

Abstract
The refinement and plasticity of neuronal connections require synaptic activity and neurotrophin signalling; their specific contributions and interplay are, however, poorly understood. We show here that brain-derived neurotrophic factor (BDNF) increased spine density in apical dendrites of CA1 pyramidal neurones in organotypic slice cultures prepared from postnatal rat hippocampal slices. This effect was observed also in the absence of action potentials, and even when miniature synaptic transmission was inhibited with botulinum neurotoxin C (BoNT/C). There were, however, marked differences in the morphology of individual spines induced by BDNF across these different levels of spontaneous ongoing synaptic activity. During both normal synaptic transmission, and when action potentials were blocked with TTX, BDNF increased the proportion of stubby, type-I spines. However, when SNARE-dependent vesicular release was inhibited with BoNT/C, BDNF increased the proportion of thin, type-III spines. Our results indicate that BDNF increases spine density irrespective of the levels of synaptic transmission. In addition, miniature synaptic transmission provides sufficient activity for the functional translation of BDNF-triggered spinogenesis into clearly defined morphological spine types, favouring those spines potentially responsible for coordinated Ca2+ transients thought to mediate synaptic plasticity. We propose that BDNF/TrkB signalling represents a mechanism of expression of both morphological and physiological homeostatic plasticity in the hippocampus, leading to a more efficient synaptic information transfer across widespread levels of synaptic activity.
The hypothesis that activity-dependent structural changes in the brain may underlie memory formation following learning was postulated more than a century ago (Tanzi, 1893; Cajal, 1909). Recent experimental observations have further extended these ideas as potential mechanisms for the refinement (Katz & Shatz, 1996) and plasticity (Yuste & Bonhoeffer, 2001) of synaptic connections throughout the CNS. As the postsynaptic compartment of most excitatory synapses, and due to their highly dynamic and plastic characteristics, dendritic spines have received a great deal of attention as the site of enduring, activity-dependent structural changes underlying learning and memory (Segal & Andersen, 2000; Sorra & Harris, 2000; Yuste & Bonhoeffer, 2001; Nimchinsky et al. 2002). In particular, variations in synaptic activity – and the ensuing Ca2+ transients – have been repeatedly demonstrated to induce pronounced effects on dendritic spine formation and morphology (Engert & Bonhoeffer, 1999; Maletic-Savatic et al. 1999; Segal & Andersen, 2000; Yuste & Bonhoeffer, 2001). Changes in spine morphology are considered relevant to synaptic function and plasticity, as thin spines with long and narrow necks may isolate Ca2+ transients from the parent dendrite; accordingly, short and stubby spines may promote more coordinated and widespread Ca2+ transients in the parent dendrite, as well as synchronize Ca2+ signalling among adjacent spines (Segal et al. 2000; Yuste et al. 2000; Nimchinsky et al. 2002). In addition, large spines with conspicuous heads have a higher density of AMPA receptors, in addition to being more stable than thinner and longer spines (Matsuzaki et al. 2001). However, the precise relationships between synaptic activity, dendritic spine formation and spine morphology remain obscure. Because of their effects on excitatory synapses (Poo, 2001; Tyler et al. 2002a, b; Vicario-Abejon et al. 2002) neurotrophins in general, and brain-derived neurotrophic factor (BDNF) in particular, are strong candidates to mediate the activity-dependent refinement of dendritic spines in the CNS.
In the classic model of activity-dependent refinement of neuronal connections – the formation of ocular dominance columns induced by visual experience (Hubel & Wiesel, 1977) – synapses are strengthened based on their ability to translate activity into structural modifications (Katz & Shatz, 1996; Bi & Poo, 2001). The structural translation of activity into appropriately segregated thalamocortical projections is mediated by the interplay between synaptic activity and neurotrophin signalling (Katz & Shatz, 1996; McAllister et al. 1999). Indeed, BDNF has been demonstrated to mediate activity-dependent refinement of synapses in the developing visual cortex (McAllister et al. 1995; Cabelli et al. 1997), and is thought to similarly regulate the formation of hippocampal synapses (Vicario-Abejon et al. 2002).
In addition, BDNF signalling has been found to promote changes in dendritic spine density and structure. Exogenous BDNF has been shown to increase spine density in cultured Purkinje neurones, whereas blockade of endogenous BDNF with TrkB-IgG in the same cells did not reduce spine density, but rather, increased the length of spine necks (Shimada et al. 1998). BDNF/TrkB signalling also increased apical dendritic spine density in CA1 pyramidal neurones of hippocampal slice cultures (Tyler & Pozzo-Miller, 2001). Further, BDNF overexpression in pyramidal neurones of slice cultures from visual cortex caused a destabilization of dendritic spines, suggesting that BDNF induces local dendritic instability, allowing activity-dependent morphological changes in dendritic spines (Horch et al. 1999; Horch & Katz, 2002). As a result of its wide effects on synaptic transmission and plasticity, BDNF may similarly regulate activity-dependent refinement of synaptic connections in the hippocampus (Schinder & Poo, 2000; Tyler et al. 2002a), a brain region of well-known involvement in learning and memory (Shapiro & Eichenbaum, 1999). The specific, activity-dependent consequences of BDNF signalling on dendritic spine size and morphology in the hippocampus, have not yet been addressed. We therefore investigated the effects of BDNF on the formation and the morphology of dendritic spines in CA1 pyramidal neurones in organotypic slice cultures maintained across widespread levels of synaptic transmission.
