin mRNA expression and elevated serum aldosterone levels (Schultheis et al. 1998; Ledoussal et al. 2001). In light of these observations, we hypothesized that the processes involved in the urinary concentrating mechanism should be stimulated in order to compensate for defective water retention in the proximal tubule, and thus minimize water loss by the kidney.
The regulation of water handling by the kidney depends on the activity of water transport proteins called aquaporins. The aquaporins are a family of transmembrane channel proteins expressed in epithelial as well as non-epithelial tissues (Sabolic et al. 1992; Brown et al. 1995; Fushimi & Marumo, 1995). AQP1 is expressed in both apical and basolateral domains of the proximal tubule and descending limb cells as well as in endothelial cells of descending vasa recta (Nielsen, et al. 1993a; Maeda et al. 1995). AQP2 is the vasopressin-regulated water channel and is predominantly expressed in the apical surface of principal cells in the connecting tubule and the entire collecting duct system (Fushimi et al. 1993; Nielsen et al. 1993b). AQP3 is located in the basolateral region of the cortical and outer medullary collecting duct. Finally, AQP4 is restricted to the basolateral domain of the inner medullary collecting duct system (Knepper et al. 1996).
In the medullary collecting duct, the AVP-stimulated water reabsorption through AQP2 is facilitated by the hypertonic medullary interstitium generated as a result of the countercurrent multiplication process (Knepper & Rector, 1996; Sands & Kokko, 1996.). This process involves active NaCl reabsorption in the medullary thick ascending limb (mTAL), which is mediated primarily via the apical Na+-K+-2Cl− cotransporter (NKCC2 or BSC1) (Burg, 1976; Hebert & Andreoli, 1984a). Recent studies reported a decrease in NKCC2 protein abundance, however, it is not clear whether this originated from the cortical or medullary thick ascending limb as the expression of NKCC2 protein was examined in whole kidney harvested from wild-type and NHE3 knockout (Brooks et al. 2001) mice.
To test our hypothesis, we examined the status of water balance and urine osmolality in Nhe3+/+ and Nhe3−/− mice, and determined the expression of the key transport pathways involved in the urinary concentrating mechanism (i.e., AQP1, AQP2 and NKCC2). In additional experiments, the mRNA expression levels of vasopressin prohormone in the brain as well as plasma vasopressin levels were measured in both Nhe3+/+ and Nhe3−/− mice.
Parts of this work have been published in abstract form (H. Amlal, C. Ledoussal, G. E. Shull & M. Soleimani, J Am Soc Nephrol11, 12A (2000). and presented at the 28th Annual Meeting of the American Society of Nephrology, October 2000, Toronto, Ontario, Canada.
Methods
Animal models
The experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati. Homozygous NHE3 knockout (Nhe3−/−) mice, heterozygous (Nhe3+/-) mice (Schultheis et al. 1998) and their wild-type (Nhe3+/+) littermates were maintained on regular mice chow and tap water ad libitum at the laboratory animal medicine service (LAMS) of University of Cincinnati.
In separate studies, normal Black Swiss mice were placed in metabolic cages and injected intraperitoneally with acetazolamide (carbonic anhydrase inhibitor, 100 mg (kg body weight)−1 (24 h−1) for 3 days. Control mice were injected with 200 µl of vehicle, and both groups were killed by peritoneal injection of sodium pentobarbital (50 mg (kg body weight)−1) after 3 days of treatment.
Water balance, urine osmolality and tissues collection
To assess the daily water balance and urine osmolality, animals were placed in mice metabolic cages. After the mice were acclimatized to metabolic cages, water intake, urine volume and urine osmolality were measured. The animals were then anaesthetized using sodium pentobarbital. Both kidneys were removed and cortex, whole medulla, inner stripe of outer medulla and inner medulla were dissected and snap-frozen in liquid nitrogen for total RNA and protein isolation. The tissues were stored at −80 °C until use.
Measurement of plasma levels of vasopressin
To measure plasma concentrations of vasopressin, wild-type and NHE3 knockout mice were decapitated and blood was collected for plasma isolation. The whole brain was dissected and rapidly frozen in liquid nitrogen and stored at −80 °C for total RNA isolation. Plasma vasopressin concentration was determined by radioimmunoassay after a peptide extraction procedure according to the manufacturer's protocol (Peninsula Laboratories, Inc., San Carlos, CA, USA).
RNA isolation and Northern hybridization
Total cellular RNA was extracted from whole kidney, cortex, outer stripe of outer medulla and whole medulla (outer + inner medulla) by the method of Chomczynski & Sacchi (1987). Total RNA samples were quantified spectrophotometrically and stored at −80 °C. Total RNA samples (30 µg lane−1) were fractionated on a 1.2 % agarose-formaldehyde gel, and transferred to nylon membranes. Membranes were cross-linked by ultraviolet light and baked for 1 h. Hybridization was performed according to Church & Gilbert (1984). AQP2- and NKCC2-specific probes (Amlal et al. 1998a, 2001) were labelled with 32P deoxynucleotides using the Rad-Prime DNA labelling kit (GIBCO-BRL). The membranes were washed, blotted dry, exposed to PhosphorImager screen for 24–72 h and read by PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA, USA). Coding sequence in vasopressin (AVP) gene (GenBank accession no. M25646) was used to design primers utilized to generate an AVP-specific probe by RT-PCR from rat brain. These primers used were 5′-CCTCACCTCTGCCTGCTACTT-3′ (forward, nucleotides 77–97) and 5′-GGGGGGCGATGGCTCAGTAGAC-3′ (reverse, nucleotides 540–519). The PCR reaction yielded a single band with expected size of 465 bp corresponding to a rat vasopressin fragment, which was verified by sequencing (DNAcore, University of Cincinnati, Cincinnati, OH, USA).
AQP1, AQP2 and NKCC2 antibodies
For AQP1 antibody, a 19-amino acid synthetic peptide within the carboxy terminal domain of rat AQP1 was used for antibody production (Alpha Diagnostic Int. Inc. San Antonio, TX, USA). AQP2 was raised and used in our laboratory as previously described (Amlal et al. 2000, 2001). Peptide-derived polyclonal antibody specific to kidney apical Na+-K+-2Cl− cotransporter (NKCC2) was raised using commercial services (Genosys Biotechnologies, Inc., The Woodlands, TX, USA). The peptide sequence used to generate the apical NKCC2 antibody was NH2-CEYYRNTGSVSGPKVNRPSLQE-COOH, which corresponds to amino acids 109–129 of the amino-terminal tail of the apical Na+-K+-2Cl− cotransporter. The sequence is identical to the one used recently to generate NKCC2 antibody (Kim et al. 1999). The specificity of NKCC2 antibody was previously demonstrated (Nielsen et al. 1998; Kim et al. 1999) and confirmed in our laboratory (Amlal et al. 2003) by competitive inhibition of the antibody by the immunizing peptide.
