

Abstract
Patch pipettes were used to record currents in whole-cell configuration to study the effects of group II metabotropic glutamate receptor (mGluR) stimulation on synaptic transmission in slices of rat subthalamic nucleus. Evoked glutamatergic excitatory postsynaptic currents (EPSCs) were reversibly reduced by the selective group II mGluR agonist (2’S,2’R,3’R)-2-(2’,3’-dicarboxycyclopropyl)glycine (DCG IV) in a concentration-dependent manner, with an IC50 of 0.19 ± 0.05 µm. DCG IV (1 µm) had no effect on inhibitory postsynaptic currents mediated by GABA. DCG IV-induced inhibition of EPSCs was reversed by the selective group II mGluR antagonist LY 341495 (100 nm) and mimicked by another selective group II agonist (2S,1’S,2’S)-2-(carboxycyclopropyl)glycine (l-CCG-I). Inhibition of EPSC amplitude by DCG IV and l-CCG-I was associated with an increase in the paired-pulse ratio of EPSCs. The protein kinase C (PKC) activator phorbol 12-myristate 13-acetate (2 µm) reduced the inhibitory effect of DCG IV on EPSCs. However, the response to DCG IV was not affected by the protein kinase A (PKA) activator forskolin (20 µm), by the adenylyl cyclase inhibitor MDL 12230A (20 µm), or by the phosphodiesterase inhibitor Ro 20–1724 (50 µm). DCG IV-induced inhibition of EPSCs was reduced by the non-selective protein kinase inhibitors H-7 (100 µm), H-8 (50 µm) and HA-1004 (100 µm). These results suggest that group II mGluR stimulation acts presynaptically to inhibit glutamate release by a PKC-dependent mechanism in the subthalamic nucleus.
The subthalamic nucleus (STN) consists of glutamate-containing neurons that play a pivotal role in regulating the output of the basal ganglia. Excitatory pathways from the STN project to the two major output nuclei of the basal ganglia: the substantia nigra reticulata and the globus pallidus interna in human brain, which is also known as the entopeduncular nucleus in rodents (Parent & Hazrati, 1995). Afferent pathways to the STN arise from the globus pallidus externa, which contains GABA, whereas excitatory glutamate-containing pathways arise from the cerebral cortex (Canteras et al. 1988; Fujimoto & Kita, 1993), the pedunculopontine tegmental nucleus (Hammond et al. 1983), and the parafascicular thalamic complex (Mouroux et al. 1995). Because of its excitatory influence on basal ganglia output, the STN is thought to play a central role in the control of normal movement and in the pathophysiology of many movement disorders. In Parkinson's disease, which is caused by chronic depletion of dopamine in the basal ganglia, increased output from the STN is thought to contribute to the rigidity, bradykinesia, and tremor that is characteristic of this disease (Wichmann & DeLong, 1996). Strategies designed to reduce STN output, such as surgical ablation and high frequency stimulation of the STN, have proved to be useful in alleviating many signs of parkinsonism in patients with Parkinson's disease (Limousin et al. 1995; Kumar et al. 1998) and in animals with experimentally induced parkinsonism (Bergman et al. 1990; Gao et al. 1999). Clearly, the STN has emerged as an important target of therapy for the treatment of Parkinson's disease.
There has also been considerable interest in ligands for metabotropic glutamate receptors (mGluRs) that may alter STN output as possible treatment of Parkinson's disease. To date, eight mGluR subtypes have been cloned, and they have been classified into three groups (I, II and III) based on the homology of the amino acid sequence, coupling to second messenger systems, and receptor pharmacology (Rouse et al. 2000). Group I mGluRs include receptor subtypes mGluR1 and mGluR5; members of this group couple to Gq and activate phospholipase C, which releases inositol trisphosphate and activates protein kinase C (PKC). Group II and III mGluRs couple to Gi/Go, and inhibit adenylyl cyclase. Group II mGluRs include receptor subtypes mGluR2 and mGluR3, whereas group III mGluRs include receptor subtypes mGluR4, mGluR6, mGluR7 and mGluR8. In situ hybridization studies have shown that the STN expresses message for members of the group I (mGluR1/5) and group II (mGluR2/3) families of receptor (Ohishi et al. 1993; Testa et al. 1994). Immunohistochemical studies also reveal expression of protein for group I (Awad et al. 2003) and group II (Testa et al. 1998) mGluRs in the STN. Behavioural studies in animals have shown that group II mGluR agonists can reverse parkinsonism produced by haloperidol (Ossowska et al. 2002) and reserpine (Dawson et al. 2000). Moreover, injection of the non-selective group I/II mGluR agonist 1-aminocyclopentane-1S,3R-dicarboxylic acid (trans-ACPD) into STN caused contralateral rotational behaviour (Kaatz & Albin, 1995). These reports support the hypothesis that group II mGluRs can play a role in the control of movement.
The activation of group II mGluRs has been shown to inhibit synaptic transmission in many regions of the basal ganglia, including the substantia nigra reticulata (Bradley et al. 2000), dopamine neurons of the ventral midbrain (Bonci et al. 1997; Wigmore & Lacey, 1998), and the striatum (Lovinger & McCool, 1995). However, Awad-Granko & Conn (2001) recently reported that activation of group I or III – but not group II – mGluRs caused depression of EPSCs in STN neurons. We now report our results showing that group II mGluRs are functionally localized at glutamatergic afferent terminals in the STN, and their activation inhibits excitatory synaptic transmission by a PKC-dependent mechanism.