Methods
Preparation of hippocampal slice cultures
Hippocampal slices were prepared aseptically and maintained in vitro as previously described (Pozzo-Miller et al. 1995). Postnatal day 7 (P7) rats were rapidly decapitated, and the brain immersed in ice-cold Gey's Balanced Salt Solution (BSS), supplemented with glucose (36 mm), and antibiotics and antimycotics (1 u ml−1 penicillin, 1 µg ml−1 streptomycin and 0.25 µg ml−1 amphotericin B). Both hippocampi were dissected and subsequently sliced transversely into 500 µm slices using a custom-made tissue slicer strung with 20 µm-thick tungsten wire (California Fine Wire Company, Grover Beach, CA, USA). After a short incubation at 4 °C, slices were individually plated on the top of tissue culture plate inserts (Millicell-CM, 0.4 µm pore size, Millipore Corporation, Bedford, MA, USA), fed with culture medium from underneath the inserts, and placed in an incubator at 36 °C, 5 % CO2, 98 % relative humidity. Culture medium contained minimum essential media (50 %), Hanks’ (or Earle's) BSS (25 %), heat-inactivated horse serum (20 %), l-glutamine (1 mm) and d-glucose (36 mm). To avoid excessive autofluorescence during fluorescence imaging, all media reagents lacked phenol red. All tissue culture reagents were acquired from Life Technologies (Gaithersburg, MD, USA). Antibiotics, antimycotics or proliferation inhibitors were never included in the culture media. All procedures on experimental animals were performed following national and international ethics guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Alabama at Birmingham.
Hippocampal slices from P7 rats were chosen for preparing the organotypic cultures in order to characterize the actions of BDNF on spine growth and form in a postnatal hippocampal network. In addition, CA1 neurones maintained in organotypic slice culture continue to differentiate with regard to their dendritic and spine morphology, as well as in terms of their synaptic connectivity within the slice with a remarkable similarity to CA1 neurones from age-matched acute hippocampal slices (De Simoni et al. 2003).
With the exception of some slice cultures kept in culture media containing 20 % horse serum (HSM), the concentration of HSM in the culture medium was titrated to reach serum-free conditions over a 48 h period starting at 6 days in vitro (div), as previously described (Tyler & Pozzo-Miller, 2001). By using serum-free medium (Neurocellular II (Biofluids, Rockville, MD, USA) plus B-27 supplement (Life Technologies)) as the control condition, the potential confounding effects due to other growth factors and hormones were eliminated.
Pharmacological treatments
Slice cultures were randomly assigned to one of three experimental conditions: (i) normal synaptic transmission; (ii) blockade of spontaneous action potential-evoked synaptic transmission with the voltage-gated Na+ channel blocker tetrodotoxin (TTX; 1 µm; Sigma, St Louis, MO, USA); and (iii) inhibition of vesicular neurotransmitter release with botulinum neurotoxin C (BoNT/C; 100 ng ml−1; Calbiochem, La Jolla, CA, USA), which cleaves syntaxin-1A (Blasi et al. 1993), a member of the SNARE protein complex required for synaptic vesicle fusion (Südhof, 2000). All treatments began on 9 div, immediately following a 24 h incubation period in control serum-free medium. Slice cultures were treated with BDNF (250 ng ml−1; provided by Amgen, Thousand Oaks, CA, USA) for 48–72 h in the presence and absence of the toxins. In order to determine the specificity of trophic actions induced by BDNF, some cultures were maintained in control medium containing 20 % HSM for 11 div.
Treatments were started at the earliest possible time in vitro (9 div) to avoid excessive recurrent axonal sprouting leading to the increased network activity observed in organotypic slice cultures after 2 weeks in vitro (McBain et al. 1989; Müller et al. 1993; Pozzo-Miller et al. 1994; Collin et al. 1997). Lastly, we decided to use a treatment protocol consistent with that used in our previous studies of the effects of BDNF on synaptic structure and function (Tyler & Pozzo-Miller, 2001).