Preparation of membrane fractions from renal cortex, outer medulla and inner medulla
A crude total membrane fraction containing plasma membrane and intracellular membrane vesicles was prepared as described (Amlal et al. 2000, 2001). Briefly, the tissue samples were homogenized in ice-cold isolation solution (250 mm sucrose and 10 mm triethanolamine, pH 7.6) containing protease inhibitors (phenazine methyl sulfonyl fluoride, 0.1 mg ml−1; leupeptine 1 µg ml−1), using a Polytron homogenizer. The homogenates were centrifuged at low speed (1000 g) for 10 min at 4 °C to remove nuclei and cell debris. Following this, the supernatants were spun at 150 000 g for 90 min at 4 °C; the pellets containing plasma membrane and intracellular vesicles were suspended in isolation solution with protease inhibitors. The total protein concentration was measured and the membrane fractions were solubilized at 60 °C for 20 min in Laemmli sample buffer.
Electrophoresis and immunoblotting
These experiments were carried out as previously described (Amlal et al. 2000, 2001). Briefly, the solubilized membrane proteins were size-fractionated on 12 % polyacrylamide minigels (Novex, San Diego, CA, USA) under denaturing conditions. Using a BioRad transfer apparatus (BioRad Laboratories, Hercules, CA, USA), the separated proteins were electrophoretically transferred to nitrocellulose membranes. The membranes were blocked with 5 % milk proteins, and then probed with affinity-purified primary antibody. The secondary antibody was donkey anti-rabbit IgG conjugated to horseradish peroxidase. The sites of antigen-antibody complexation on the nitrocellulose membranes were visualized using a chemiluminescence method (SuperSignal Substrate, Pierce) and captured on light-sensitive imaging film (Kodak). Bands corresponding to AQP1, AQP2 and NKCC2 proteins were quantified by densitometric analysis (UN-SCAN-IT gel software, Silk Scientific, Inc., Orem, Utah, USA) and were expressed as percentages of control. The equity in protein loading in all blots was first verified by gel staining using the coomassie brilliant blue R-250 (Bio-Rad, Hercules, CA, USA).
Materials
32P-dCTP was purchased from New England Nuclear (Boston, MA, USA). RadPrime DNA labelling kit was purchased from Gibco BRL, USA. Nitrocellulose membrane used for RNA transfer was purchased from Schleicher & Schuell (Keene, NH, USA). Sequi-blot PVDF membrane for protein transfer and blotting paper were purchased from Bio-Rad (Hercules, CA, USA). All other chemicals were purchased from Sigma Chemical Co. (St Louis, MO, USA). AQP1 antibody was purchased from Alpha Diagnostic Int. Inc. (San Antonio, TX, USA).
Statistic analyses
Semi-quantification of immunoblot and Northern hybridization band densities was determined by densitometry using a scanner (ScanJet ADF, Hewlett Packard) and UN-SCAN-IT gel software (Silk Scientific, Inc., Orem, Utah, USA) and ImageQuaNT software (Molecular Dynamics, Sunnyvale, CA, USA), respectively. Data were expressed as percentages of control. Results were presented as means ± s.e.m. Statistical significance between control and experimental groups was determined by one-way ANOVA or Student's unpaired t-test using GraphPad InStat software (http://www.graphpad.com) as needed. P < 0.05 was considered significant.
Results
Water balance and urine osmolality
To determine the status of water balance and urine osmolality, Nhe3+/+, Nhe3−/− and Nhe3+/- mice were placed in metabolic cages. After 3 days of adjustment to metabolic cages, water intake, urine volume and urine osmolality were measured for 3 consecutive days. The results are summarized in Fig. 1 and showed a significant increase in water intake in Nhe3+/- as compared to Nhe3+/+ mice (6.22 ± 1.19 ml (24 h)−1, n = 8, P < 0.001, Fig. 1A). The water intake was comparable between Nhe3+/+ and Nhe3+/- (2.71 ± 0.35 ml (24 h)−1, n = 8, vs. 3.5 ± 0.48 ml (24 h)−1, n = 4, P > 0.05, Fig. 1A). Urine volume was significantly increased in Nhe3−/− vs. Nhe3+/+ mice (1.82 ± 0.06 ml (24 h−1), n = 8, vs. 1.21 ± 0.35 ml (24 h)−1, n = 8, P < 0.01, Fig. 1B). Urine volume was comparable in Nhe3+/- and Nhe3+/+ mice (1.15 ± 0.10 ml (24 h)−1, n = 4, vs. 1.21 ± 0.09 ml (24 h−1, n = 8, P > 0.05, Fig. 1B). Urine osmolality was not significantly different between Nhe3+/+ and Nhe3+/- mice (2906 ± 235 mosmol (kg H2O)−1, n = 8, vs. 2598 ± 158 mosmol (kg H2O)−1, n = 4, P > 0.05, Fig. 1C). However, urine osmolality was significantly decreased (1737 ± 113 mosmol (kg H2O)−1, n = 6, P < 0.001, Fig. 1C) in Nhe3−/− vs. Nhe3+/+ mice). These results indicate that mice with a null mutation in the Nhe3 gene develop a polyuria/polydipsia syndrome associated with decreased urine osmolality, indicating a basal defect in their urinary concentrating mechanism.
Figure 1. Water balance and urine osmolality in Nhe3+/+, Nhe3+/- and Nhe3−/− mice.
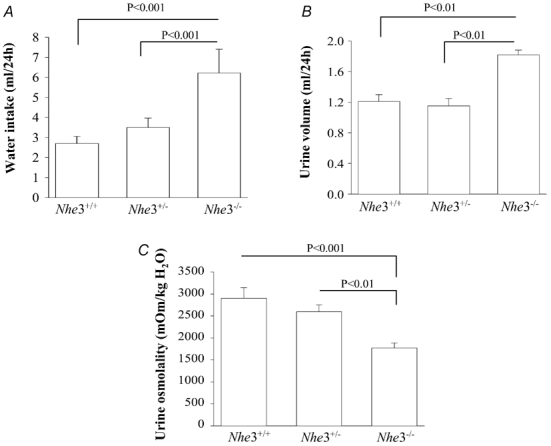
Mice were placed in metabolic cages and had access to food and water ad libitum. Water intake (A) and urine volume (B) were measured daily for 3 days. Urine osmolality (C) was measured in urine that was collected under mineral oil. n = 8 mice for Nhe3+/+ and Nhe3−/−; and n = 4 for Nhe3+/-.