Methods
Tissue preparation
Horizontal slices of midbrain (300 µm thick) were prepared from male Sprague-Dawley rats (120–180 g; Simonsen, Gilroy, CA, USA) as described previously (Shen & Johnson, 1997). Briefly, rats were anaesthetized with halothane and killed by severing major thoracic vessels, in accordance with institutional guidelines. The brain was rapidly removed and slices containing caudal diencephalon and rostral midbrain were cut in cold physiological saline with a vibratome. A slice containing the STN was then placed on a supporting net and submerged in a continuously flowing solution (2 ml min−1) of the following composition (mm): NaCl, 126; KCl, 2.5; CaCl2, 2.4; MgCl2, 1.2; NaH2PO4, 1.2; NaHCO3, 19; glucose, 11; gassed with 95 % O2 and 5 % CO2 (pH 7.4) at 36 °C. Using a dissection microscope for visual guidance, the STN was located as grey matter approximately 2.7 mm lateral to the midline and 2 mm rostral to the centre of the substantia nigra pars reticulata.
Electrophysiological recordings
Whole-cell recordings were made with pipettes containing (mm): potassium gluconate, 130; MgCl2, 2; CaCl2, 1; EGTA, 11; Hepes, 10; ATP, 1.5; GTP, 0.3 (pH 7.3). Membrane currents were recorded under voltage clamp (−70 mV) and amplified with an Axopatch-1D amplifier. Data were acquired using a personal computer with a Digidata analog/digital interface and analysed using pCLAMP software (Axon Instruments, Foster City, CA, USA). Holding currents were recorded continuously using a MacLab analog/digital interface and Chart software (AD Instruments, Castle Hill, Australia) and a Macintosh computer. Series resistance was compensated electronically 50–80 % to 10–30 MΩ; membrane potentials have been corrected for the liquid junction potential (10 mV).
Synaptic currents
Bipolar stimulation electrodes (tip separation 300–500 µm) were placed in the slice 300 µm rostral to the STN. Synaptic currents were evoked by focal electrical stimulation of the brain slice. A single rectangular pulse (0.1 ms duration) of constant current was used to evoke a synaptic current every 10 s. The amplitude of evoked synaptic currents was measured from the average of three responses. An IPSC mediated by GABAA receptors was isolated pharmacologically by recording in the presence of (±)-2-amino-5-phosphonopentanoic acid (AP5, 50 µm) and 6-cyano-7-nitro-quinoxalone (CNQX, 10 µm) in order to block NMDA and non-NMDA receptors. An EPSC mediated by glutamate receptors was recorded in the presence of picrotoxin (100 µm) to block GABAA receptor-mediated currents.
Drugs
All drugs were dissolved as stock solutions either in water or in dimethyl sulfoxide. Stock solutions of drugs were diluted at least 1:1000 to the desired concentration in superfusate immediately prior to their use. Dimethyl sulfoxide, diluted 1:1000 in artificial spinal fluid, had no effect on either holding current or synaptic currents. Approximately 30 s were required for the drug solution to enter the recording chamber. This delay was due to passage of the perfusate through a heat exchanger. (2′S,2′R,3′R)-2-(2′,3′-Dicarboxycyclopropyl)glycine (DCG IV), 4-(3-butoxy-4-methoxyphenyl)methyl-2-imidazolidone (Ro 20–1724), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY 341495), (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (l-CCG-I), forskolin, phorbol 12-myristate 13-acetate (PMA), and (±)-1-(5-isoquinolinesulfonyl)-2-methylpiperazine dihydrochloride (H-7) were from Tocris Cookson Inc. (Ellisville, MO, USA). N-(2-Guanidinoethyl)-5-isoquinolinesulfonamide hydrochloride (HA-1004), N-(2-[methylamino]ethyl)-5-isoquinolinesulfonamide hydrochloride (H-8), cis-N-(2-phenylcyclopentyl)-azacyclotridec-1-en-2-amine monohydrochloride (MDL 12330A), picrotoxin, AP5 and CNQX were obtained from Sigma Chemical Co. (St Louis, MO, USA).
Data analysis
Numerical data in the text and error bars in the figures are expressed as mean ±s.e.m. Student's two-tailed t tests were used to test for significant differences using SigmaStat software (Jandel Scientific, San Rafael, CA, USA) run on a personal computer. A significant difference was accepted when P < 0.05. Prior to analysing data for significance, a Kolmogorov-Smirnov test was performed to ensure that data were distributed normally. In evaluating concentration-dependent drug effects, an IC50 was calculated for the responses from each neuron using the KaleidaGraph curve-fitting program (Synergy Software) on a Macintosh computer. Concentration-response data (expressed as percentage of control) were fitted to the Hill-Langmuir equation: y = ax/(x + b), where y is the percentage effect, a is percentage of maximum effect, x is the drug concentration and b is the concentration that inhibits the effects by 50 % (IC50).