AQP2 protein abundance
AQP2 is the only apically expressed water channel in the collecting duct, and plays an important role in the final tuning of water reabsorption in the kidney. Therefore AQP2 represents a limiting barrier for water reabsorption in the collecting duct system. Hence, the following experiments were undertaken to examine the abundance of AQP2 protein in microsomes harvested from the cortex, outer medulla and inner medulla of Nhe3+/+, Nhe3+/- and Nhe3−/− mice. Semi-quantitative immunoblotting studies indicated that the abundance of AQP2 protein was significantly reduced in the cortex of Nhe3−/− mice (26 ± 6 % for Nhe3−/− vs. 100 ± 3 % for Nhe3+/+, n = 5 for each, P < 0.0003, Fig. 2). In the inner medulla, the abundance of AQP2 decreased by ≈53 % in Nhe3−/− mice (47 ± 5 %, vs. 100 ± 7 % in Nhe3+/+, n = 5 for each, P < 0.002, Fig. 3). The abundance of AQP2 protein in the outer medulla of Nhe3−/− mice was not altered (86 ± 19 %, vs. 100 ± 17 % in Nhe3+/+, n = 5 for each, P > 0.05, Fig. 4). AQP2 protein expression was not different between Nhe3+/+ and Nhe3+/- in all three regions of the kidney (P > 0.05, Figs 2, 3 and 4).
Figure 2. AQP2 protein abundance in the kidney cortex of Nhe3+/+, Nhe3+/- and Nhe3−/− mice.
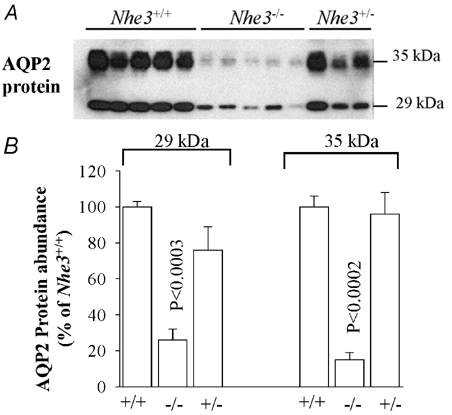
A, immunoblot of AQP2 protein level in the cortex. Both 29 and 35 kDa bands are detected by the anti-AQP2 antibody in the kidney cortex. B, densitometry of AQP2 protein (29 and 35 kDa) expression in the cortex showing a decrease in the abundance of both 29 kDa (n = 5) and 35 kDa (n = 5) bands in Nhe3−/− as compared to Nhe3+/+ mice. These two bands were not significantly altered in Nhe3+/-(n = 3) as compared to Nhe3+/+ (n = 5); 16 µg of total protein from the cortex was loaded per lane.
Figure 3. AQP2 protein abundance in the kidney inner medulla of Nhe3+/+, Nhe3+/- and Nhe3−/− mice.
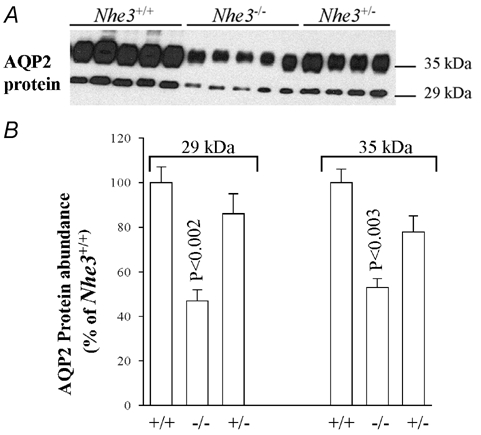
A, immunoblot of AQP2 protein level in the inner medulla. Both 29 and 35 kDa bands are expressed in the mouse kidney inner medulla. B, densitometry of AQP2 protein (29 kDa and 35 kDa bands) in the inner medulla showing a decrease in the abundance of both 29 kDa (n = 5) and 35 kDa (n = 5) bands in Nhe3−/− as compared to Nhe3+/+ mice. These two bands were not significantly altered in Nhe3+/- (n = 4) vs. Nhe3+/+ (n = 5); 3 µg of total protein from the inner medulla were loaded per lane.
Figure 4. AQP2 protein abundance in the kidney outer medulla of Nhe3+/+, Nhe3+/- and Nhe3−/− mice.
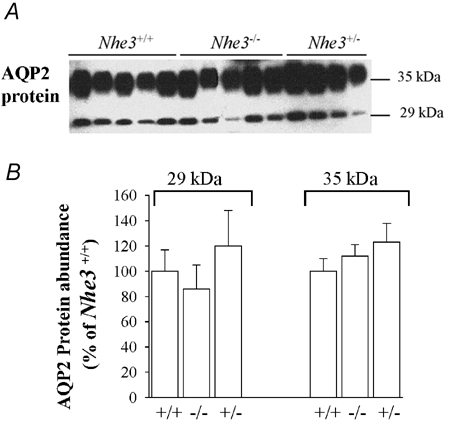
A, immunoblot of AQP2 protein level in the outer medulla. The 29 and 35 kDa AQP2 bands are shown. B, densitometry of AQP2 protein (29 and 35 kDa bands) in the outer medulla showing that the expression of these two bands was not altered in Nhe3−/− (n = 5, P > 0.05) and Nhe3+/- (n = 4, P > 0.05) mice vs. Nhe3+/+ mice (n = 5); 10 µg of total protein from the outer medulla was loaded per lane.
AQP2 mRNA expression
We next examined the expression levels of AQP2 mRNA in Nhe3−/− and Nhe3+/+ mice. Total RNA was isolated from whole kidneys, cortex, inner stripe of outer medulla and whole medulla (outer + inner medulla) and utilized for Northern hybridization. The results indicated that AQP2 mRNA expression levels decreased in Nhe3−/− mice by 36 % in whole kidney (P < 0.03, Fig. 5A and B), and by 52 % in the cortex (P < 0.003, Fig. 5C and D) as compared to Nhe3+/+ mice, n = 3 to 4 for each group. In addition, AQP2 mRNA expression was reduced by 53 % in whole medulla (Fig. 6A and B), but did not change in the inner stripe of outer medulla of Nhe3−/− as compared to Nhe3+/+ mice (Fig. 6C and D), indicating that the reduction in AQP2 mRNA expression in whole medulla is originating from the inner medulla of the kidney.
Figure 5. Northern hybridization of kidney AQP2 mRNA in Nhe3+/+ and Nhe3−/− mice.
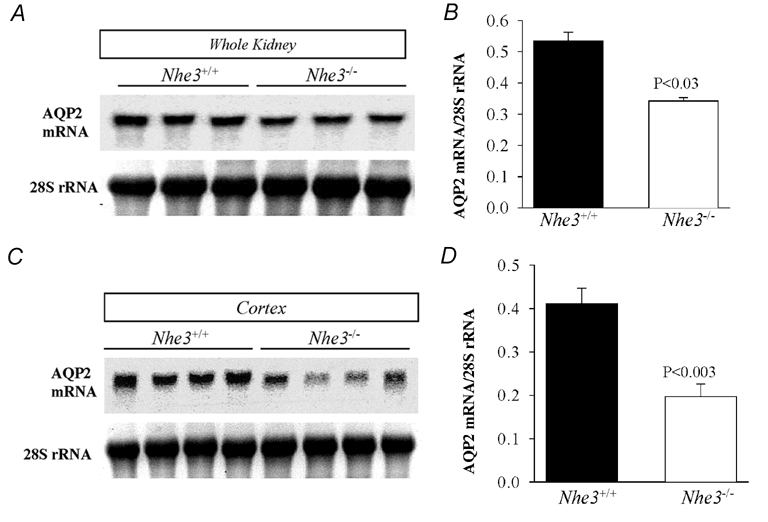
Northern blots of AQP2 mRNA expression in whole kidney (A) and cortex (C). B and D are ratios of AQP2 mRNA to 28S rRNA for A and C, respectively. The results indicate a significant decrease in the expression of AQP2 mRNA in whole kidney (n = 3) and cortex (n = 4) of Nhe3−/− as compared to Nhe3+/+ mice (n = 3 or 4); 30 µg RNA was loaded on each lane.
Figure 6. Northern hybridization of AQP2 mRNA in kidney medulla of Nhe3+/+ and Nhe3−/− mice.
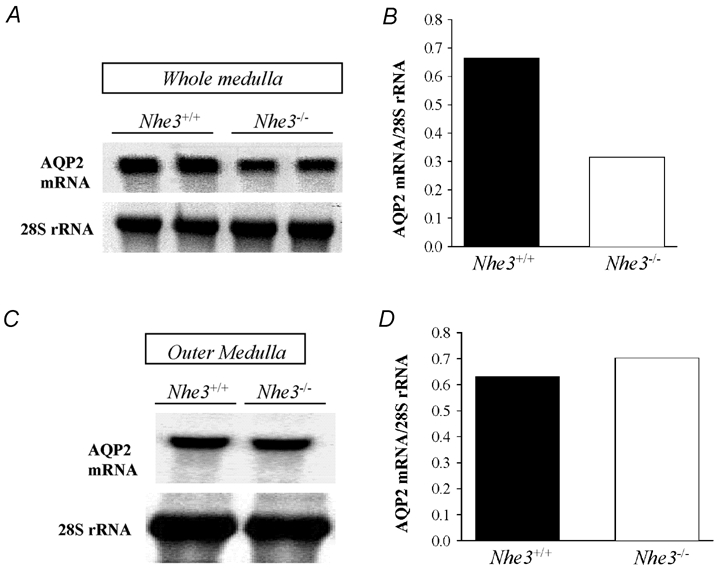
Northern blots of AQP2 mRNA expression in whole medulla (outer + inner medulla) (A) and outer medulla (C). B and D are ratios of AQP2 mRNA to 28S rRNA for A and C, respectively. Results show a decrease (−54 %) in the expression of AQP2 mRNA in whole medulla of Nhe3−/− mice (B). In the outer medulla, the AQP2 mRNA expression was not altered in Nhe3−/− mice (D) vs. Nhe3+/+ mice. Each lane was loaded with 30 µg RNA isolated from pooled tissues harvested from two (whole medulla), or three mice (outer medulla).
Taken together, these results indicate that the reduction in AQP2 protein abundance correlates with a decrease in its mRNA expression levels in the cortex and inner medulla of NHE3 null mice.
AQP1 protein abundance and Na+-K+ATPase expression in the kidney
In the next experiments, the expression of AQP1 protein was examined by semi-quantitative immunoblotting studies in Nhe3+/+, Nhe3+/- and Nhe3−/− mice. The results indicate that AQP1 protein abundance was comparable in the cortex, outer medulla and inner medulla of all three groups (P > 0.05, data not shown). In addition, the expression levels of alpha subunit of Na+-K+-ATPase remained unchanged in the cortex and whole medulla of Nhe3−/− vs. Nhe3+/+ mice (P > 0.05, data not shown).
Expression of mTAL apical Na+–K+–2Cl− cotransporter (NKCC2)
The mTAL apical Na+-K+-2Cl− cotransporter or NKCC2 plays a crucial role in the process of urinary concentrating mechanism as it mediates active NaCl reabsorption in the thick ascending limb with no net movement of water. This promotes the generation of a hypertonic medullary interstitium, which drives water reabsorption by the adjacent collecting duct system. In the following experiments we examined the protein abundance and mRNA expression levels of NKCC2 by immunoblotting and Northern hybridization experiments, respectively. The results depicted in Fig. 7A, indicate that NKCC2 protein abundance was decreased by 44 % in Nhe3−/− (n = 5) vs. Nhe3+/+ mice (n = 4) (P < 0.02, Fig. 7A and B). NKCC2 protein abundance did not change in Nhe3+/- as compared to wild-type mice (Fig. 7A and B). The decrease in NKCC2 protein in Nhe3−/− mice correlates with a 52 % reduction in its mRNA expression levels (P < 0.01, vs. Nhe3+/+ mice, n = 3 for each, Fig. 7C and D).
Figure 7. NKCC2 protein abundance and mRNA expression in Nhe3−/− and Nhe3+/+ mice.