Results
Activation of group II mGluRs inhibits excitatory transmission in the STN
Bipolar stimulation electrodes placed in the rostral region of the slice were used to evoke synaptic currents. In the presence of picrotoxin (100 µm), a single electrical stimulus evoked an inward synaptic current at a holding potential of −70 mV, which was mediated by glutamate receptors because it was completely blocked by CNQX (10 µm) plus AP5 (50 µm). In the presence of AP5 (50 µm) and CNQX (10 µm), focal stimulation evoked an outward synaptic current, which was mediated by GABAA receptors because it was completely blocked by picrotoxin (100 µm). As seen in Fig. 1A, the selective group II mGluR agonist DCG IV (1 µm) had no significant effect on IPSC amplitude (n = 5), whereas this concentration of DCG IV reduced EPSCs by 49 ± 2 % (n = 24). Therefore, further studies were carried out only on EPSCs. As seen in Fig. 1B, DCG IV reversibly reduced the amplitude of evoked EPSCs in a concentration-dependent manner. The time course of the action of DCG IV is shown in Fig. 1C. The effect of DCG IV could be seen within 1 min of starting perfusion, reached its peak within 4 min, and was completely reversed 10–15 min after washout. The concentration-response curve in Fig. 1D shows that DCG IV inhibited EPSCs with an IC50 of 0.19 ± 0.05 µm (n = 4). DCG IV (1 µm) did not significantly affect the holding current (mean = −1.4 ± 0.9 pA at −70 mV; n = 34).
Figure 1. DCG IV reduces excitatory synaptic transmission in the STN.
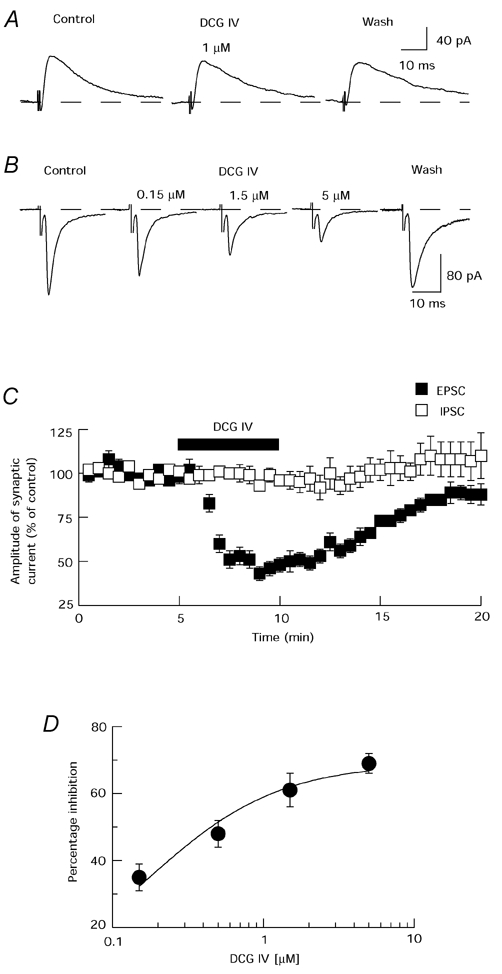
A, the selective group II mGluR agonist DCG IV (1 µm) has no effect on IPSCs. B, DCG IV produces a reversible and concentration-dependent reduction in amplitude of EPSCs. C, averaged data show that DCG IV (1 µm) time-dependently inhibits EPSCs, but has no effect on IPSCs. Each point is the mean of 11 cells for EPSCs and 5 cells for IPSCs. D, concentration- response curve for percentage inhibition of EPSC amplitude by DCG IV. Each data point represents the average of 4 cells.
Interactions between DCG IV and LY 341495, a selective group II mGluR antagonist, are illustrated in Fig. 2A and B. Under control conditions, 5 µm DCG IV reduced the amplitude of EPSCs by 51 ± 6 % (n = 6). However, in the presence of LY 341495 (100 nm), DCG IV only reduced EPSC amplitude by 9 ± 10 % in the same six neurons. LY 341495, when applied alone, had no effect on either EPSC amplitude or holding current. These data support the conclusion that inhibition of EPSCs by DCG IV is mediated by the activation of group II mGluRs.
Figure 2. Antagonism of DCG IV-induced inhibition of EPSCs by LY 341495.
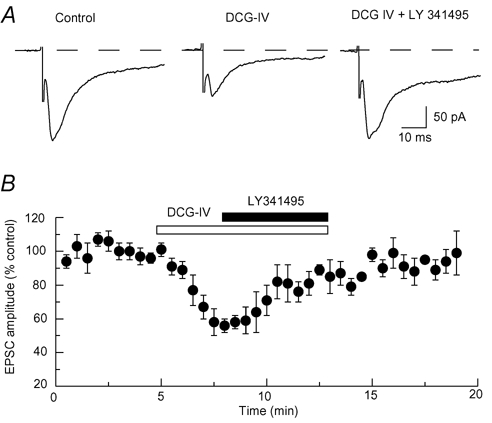
A, representative traces show that the selective group II mGluR antagonist LY 341495 (100 nm) reverses the inhibitory effect of DCG IV (5 µm) on EPSCs. B, summarized results of 6 cells showing that LY 341495 (100 nm) reverses the reduction in EPSCs induced by DCG IV (5 µm).
As demonstrated in Fig. 3A, the selective group II mGluR agonist l-CCG-I mimicked the ability of DCG IV to inhibit EPSCs. This effect of l-CCG-I was reversible, with nearly complete recovery occurring 10 min after stopping perfusion (Fig. 3B). As seen in Fig. 3C, l-CCG-I reduced the peak amplitude of evoked EPSCs in a concentration-dependent manner, with an IC50 value of 1.8 ± 0.3 µm (n = 7). At 10 µm, l-CCG-I reduced EPSCs by 52 ± 5 % (n = 8). These data further support the conclusion that group II mGluR stimulation inhibits glutamate neurotransmission in the STN.
Figure 3. l-CCG-I mimics the action of DCG IV.