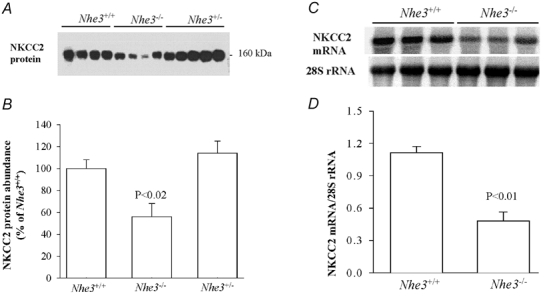
A, immunoblot of NKCC2 protein level in the outer medulla harvested from Nhe3+/+, Nhe3+/- and Nhe3−/− mice. B, corresponding densitometric analysis showing 44 % decrease in the abundance of NKCC2 protein (n = 5) in Nhe3−/− as compared to Nhe3+/+ (n = 4) mice. NKCC2 protein was not significantly altered in Nhe3+/- (n = 4) as compared to Nhe3+/+ (n = 5); 12 µg of total protein from the outer medulla was loaded per lane. C, Northern hybridization of NKCC2 mRNA in whole medulla of Nhe3+/+ and Nhe3−/− mice. D, ratio of NKCC2 mRNA to 28S rRNA for Northern blot shown in C. The results show a significant decrease (−52 %) in the expression of NKCC2 mRNA in Nhe3−/− mice as compared to Nhe3+/+ mice (n = 3 for each); 30 µg RNA was loaded on each lane.
mRNA expression and plasma levels of vasopressin
Vasopressin (AVP) or antidiuretic hormone plays a vital role in the regulation of water homeostasis and maintenance of the tonicity of body fluid. AVP regulates the expression and activity of both NKCC2 in mTAL and AQP2 in the collecting duct system through its V2 receptor. To determine whether a reduction in plasma AVP might be responsible for AQP2 and NKCC2 downregulation in NHE3 mutant mice, the circulating level of vasopressin was measured by radioimmunoassay. The results depicted in Fig. 8A indicate that the plasma levels of AVP are actually significantly increased in NHE3 knockout vs. wild-type mice (16 ± 3.6 vs. 3.58 ± 0.16 pg ml−1, P < 0.05, Fig. 8A). These results are further confirmed by Northern hybridization experiment (Fig. 8B) indicating a significant increase in the mRNA expression levels of AVP prohormone in Nhe3−/− mice (236 % ± 34 vs. 100 % ± 11 in Nhe3+/+ mice, P < 0.05, Fig. 8B).
Figure 8. Plasma and mRNA expression levels of vasopressin in Nhe3+/+ and Nhe3−/− mice.
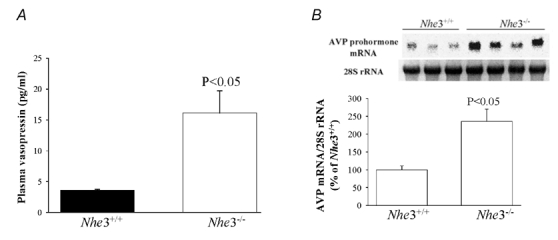
A, plasma concentration of vasopressin in Nhe3+/+ (n = 3) and Nhe3−/− (n = 4). B, Northern hybridization of vasopressin prohormone mRNA expression and 28S rRNA in Nhe3+/+ and Nhe3−/− mice (upper panel) and corresponding densitometric analysis showing AVP mRNA:28S rRNA ratio expressed as a percentage of Nhe3+/+ mice; 30 µg RNA was loaded on each lane. n = 3–4 mice in each group.
Effects of increased delivery of fluid and bicarbonate to distal nephron on NKCC2 and AQP2 expression
The lack of NHE3 is associated with a significant inhibition of fluid and bicarbonate reabsorption in the proximal tubule. To test whether an increase in fluid and bicarbonate delivery to the distal nephron could mediate the downregulation of NKCC2 and AQP2 in NHE3 knockout mice, normal Black Swiss mice were treated with acetazolamide (ACTZ), a carbonic anhydrase inhibitor that inhibits fluid and bicarbonate reabsorption in the proximal tubule. As expected, the results depicted in Table 1 indicate that mice treated with acetazolamide exhibited a significant polyuria, natriuresis and bicarbonaturia as shown by higher urine volume, increased Na+ excretion and elevated urine pH in acetazolamide- vs. vehicle- treated mice (Table 1). In addition, urine osmolality decreased significantly, and water intake increased slightly but not significantly (Table 1).
Table 1.
Effects of acetazolamide treatment for 72 h vs. its vehicle on water balance, urine osmolality, urine pH and sodium excretion in mice
| Vehicle (n = 5) | Acetazolamide (n = 5) | P | |
|---|---|---|---|
| Urine volume (ml (24 h)−1) | 1.22 ± 0.31 | 2.88 ± 0.49 | <0.02 |
| Na+ excretion (mequiv (24 h)−1) | 0.24 ± 0.08 | 0.481 ± 0.03 | <0.04 |
| Urine pH (pH unit) | 7.03 ± 0.04 | 8.16 ± 0.07 | <0.0001 |
| Urine osmolality (mosmol (kg H2 O)−1) | 2338 ± 201 | 1528 ± 174 | <0.02 |
| Water intake (ml (24 h)−1) | 3.45 ± 0.45 | 5.18 | n.s. |
n.s., not significant. Results are means ± s.e.m.
Effects of acetazolamide treatment on AQP2 and NKCC2 expression
The mRNA expression and protein abundance of AQP2 was examined in the cortex, whole medulla, outer medulla, or inner medulla by Northern hybridization and immunoblotting, respectively. The results depicted in Table 2 indicate that cortical and medullary AQP2 mRNA expression was not altered in ACTZ- vs. vehicle-treated mice (Table 2). Moreover, AQP2 protein abundance did not change in the cortex (Fig. 9A and Table 2) and inner medulla (Table 2) but did decrease with a borderline significance (P = 0.05) in the outer medulla of ACTZ-treated mice as compared to vehicle (Fig. 9B and Table 2). The increase in osmotic diuresis and the downregulation in AQP2 protein in outer medulla probably explain the decrease in urine osmolality in ACTZ-treated mice (Table 1).
Table 2.
Effects of acetazolamide treatment for 72 h vs. its vehicle on AQP2 mRNA expression and total AQP2 protein abundance in the different regions of the mouse kidney
| AQP2 mRNA/28S rRNA | Total AQP2 protein abundance | |||||
|---|---|---|---|---|---|---|
| Vehicle | Acetazolamide | P | Vehicle | Acetazolamide | P | |
| Cortex | 100 ± 6 | 113 ± 7 | n.s. | 100 ± 3 | 80 ± 10 | n.s. |
| Outer medulla | n.d. | n.d. | — | 100 ± 8 | 56 ± 15 | 0.05 |
| Whole medulla | 100 ± 12 | 96 ± 4 | n.s. | n.d. | n.d. | — |
| Inner medulla | n.d. | n.d. | — | 100 ± 13 | 91 ± 19 | n.s. |
n.s., not significant; n.d., not determined. Total AQP2 protein = densitometry of 29 + 35 kDa bands. Results are means ± s.e.m. and are expressed as a percentage of vehicle (n = 4 or 5 mice in each group).
Figure 9. Effect of acetazolamide on AQP2 expression in the cortex and outer medulla.