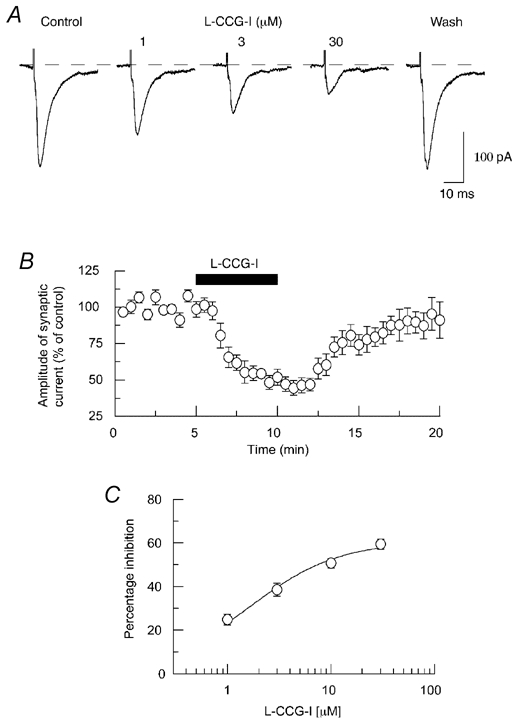
A, traces show that l-CCG-I, another selective group II mGluR agonist, produces a reversible and concentration-dependent reduction in amplitude of EPSCs. B, averaged data show that l-CCG-I (10 µm) inhibits EPSCs in a time-dependent and reversible fashion. Each point is the mean of 8 cells. C, concentration-response curve for percentage inhibition of EPSC amplitude by l-CCG-I. Each data point represents the average of 7 cells.
Group II mGluR agonists increase the EPSC paired-pulse ratio
In order to examine site of action, we tested the effect of DCG IV on EPSCs evoked by pairs of stimuli. Intervals between stimuli (30–50 ms) were adjusted so that the second EPSC was approximately equal to that of the first EPSC. Under control conditions, the ratio of the amplitude of the second EPSC divided by the first one (paired-pulse ratio) was 1.09 ± 0.10 (n = 8). Although DCG IV (5 µm) reduced the amplitude of the first EPSC, DCG IV significantly increased the EPSC paired-pulse ratio to 1.39 ± 0.08 (n = 8), as can be seen in Fig. 4A-C (P = 0.0034, Student's paired t test). l-CCG-I (3 µm) mimicked DCG IV by increasing the EPSC paired-pulse ratio from 1.02 ± 0.07 to 1.22 ± 0.08 (P = 0.00216, Student's paired t test; n = 7; Fig. 4D-F). Paired-pulse facilitation of synaptic transmission suggests that DCG IV acts at a presynaptic site of action (Harris & Cotman, 1985; Dunwiddie & Haas, 1985). For simplicity, further experiments focused on the effects of DCG IV.
Figure 4. Activation of mGluRs increases paired-pulse ratio of EPSCs.
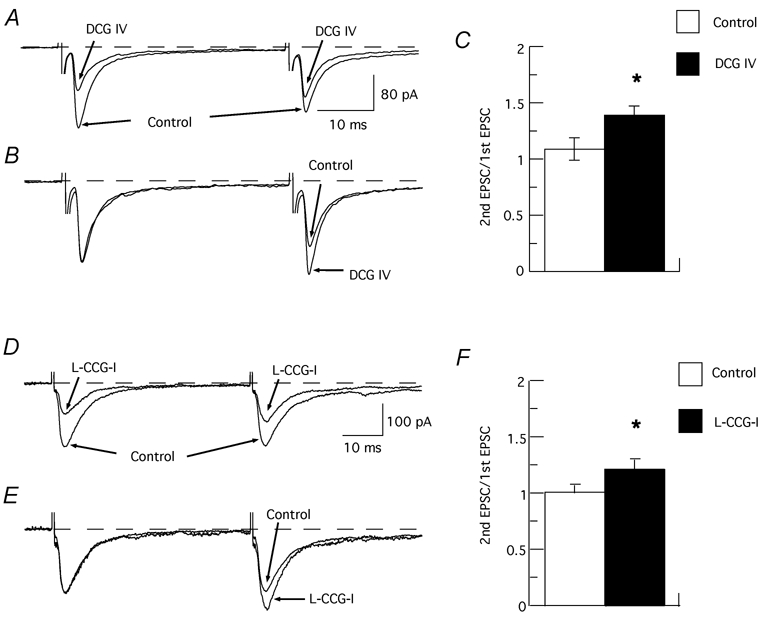
A, superimposed EPSCs evoked by pairs of identical stimuli (40 ms apart) in the absence (control) and presence of DCG IV (1 µm). B, traces are the same as those shown in (A) except that the amplitude of the first EPSC recorded in DCG IV has been scaled to match the amplitude of the first control EPSC. C, bar graph showing an increase in the paired-pulse ratio of EPSCs produced by DCG IV (n = 8). D, superimposed EPSCs evoked by pairs of identical stimuli (50 ms apart) in the absence (control) and presence of l-CCG-I (3 µm). E, traces are the same as those shown in (D) except that the amplitude of the first EPSC recorded in l-CCG-I has been scaled to match the amplitude of the first control EPSC. F, bar graph showing an increase in the paired-pulse ratio of EPSCs produced by l-CCG-I (n = 7). * Significant difference from control (P < 0.01).