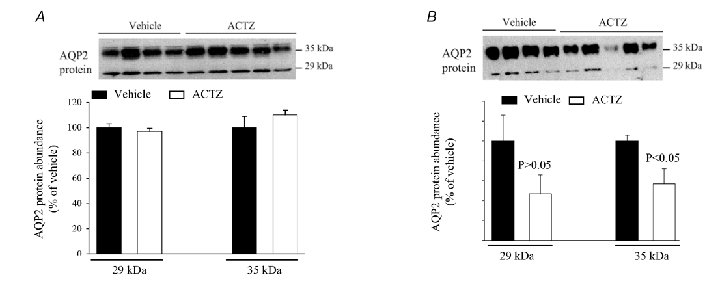
Total cellular protein was isolated from the kidney cortex and outer medulla of acetazolamide- and vehicle-treated mice and used for immunoblotting experiments. A, immunoblot of AQP2 (upper panel) and corresponding densitometric analysis (lower panel) in the cortex. B, immunoblot of AQP2 (upper panel) and corresponding densitometric analysis (lower panel) in the outer medulla; 16 and 10 µg of total protein from the cortex and outer medulla, respectively, was loaded per lane. n = 4–5 mice in each group.
In addition to AQP2, the expression of mTAL NKCC2 was examined in the outer medulla of ACTZ- and vehicle-treated mice. The results indicate that NKCC2 mRNA expression (87 ± 2 vs. 100 % ± 13 for ACTZ and vehicle, respectively, n = 4 mice in each, P > 0.05, Fig. 10A) and NKCC2 protein abundance (111 ± 16 vs. 100 % ± 13 for ACTZ and vehicle, respectively, n = 4 mice in each, P > 0.05, Fig. 10B) remained unchanged in acetazolamide-treated mice.
Figure 10. Effect of acetazolamide on NKCC2 expression in the outer medulla.
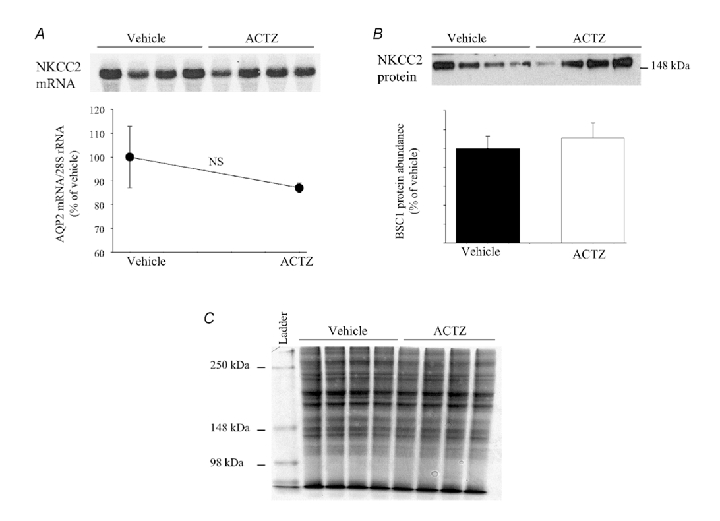
Total RNA and total cellular protein were isolated from kidney medulla of acetazolamide- and vehicle-treated mice and used for Northern hybridization and immunoblotting, respectively. A, NKCC2 mRNA expression in whole medulla (upper panel) and the densitometry analysis of NKCC2 mRNA/28S rRNA (lower panel). B, NKCC2 protein abundance in the outer medulla (upper panel) and the densitometry analysis corresponding to NKCC2 protein band (lower panel); 30 µg of total RNA or 12 µg of total protein was loaded per lane. n = 4–5 mice in each group. C, gel was loaded with 15 µg of total protein from the outer medulla (similar protein used for AQP2 and BSC1) and stained with Coomassie brilliant blue to confirm the equity of protein loading.
Taken together, these results indicate that ACTZ increased fluid and bicarbonate delivery to the distal nephron but did not alter the expression of mTAL NKCC2 and did not generate the distinct zonal regulation of AQP2 that is observed in NHE3 null mice. Hence it is unlikely that the downregulation of these transport pathways in NHE3 knockout mice is mediated via increased fluid and/or bicarbonate delivery to the distal nephron.
Discussion
The status of water balance and urine osmolality in mice lacking the apical Na+-H+ exchanger (NHE3) was examined. The results demonstrated that Nhe3−/− mice developed a basal defect in urine concentration as shown by the presence of polyuria and polydipsia associated with decreased urine osmolality (Fig. 1). A significant difference in net water balance was observed in Nhe3−/− vs. Nhe3+/+ mice, which is probably secondary to severe intestinal absorptive defect and fluid loss in the gastrointestinal tract (Schultheis et al. 1998; Ledoussal et al. 2001). Immunoblot analysis and Northern hybridization experiments demonstrated that expression of the collecting duct apical water channel AQP2 was significantly downregulated at both protein and mRNA levels in the cortex (Fig. 2 and Fig. 5) and inner medulla (Fig. 3 and Fig. 6) of Nhe3−/− mice. Interestingly, AQP2 expression remained unchanged in the outer medulla (Fig. 4). This region-specific regulation of AQP2 has also been observed in food-deprived rats, which exhibited a decrease in the expression levels of cortical and outer medullary but not inner medullary AQP2 (Amlal et al. 2001), indicating that AQP2 protein can be differentially regulated in pathophysiologic states. In addition to AQP2, the results show a significant decrease in the mRNA expression levels and protein abundance of the apical Na+-K+-2Cl− cotransporter NKCC2 in mTAL of Nhe3−/− as compared to Nhe3+/+ mice (Fig. 7). The downregulation of NKCC2 could contribute to decreased baseline urine concentration in Nhe3−/− mice. The downregulation of NKCC2 results in decreased NaCl reabsorption in mTAL, which will reduce the medullary interstitial osmolality and as a result decreases the driving force for water retention in the outer medullary collecting duct. Interestingly, the downregulation of AQP2 and BSC1 occurred despite a significant increase in the circulating levels as well as the mRNA expression of vasopressin (Fig. 8). The increase in AVP synthesis and secretion is expected in Nhe3−/− mice as these animals exhibit higher plasma osmolality (Brooks et al. 2001) and develop significant congenital volume depletion predicted by low blood pressure, increased kidney renin mRNA and higher serum aldosterone levels (Schultheis et al. 1998; Ledoussal et al. 2001). The volume depletion status of Nhe3−/− mice is presumably due to fluid and electrolyte loss through the gastrointestinal tract.