Second messengers and group II mGluR regulation of glutamatergic synaptic transmission
Because group II mGluRs are known to be negatively coupled to adenylyl cyclase (Rouse et al. 2000), we investigated the possibility that PKA played a role in DCG IV-induced inhibition of EPSCs. Pretreatment of the slice with forskolin (20 µm), an activator of adenylyl cyclase, failed to affect inhibition of EPSC amplitude by DCG IV (1 µm). Thus, under control conditions, DCG IV reduced the EPSC amplitude by 46 ± 4 %, which was not significantly different when DCG IV was superfused in the presence of forskolin (42 ± 1 %; n = 11; P = 0.66). When slices were preincubated with the adenylyl cyclase inhibitor MDL 12330A (20 µm), DCG IV (1 µm) reduced EPSC amplitude by 48 ± 5 %, which was not different from the control value for DCG IV alone (50 ± 3 %, n = 4; P = 0.686). Furthermore, inhibition of EPSC amplitude by 5 µm DCG IV (63 ± 4 %) was not significantly different from inhibition of EPSCs recorded in the presence of Ro 20–1724 (50 µm), an inhibitor of phosphodiesterase (54 ± 5 %, n = 8; P = 0.115). These data suggest that PKA is not involved in the inhibition of glutamate synaptic transmission by group II mGluR stimulation.
We next examined whether or not PKC played a role in the action of DCG IV. The PKC activator PMA (2 µm) alone increased the amplitude of EPSCs. The mean EPSC amplitude reached 120 ± 10 % of control (n = 6) after treatment with PMA for 20 min. As seen in Fig. 5, PMA (2 µm) significantly reduced the DCG IV-induced inhibition of EPSCs. Under control conditions, DCG IV (1 µm) reduced the EPSC amplitude by 46 ± 3 % (n = 12). However, in the presence of PMA, DCG IV reduced EPSC amplitude by only 22 ± 2 % (n = 12), which was significantly less than control (P = 0.002). This result is consistent with the hypothesis that DCG IV reduces EPSCs by inhibiting PKC activity, and that the inhibition of PKC activity can be successfully opposed by a phorbol ester. To test the role of PKC further, we examined the effects of protein kinase inhibitors on EPSC amplitude. Although neither of the protein kinase inhibitors H-7 (100 µm) or H-8 (50 µm) altered the control amplitude of EPSCs, both agents significantly reduced the effect of DCG IV on EPSCs. As shown in Fig. 6, DCG IV (1 µm) only reduced EPSC amplitude by 20 ± 4 % in the presence of H-7 (n = 7), and by 20 ± 3 % in H-8 (n = 10), both of which were significantly less than inhibition of EPSCs by DCG IV alone (P < 0.001). We also examined the effects of two different concentrations of the protein kinase inhibitor HA-1004, which has a high affinity for PKA (Ki value of 2.3 µm) and a low affinity for PKC (Ki value of 40 µm) (Hidaka et al. 1984). In the presence of 100 µm HA-1004, DCG IV (1 µm) reduced EPSC amplitude by only 26 ± 3 % (n = 14), which was significantly less than the 48 ± 3 % reduction in amplitude produced by DCG IV alone (P < 0.001). In contrast, DCG IV (1 µm), in the presence of 10 µm HA-1004, reduced EPSCs by 52 ± 3 % (n = 12), which was not different from the average control value. These data suggest that inhibition of PKC impairs the inhibitory action of DCG IV on excitatory transmission.
Figure 5. Activation of PKC reduces the EPSC inhibition induced by DCG IV.
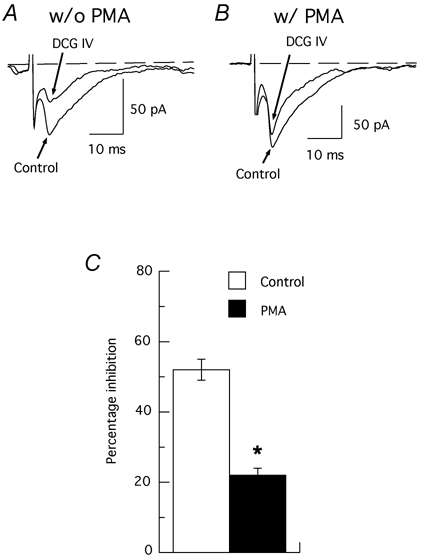
A and B, superimposed traces obtained from one experiment showing the inhibition of EPSCs by DCG IV (1 µm) in the absence (w/o PMA, A) and presence (w/ PMA, B) of PMA (2 µm). Note that PMA markedly prevents DCG IV-induced inhibition of EPSCs. C, summary bar graph indicating that the PKC activator PMA partially prevents DCG IV-induced inhibition of EPSCs. * Significant difference from control (n = 12; P = 0.002).
Figure 6. Effects of protein kinase inhibitors on DCG IV-induced inhibition of EPSCs.

A, summarized results demonstrating the inhibition of EPSCs by DCG IV (1 µm) in the absence (control) and presence of protein kinase inhibitors H-8 (50 µm) and HA-1004 (10 and 100 µm). H-8 and 100 µm HA-1004 significantly reduce the DCG IV-induced inhibition of EPSCs. Note that 10 µm HA-1004 does not reduce the effect of DCG IV. B, bar graph of summarized data indicating that the non-selective protein kinase inhibitors H-8 (50 µm), H-7 (100 µm) and HA-1004 (100 µm) reduce DCG IV-induced inhibition of EPSCs, whereas 10 µm HA-1004 has no effect. * Significant difference from control (P < 0.001).