The mechanism underlying the downregulation of AQP2 and NKCC2 expression with subsequent decreased urinary concentration in Nhe3−/− mice remains unclear. Several studies have shown that vasopressin (AVP) stimulates both NaCl and water reabsorption in the medullary thick ascending limb (Hebert et al. 1981; Hebert & Andreoli, 1984b) and collecting duct (Knepper, 1997), respectively. Acutely, the regulation of both transporters by AVP is mediated via an increase in cAMP content, which through protein kinase A (PKA) increases the trafficking of AQP2 to the plasma membrane (Nielsen et al. 1995) or stimulates existing plasma membrane NKCC2 protein and thus increases their activity (Hebert et al. 1981; Amlal et al. 1996). Chronically, long-term exposure of mTAL or collecting duct principal cells to AVP increases the expression of both NKCC2 (Kim et al. 1999) and AQP2 (Knepper, 1997), respectively. This effect is probably mediated via a cAMP response element present in the 5′ flanking region of their respective genes (Uchida et al. 1994; Igarashi et al. 1996).
The downregulation of AQP2 and the resulting polyuria and decreased urine osmolality in Nhe3−/− mice occur despite a significant increase in circulating AVP levels. The increase in AVP levels reflects severe volume depletion in NHE3 deficient mice and is in agreement with enhanced renin expression and increased plasma aldosterone levels (Schultheis et al. 1998). The volume depletion is in large part due to defective salt and water absorption in the gastrointestinal tract (Schultheis et al. 1998). Increased AVP levels in pathophysiological states such as water deprivation have been shown to upregulate AQP2 expression and increase urine osmolality. The current results are the first reported ones demonstrating the downregulation of AQP2 in the kidney despite the presence of volume depletion and elevated AVP levels. Whether a decrease in adenylate cyclase sensitivity to vasopressin, such the one that is observed in hypokalemia- or hypercalcemia-induced nephrogenic diabetes insipidus (Beck et al. 1974; Beck & Webster, 1976), plays a role in AQP2 downregulation in NHE3 mutant mice remains speculative. Alternatively, the possibility of a circulating hormone/factor blocking the action of AVP cannot be excluded.
The lack of apical Na+-H+ exchanger NHE3 is associated with decreased fluid and bicarbonate reabsorption in the proximal tubule, which leads to increased fluid and bicarbonate delivery to the rest on the nephron (Schultheis et al. 1998). The treatment of normal mice with the carbonic anhydrase inhibitor acetazolamide, which caused a significant fluid and bicarbonate wasting (Table 1) did not downregulate mTAL NKCC2 expression (Fig. 10), and did not alter the cortical or inner medullary AQP2 expression (Fig. 9A and Table 2). These findings clearly indicate that the downregulation of AQP2 and NKCC2 in Nhe3−/− mice is not mediated via increased fluid and bicarbonate load to the thick ascending limb and collecting duct system.
Nhe3−/− mice exhibit higher circulating levels of aldosterone and normal serum potassium concentration (Schultheis et al. 1998; Ledoussal et al. 2001). It is unlikely that increased aldosterone levels play a role in the downregulation of AQP2 and NKCC2 proteins, as aldosterone potentiates the hydroosmotic action of AVP in the collecting tubule (Schwartz & Kokko, 1980) and stimulates NaCl reabsorption in the thick ascending limb (Work & Jamison, 1987). However, whether increased angiotensin II (ANG II) is responsible for some of the observed findings in these studies remain speculative. ANG II was reported to acutely regulate the mTAL apical Na+-K+-2Cl− cotransporter (Amlal et al. 1998b), however, its chronic effects on this carrier and on AQP2 expression remain unknown.
In conclusion, mice lacking the apical Na+-H+ exchanger NHE3 isoform develop polyuria and polydipsia associated with decreased urine osmolality at steady state. This syndrome correlates with the downregulation of the collecting duct apical water channel AQP2 and the mTAL apical Na+ -K+-2Cl− cotransporter despite an increase in vasopressin synthesis and secretion. These findings do not support our hypothesis that the distal nephron may compensate for the fluid and salt reabsorption defect in the proximal tubule in NHE3 deficient mouse. Rather, they indicate an apparently maladaptive regulation of AQP2 and NKCC2 with resulting impairment in water retention by the distal nephron, which could contribute to volume depletion in NHE3 knockout mice.
Acknowledgments
These studies were supported by the University of Cincinnati Academic Development Fund and by the Paul Teschan Research Fund, Dialysis Clinic, Inc. (H.A.), by the National Institute of Diabetes and Digestive and Kidney Disease Grants DK 46789 (M.S.) and DK 50594 (G.S.).
References
- Amlal H, Chen Q, Habo K, Wang Z, Soleimani M. Fasting downregulates renal. water channel AQP-2 and causes polyuria. Am J Physiol Renal Physiol. 2001;280:F513–523. doi: 10.1152/ajprenal.2001.280.3.F513. [DOI] [PubMed] [Google Scholar]
- Amlal H, Krane CM, Chen Q, Soleimani M. Early polyuria and urinary concentrating defect in potassium deprivation. Am J Physiol Renal Physiol. 2000;279:F655–663. doi: 10.1152/ajprenal.2000.279.4.F655. [DOI] [PubMed] [Google Scholar]
- Amlal H, Legoff C, Vernimmen C, Paillard M, M Bichara. Na+ -K+ (NH4+)-2Cl− cotransport in medullary thick ascending limb: control by PKA, PKC, and 20-HETE. Am J Physiol. 1996;271:C455–463. doi: 10.1152/ajpcell.1996.271.2.C455. [DOI] [PubMed] [Google Scholar]
- Amlal H, Legoff C, Vernimmen C, Soleimani M, Paillard M, Bichara M. Ang II controls Na+ -K+-(NH4+)-2Cl− cotransport via 20-HETE and PKC in medullary thick ascending limb. Am J Physiol. 1998b;274:C1047–1056. doi: 10.1152/ajpcell.1998.274.4.C1047. [DOI] [PubMed] [Google Scholar]
- Amlal H, Wang Z, Soleimani M. Potassium depletion downregulates chloride-absorbing transporters in rat kidney. J Clin Invest. 1998a;101:1045–1054. doi: 10.1172/JCI686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlal H, Wilke C. Resistance of mTAL Na+-dependent transporters and collecting duct aquaporins to dehydration in 7-month-old rats. Kidney Int. 2003;64:544–554. doi: 10.1046/j.1523-1755.2003.00110.x. [DOI] [PubMed] [Google Scholar]