Discussion
The principal findings in the present study are: (1) group II mGluR stimulation inhibits glutamate-mediated, but not GABA-mediated, synaptic transmission in the STN by a presynaptic mechanism of action, and (2) PKC, but not PKA, plays a role in mediating this action of group II mGluRs.
Our finding that DCG IV and l-CCG-I increased the paired-pulse ratio of evoked glutamatergic EPSCs suggests that group II mGluR agonists act presynaptically to inhibit glutamate release in the STN. Presynaptic inhibition of glutamatergic synaptic transmission by group II mGluRs has been shown in a variety of central neurons, including those in the hippocampus (Vignes et al. 1995; Ugolini & Bordi, 1995; Bushell et al. 1996), dentate gyrus (Kamiya et al. 1996), striatum (Lovinger & McCool, 1995), and dopamine neurons of the ventral midbrain (Bonci et al. 1997; Wigmore & Lacey, 1998; Manzoni & Williams, 1999). However, our results differ from those of Awad-Granko & Conn (2001). who reported that group II mGluR stimulation had no effect on electrically evoked EPSCs recorded in STN neurons of the rat brain slice. There were several differences in experimental procedure that might contribute to these different results, such as their use of parasagittal (rather than horizontal) slices, recording at room temperature (rather than 36 °C), and the use of LY 354740 as a selective group II mGluR agonist (compared to our use of DCG IV and l-CCG-I). But perhaps the most significant difference between the study by Awad-Granko & Conn and ours is their use of young, 15- to 18-day-old rats, compared to our use of adult rat tissue. This is a consideration, because expression of mGluRs in the brain is known to change dramatically during development (Condorelli et al. 1992), with more extensive expression of group II mGluRs in adulthood compared to perinatal rats (Jokel et al. 2001). Although our study used the selective group II agonist DCG IV rather than LY 354740, we showed that DCG IV reduced glutamate transmission effectively and with high potency (IC50 0.19 µm), and that this effect was sensitive to the selective group II antagonist LY 341495 and mimicked by another selective group II agonist l-CCG-I. These results clearly support the conclusion that group II mGluR stimulation inhibits glutamate-mediated synaptic transmission in the adult rat STN.
Our findings that the DCG IV-induced inhibition of EPSCs was not affected by forskolin, MDL 12330A, or Ro 20–1724 – all of which modify levels of cAMP – suggests that PKA does not play a role in this action of group II mGluRs. This is surprising, given that group II mGluRs are known to inhibit adenylyl cyclase (Conn & Pin, 1997), and inhibition of PKA activity has been implicated as a mechanism for the inhibition of glutamate release at glutamatergic synapses in hippocampus (Kamiya & Yamamoto, 1997; Schaffhauser et al. 2000). Instead, our data showing that the PKC activator PMA caused a significant reduction in the effect of DCG IV on EPSCs suggest that inhibition of glutamate release by group II mGluR stimulation may be mediated by PKC in the STN. This result agrees with those of others who showed that phorbol esters impair the ability of presynaptic group II mGluRs to reduce glutamatergic synaptic transmission in the hippocampus (Kamiya & Yamamoto, 1997; Macek et al. 1998) and striatum (Tyler & Lovinger, 1995). Possible explanations for how PKC activation inhibits group II mGluR-mediated effects include direct phosphorylation of group II mGluRs, which causes G protein uncoupling (Chavis et al. 1994; Macek et al. 1998), or potentiation of presynaptic voltage-dependent calcium channels that could otherwise be inhibited by mGluR2/3 stimulation (Swartz et al. 1993; Choi & Lovinger, 1996). However, we should note that our results also showed that the PKC inhibitors H-7, H-8 and HA-1004 reduced the inhibitory effect of DCG IV on EPSCs, and that this result is not consistent with the hypothesis that group II mGluRs reduce glutamate release by inhibiting PKC activity. It is possible that these PKC inhibitors reduce the action of DCG IV via an alternate site of action, which causes presynaptic inhibition via a diffusible second messenger. A similar mechanism has been proposed to explain the PKC-dependent inhibition of glutamate release caused by stimulation of dopamine D1-like receptors in slices of rat nucleus accumbens (Chergui & Lacey, 1999).
Because increased output from the STN is thought to underlie many of the symptoms of Parkinson's disease (Wichmann & DeLong, 1996), there has been considerable interest in developing pharmacological methods for reducing STN output as a treatment of this disease. Much recent work has focused on the group I family of mGluRs, because mGluR5 stimulation excites STN neurons (Abbott et al. 1997; Awad et al. 2003). Moreover, treatment with mGluR5 antagonists has been shown to improve motor function in animal models of parkinsonism (Breysse et al. 2002; Ossowska et al. 2002). However, results of our present study suggest that mGluR2/3 stimulation, by reducing glutamate-mediated EPSCs in STN neurons, may also inhibit STN output and could thereby improve signs of parkinsonism. Moreover, group II mGluR agonists may be relatively selective for reducing STN output because our work showed that the mGluR2/3 agonist DCG IV had no effect on GABA-mediated inhibitory input to STN neurons. Previous studies in awake rats showed that injection of a group II mGluR agonist into the STN produced contralateral rotatory behaviour consistent with activation of STN output (Kearney & Albin, 2000), whereas other studies showed that systemic or intraventricular injection of mGluR2/3 agonists alleviates parkinsonism produced by reserpine (Dawson et al. 2000) or haloperidol (Ossowska et al. 2002). Picconi et al. (2002) found that dopamine denervation selectively potentiated the presynaptic inhibitory effect of group II mGluRs on corticostriatal excitatory synaptic transmission without affecting responses mediated by group I and III mGluRs, which further supports a significant role for group II mGluRs in the pathophysiology of Parkinson's disease. Further studies using animal models of parkinsonism are warranted to test the hypothesis that group II mGluR agonists might be effective in the treatment for Parkinson's disease.
Acknowledgments
This study was supported by USPHS grant NS 38175, the Roger Duvoisin, M.D. Fellowship of The American Parkinson Disease Association, Inc. (K.-Z. S.), and the Portland VA Parkinson's Disease Research, Education, and Clinical Center.
References
- Abbott A, Wigmore MA, Lacey MG. Excitation of rat subthalamic nucleus neurones in vitro by activation of a group I metabotropic glutamate receptor. Brain Res. 1997;766:162–167. doi: 10.1016/s0006-8993(97)00550-7. [DOI] [PubMed] [Google Scholar]
- Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2003;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad-Granko H, Conn PJ. Activation of groups I or III metabotropic glutamate receptors inhibits excitatory transmission in the rat subthalamic nucleus. Neuropharmacology. 2001;41:32–41. doi: 10.1016/s0028-3908(01)00047-8. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bonci A, Grillner P, Siniscalchi A, Mercuri NB, Bernardi G. Glutamate metabotropic receptor agonists depress excitatory and inhibitory transmission on rat mesencephalic dopamine neurons. Eur J Neurosci. 1997;9:2359–2369. doi: 10.1111/j.1460-9568.1997.tb01653.x. [DOI] [PubMed] [Google Scholar]
- Bradley SR, Marino MJ, Wittmann M, Rouse ST, Awad H, Levey AI, Conn PJ. Activation of group II metabotropic glutamate receptors inhibits synaptic excitation of the substantia nigra pars reticulata. J Neurosci. 2000;20:3085–3094. doi: 10.1523/JNEUROSCI.20-09-03085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breysse N, Baunez C, Spooren W, Gasparini F, Amalric M. Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of parkinsonism. J Neurosci. 2002;22:5669–5678. doi: 10.1523/JNEUROSCI.22-13-05669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell TJ, Jane DE, Tse HW, Watkins JC, Garthwaite J, Collingridge GL. Pharmacological antagonism of the actions of group II and III mGluR agonists in the lateral perforant path of rat hippocampal slices. Br J Pharmacol. 1996;117:1457–1462. doi: 10.1111/j.1476-5381.1996.tb15306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canteras NS, Shammah-Lagnado SJ, Silva BA, Ricardo JA. Somatosensory inputs to the subthalamic nucleus: a combined retrograde and anterograde horseradish peroxidase study in the rat. Brain Res. 1988;458:53–64. doi: 10.1016/0006-8993(88)90495-7. [DOI] [PubMed] [Google Scholar]
- Chavis P, Shinozaki H, Bockaert J, Fagni L. The metabotropic glutamate receptor types 2/3 inhibit L-type calcium channels via a pertussis toxin-sensitive G-protein in cultured cerebellar granule cells. J Neurosci. 1994;14:7067–7076. doi: 10.1523/JNEUROSCI.14-11-07067.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chergui K, Lacey MG. Modulation by dopamine D1-like receptors of synaptic transmission and NMDA receptors in rat nucleus accumbens is attenuated by the protein kinase C inhibitor Ro 32–0432. Neuropharmacology. 1999;38:223–231. doi: 10.1016/s0028-3908(98)00187-7. [DOI] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J Neurosci. 1996;16:36–45. doi: 10.1523/JNEUROSCI.16-01-00036.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli DF, Dell'Albani P, Amico C, Casabona G, Genazzani AA, Sortino MA, Nicoletti F. Development profile of metabotropic glutamate receptor mRNA in rat brain. Mol Pharmacol. 1992;41:660–664. [PubMed] [Google Scholar]
- Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Dawson L, Chadha A, Megalou M, Duty S. The group II metabotropic glutamate receptor agonist, DCG-IV, alleviates akinesia following intranigral or intraventricular administration in the reserpine-treated rat. Br J Pharmacol. 2000;129:541–546. doi: 10.1038/sj.bjp.0703105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Haas HL. Adenosine increases synaptic facilitation in the in vitro rat hippocampus: evidence for a presynaptic site of action. J Physiol. 1985;369:365–377. doi: 10.1113/jphysiol.1985.sp015907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Kita H. Response characteristics of subthalamic neurons to the stimulation of the sensorimotor cortex in the rat. Brain Res. 1993;609:185–192. doi: 10.1016/0006-8993(93)90872-k. [DOI] [PubMed] [Google Scholar]
- Gao DM, Benazzouz A, Piallat B, Bressand K, Ilinsky IA, Kultas-Ilinsky K, Benabid AL. High-frequency stimulation of the subthalamic nucleus suppresses experimental resting tremor in the monkey. Neuroscience. 1999;88:201–212. doi: 10.1016/s0306-4522(98)00235-8. [DOI] [PubMed] [Google Scholar]
- Hammond C, Rouzaire-Dubois B, Feger J, Jackson A, Crossman AR. Anatomical and electrophysiological studies on the reciprocal projections between the subthalamic nucleus and nucleus tegmenti pedunculopontinus in the rat. Neuroscience. 1983;9:41–52. doi: 10.1016/0306-4522(83)90045-3. [DOI] [PubMed] [Google Scholar]