- Beck N, Singh H, Reed SW, Murdaugh HV, Davis BB. Pathogenic role of cyclic AMP in the impairment of urinary concentrating ability in acute hypercalcemia. J Clin Invest. 1974;54:1049–1055. doi: 10.1172/JCI107848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck N, Webster SK. Impaired urinary concentrating ability and cyclic AMP in K+-depleted rat kidney. Am J Physiol. 1976;231:1204–1208. doi: 10.1152/ajplegacy.1976.231.4.1204. [DOI] [PubMed] [Google Scholar]
- Brooks HL, Sorensen AM, Terris J, Schultheis PJ, Lorenz JN, Shull GE, Knepper MA. Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. J Physiol. 2001;530:359–366. doi: 10.1111/j.1469-7793.2001.0359k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Katsura T, Kawashima M, Verkman AS, Sabolic I. Cellular distribution of the aquaporins: a family of water channel proteins. Histochem Cell Biol. 1995;104:1–9. doi: 10.1007/BF01464780. [DOI] [PubMed] [Google Scholar]
- Burg MB. Tubular chloride transport and the mode of action of some diuretics. Kidney Int. 1976;9:189–197. doi: 10.1038/ki.1976.20. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocynate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci U S A. 1984;81:991–995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi K, Marumo F. Water channels. Curr Opin Nephrol Hyper. 1995;4:392–397. doi: 10.1097/00041552-199509000-00003. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Uchida S, Hara Y, Hirata Y, Marumo F, Sasaki S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature. 1993;361:549–552. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- Hebert SC, Andreoli TE. Control of NaCl transport in the thick ascending limb. Am J Physiol. 1984a;253:F745–756. doi: 10.1152/ajprenal.1984.246.6.F745. [DOI] [PubMed] [Google Scholar]
- Hebert SC, Andreoli TE. Effect of antidiuretic hormone on cellular conductive pathways in mouse medullary thick ascending limbs of Henle: II. Determinants of ADH-mediated increase in transepithelial voltage and in net Cl− absorption. J Membr Biol. 1984b;80:221–233. doi: 10.1007/BF01868440. [DOI] [PubMed] [Google Scholar]
- Hebert SC, Culpepper RM, Andreoli TE. NaCl transport in mouse medullary thick ascending limbs. I. Functional nephron heterogeneity and ADH-stimulated NaCl cotransport. Am J Physiol. 1981;241:F412–431. doi: 10.1152/ajprenal.1981.241.4.F412. [DOI] [PubMed] [Google Scholar]
- Igarashi P, Whyte DA, Li K, Nagami GT. Cloning and kidney cell-specific activity of the promoter of the murine renal Na-K-C1 cotransporter gene. J Biol Chem. 1996;271:9666–9674. doi: 10.1074/jbc.271.16.9666. [DOI] [PubMed] [Google Scholar]
- Kim G, Ecelbarger CA, Mitchell C, Packer RK, Wade JB, Knepper MA. Vasopressin increases Na-K-2Cl cotransporter expression in thick ascending limb of Henle's loop. Am J Physiol. 1999;276:F96–103. doi: 10.1152/ajprenal.1999.276.1.F96. [DOI] [PubMed] [Google Scholar]
- Knepper MA. Molecular physiology of urinary concentrating mechanism: Regulation of aquaporin water channels by vasopressin. Am J Physiol. 1997;272:F3–12. doi: 10.1152/ajprenal.1997.272.1.F3. [DOI] [PubMed] [Google Scholar]
- Knepper MA, Rector FC. Urine concentration and dilution. In: Brenner BM, Rector FC, editors. The Kidney: Diagnosis and Treatment. 5. Vol. 1. Philadelphia: Saunders WB; 1996a. pp. 532–570. [Google Scholar]
- Knepper MA, Wade JB, Terris J, Ecelbarger CA, Marples D, Mandon B, Chou CL, Kishore BK, Nielsen S. Renal aquaporins. Kidney Int. 1996b;49:1712–1717. doi: 10.1038/ki.1996.253. [DOI] [PubMed] [Google Scholar]
- Ledoussal C, Woo AL, Miller ML, Shull GE. Loss of the NHE2 Na+/H+ exchanger has no apparent effect on diarrheal state of NHE3-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1385–1396. doi: 10.1152/ajpgi.2001.281.6.G1385. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Smith BL, Agre P, Knepper MA. Quantification of Aquaporin-CHIP water channel protein in microdissected renal tubules by fluorescence-based ELISA. J Clin Invest. 1995;95:422–428. doi: 10.1172/JCI117672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Amlal H, Schultheis PJ, Galla JH, Shull GE, Soleimani M. HCO3− reabsorption in renal collecting duct of NHE-3-deficient mouse: a compensatory response. Am J Physiol. 1999;276:F914–921. doi: 10.1152/ajprenal.1999.276.6.F914. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Chou CL, Marples D, Christensen EI, Kishore BK, Knepper MA. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci U S A. 1995;92:1013–1017. doi: 10.1073/pnas.92.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Digiovanni SR, Christensen EI, Knepper MA, Harris HW. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci U S A. 1993b;90:11663–11667. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Maunsbach AB, Ecelbarger CA, Knepper MA. Ultrastructural localization of Na-K-2Cl cotransporter in thick ascending limb and macula densa of rat kidney. Am J Physiol. 1998;275:F885–893. doi: 10.1152/ajprenal.1998.275.6.F885. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Smith BL, Christensen EI, Knepper MA, Agre P. CHIP28 water channels are localized in constitutively water-permeable segments of the nephron. J Cell Biol. 1993a;120:371–383. doi: 10.1083/jcb.120.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabolic I, Valenti G, Verbavatz JM, Van Hoek AN, Verkman AS, Ausiello DA, Brown D. Localization of the CHIP28 water channel in rat kidney. Am J Physiol. 1992;263:C1225–1233. doi: 10.1152/ajpcell.1992.263.6.C1225. [DOI] [PubMed] [Google Scholar]
- Sands JM, Kokko JP. Current concepts of the countercurrent multiplication system. Kidney Int Suppl. 1996;57:S93–99. [PubMed] [Google Scholar]
- Schultheis PJ, Clark LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle JJ, Duffy TM, Doetschman T, Wang T, Giebish G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- Schwartz MJ, Kokko JP. Urinary concentrating defect of adrenal insufficiency. Permissive role of adrenal steroids on the hydroosmotic response across the rabbit cortical collecting tubule. J Clin Invest. 1980;166:234–242. doi: 10.1172/JCI109849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Sasaki S, Fushimi K, Marumo F. The isolation of human aquaporin-CD gene. J Biol Chem. 1994;269:23451–23455. [PubMed] [Google Scholar]
- Work J, Jamison RL. Effect of adrenalectomy on transport in the rat medullary thick ascending limb. J Clin Invest. 1987;80:1160–1164. doi: 10.1172/JCI113174. [DOI] [PMC free article] [PubMed] [Google Scholar]