- Harris EW, Cotman CW. Effects of synaptic antagonists on perforant path paired-pulse plasticity: differentiation of pre- and postsynaptic antagonism. Brain Res. 1985;334:348–353. doi: 10.1016/0006-8993(85)90230-6. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Jokel ES, Garduno ER, Ariano MA, Levine MS. Metabotropic glutamate receptors mGluR1α and mGluR2/3 display dynamic expression patterns in developing rat striatum. Dev Neurosci. 2001;23:1–6. doi: 10.1159/000048690. [DOI] [PubMed] [Google Scholar]
- Kaatz KW, Albin RL. Intrastriatal and intrasubthalamic stimulation of metabotropic glutamate receptors: a behavioral and Fos immunohistochemical study. Neuroscience. 1995;66:55–65. doi: 10.1016/0306-4522(94)00568-p. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fibre synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Albin RL. Intrasubthalamic nucleus metabotropic glutamate receptor activation: a behavioral, Fos immunohistochemical and [14C]2-deoxyglucose autoradiographic study. Neuroscience. 2000;95:409–416. doi: 10.1016/s0306-4522(99)00439-x. [DOI] [PubMed] [Google Scholar]
- Kumar R, Lozano AM, Kim YJ, Hutchison WD, Sime E, Halket E, Lang AE. Double-blind evaluation of subthalamic nucleus deep brain stimulation in advanced Parkinson's disease. Neurology. 1998;51:850–855. doi: 10.1212/wnl.51.3.850. [DOI] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Benazzouz A, Hoffmann D, Le Bas JF, Broussolle E, Perret JE, Benabid AL. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995;345:91–95. doi: 10.1016/s0140-6736(95)90062-4. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, McCool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGluR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci. 1998;18:6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouroux M, Hassani OK, Feger J. Electrophysiological study of the excitatory parafascicular projection to the subthalamic nucleus and evidence for ipsi- and contralateral controls. Neuroscience. 1995;67:399–407. doi: 10.1016/0306-4522(95)00032-e. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience. 1993;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- Ossowska K, Konieczny J, Wardas J, Golembiowska K, Wolfarth S, Pilc A. The role of striatal metabotropic glutamate receptors in Parkinson's disease. Amino Acids. 2002;23:193–198. doi: 10.1007/s00726-001-0128-0. [DOI] [PubMed] [Google Scholar]
- Parent A, Hazrati L-N. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- Picconi B, Pisani A, Centonze D, Battaglia G, Storto M, Nicoletti F, Bernardi G, Calabresi P. Striatal metabotropic glutamate receptor function following experimental parkinsonism and chronic levodopa treatment. Brain. 2002;125:2635–2645. doi: 10.1093/brain/awf269. [DOI] [PubMed] [Google Scholar]
- Rouse ST, Marino MJ, Bradley SR, Awad H, Wittmann M, Conn PJ. Distribution and roles of metabotropic glutamate receptors in the basal ganglia motor circuit: implications for treatment of Parkinson's disease and related disorders. Pharmacol Ther. 2000;88:427–435. doi: 10.1016/s0163-7258(00)00098-x. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Cai Z, Hubalek F, Macek TA, Pohl J, Murphy TJ, Conn PJ. cAMP-dependent protein kinase inhibits mGluR2 coupling to G-proteins by direct receptor phosphorylation. J Neurosci. 2000;20:5663–5670. doi: 10.1523/JNEUROSCI.20-15-05663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K-Z, Johnson SW. Presynaptic GABAB and adenosine A1 receptors regulate synaptic transmission to rat substantia nigra reticulata neurones. J Physiol. 1997;505:153–163. doi: 10.1111/j.1469-7793.1997.153bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- Testa CM, Friberg IK, Weiss SW, Standaert DG. Immunohistochemical localization of metabotropic glutamate receptors mGluR1a and mGluR2/3 in the rat basal ganglia. J Comp Neurol. 1998;390:5–19. [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler EC, Lovinger DM. Metabotropic glutamate receptor modulation of synaptic transmission in corticostriatal co-cultures: role of calcium influx. Neuropharmacology. 1995;34:939–952. doi: 10.1016/0028-3908(95)00066-f. [DOI] [PubMed] [Google Scholar]
- Ugolini A, Bordi F. Metabotropic glutamate group II receptors are responsible for the depression of synaptic transmission induced by ACPD in the dentate gyrus. Eur J Pharmacol. 1995;294:403–410. doi: 10.1016/0014-2999(95)00560-9. [DOI] [PubMed] [Google Scholar]
- Vignes M, Clarke VR, Davies CH, Chambers A, Jane DE, Watkins JC, Collingridge GL. Pharmacological evidence for an involvement of group II and group III mGluRs in the presynaptic regulation of excitatory synaptic responses in the CA1 region of rat hippocampal slices. Neuropharmacology. 1995;34:973–982. doi: 10.1016/0028-3908(95)00093-l. [DOI] [PubMed] [Google Scholar]
- Wichmann T, DeLong MR. Functional and pathophysiological models of the basal ganglia. Curr Opin Neurobiol. 1996;6:751–758. doi: 10.1016/s0959-4388(96)80024-9. [DOI] [PubMed] [Google Scholar]
- Wigmore MA, Lacey MG. Metabotropic glutamate receptors depress glutamate-mediated synaptic input to rat midbrain dopamine neurones in vitro. Br J Pharmacol. 1998;123:667–674. doi: 10.1038/sj.bjp.0701662. [DOI] [PMC free article] [PubMed] [Google Scholar]