Electrophysiology
Whole-cell voltage-clamp recordings of miniature excitatory postsynaptic currents (mEPSCs) were performed from visually identified CA1 pyramidal neurones in 11–12 div slice cultures using differential interference contrast imaging with infrared illumination (IR-DIC) using a fixed-stage upright microscope (Zeiss Axioskop FS, Thornwood, NY, USA), as previously described (Pozzo-Miller et al. 1995, 1999). Slice cultures were continuously perfused (1 ml min−1) with artificial cerebrospinal fluid (aCSF) containing (mm): NaCl 124; KCl 2; KH2PO4 1.24; MgSO4 1.3; NaHCO3 17.6; CaCl2 2.5; d-glucose 10; osmolarity was adjusted to 310 mosmol l−1with sucrose; continuously bubbled with 95 % O2/5 % CO2. To record AMPA-mediated mEPSCs, the aCSF contained (mm): TTX 0.5; d-aminophosphonovalerate (APV) 80; and picrotoxin 50. For subsequent spine visualization, cells were filled with the fluorescent dye Alexa-594 included in the patch pipette (absorption at 588 nm, emission at 613 nm; Molecular Probes, Eugene, OR, USA), as previously described (Tyler & Pozzo-Miller, 2001). Patch electrode pipettes were filled with an intracellular solution containing (mm): Alexa-594 0.132; cesium gluconate 120; CsCl 17.5; NaCl 10; Na-Hepes 10; EGTA 0.2; Mg-ATP 2; Na-GTP 0.2; N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride) QX-314 20; 280- 290 mosmol l−1; pH 7.2; the final resistance of these unpolished patch electrodes was 8–10 MΩ. Typical values of access resistances after whole-cell access were <25 MΩ, and whole-cell capacitances were ≈110 pF in all CA1 pyramidal neurones. Membrane currents were acquired and filtered (2 kHz, low-pass Bessel filter) with an Axopatch-200B amplifier (Axon Instruments, Foster City, CA, USA) and digitized (4 kHz; ITC-18, Instrutech, Port Washington, NY, USA) using custom-written acquisition software (TI WorkBench, kindly provided by Dr T. Inoue, Tokyo University, Japan). Digitized data were analysed off-line with Mini-Analysis Program (Synaptosoft, Leonia, NJ, USA) in 5 min epochs.
Dendritic spine imaging
Following 15–20 min of whole-cell access, the patch electrode was gently removed, and slices were fixed overnight in 4 % paraformaldehyde in phosphate-buffered saline (PBS) at room temperature, and subsequently rinsed in PBS prior to mounting for fluorescence microscopy with Vectashield (Vector Laboratories, Burlingame, CA, USA). Distal secondary and tertiary branches of CA1 pyramidal apical dendrites were imaged using an Olympus Fluoview laser scanning confocal microscope (Olympus Fluoview, Mellville, NY, USA) and a ×100 PlanApo (NA 1.4, Olympus) oil immersion lens. Alexa-594 fluorescence was excited with a krypton laser (647 nm), and detected with standard Texas Red filters (Olympus). Confocal images were acquired as a series of optical z-sections at 0.1 µm intervals through each apical dendritic branch. Images of individual optical sections were digitally stored for later quantitative analysis.
Analyses of spine density and morphology
A general concern when using light microscopy to examine small structures such as dendritic spines is a potential lack of sufficient resolving power. Additionally, in relatively thick preparations such as organotypic slice cultures (≈200 µm after 10 div), where objects of interest may be deep into light-scattering tissue, resolution decreases due to the diffraction of excitation and emission light. Therefore, we strictly selected the most superficial pyramidal neurones for all experiments. The majority of dendritic segments imaged were less than 30 µm below the surface of slice cultures. We observed minimal spherical aberration, and found that dendritic spines are readily observable and measurable in these superficial neurones. However, because of the constraints of the optical methods used here, our observations may be biased towards larger spines. Since we focused on relative comparisons across populations of spines, the bias remains constant in all treatments, making the comparisons scientifically valid. Thus, all measurements of spine dimensions presented here should be considered relative and not absolute. Lastly, our measurements of dendritic spine dimensions are in excellent agreement with observations using electron microscopy (Sorra & Harris, 2000).
Dendritic spines were identified as small protrusions extending ≤3.0 µm from the parent dendrite. Fine dendritic processes longer than 3.0 µm were rarely observed in 11–12 div slice cultures; since they are likely to represent filipodia, they were not included in our dendritic spine analyses. Prior to dendritic spine densitometry and morphometry, image stacks were volume-rendered to produce three-dimensional reconstructions using Imaris (Bitplane AG, Zurich, Switzerland). The three-dimensional reconstructions were rotated about the dendritic axis to confirm that no spines extending perpendicular to the parent dendrite were excluded from the analyses. Since no spines were detected along the perpendicular axis, probably because of limited axial resolution, spine densitometry and morphometry were performed in two-dimensional, maximum-intensity z-projections of the individual optical sections.
Individual spines and lengths of dendritic segments were measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA). By reconstructing fluorescent microspheres, we determined that our effective lateral (x-y axis) pixel resolution is 0.09 µm pixel−1. Only spines appearing continuous with their parent dendrites in maximum-intensity z-projections were used for quantitative analyses. Spine density was calculated by quantifying the number of spines per unit length of parent dendrite, and normalized to 10 µm of dendrite. The total dendritic length used for spine densitometry was as follows: serum-free 2200.8 µm; BDNF 2015.9 µm; 20 % HSM 896.0 µm; TTX 2360.1 µm; TTX-BDNF 1871.8 µm; BoNT/C 1612.9 µm; and BoNT/C-BDNF 1267.7 µm.
Classification of spine categories
Dendritic spines were classified following the categories defined by Peters and colleagues (Peters & Kaiserman-Abramof, 1969, 1970), as stubby (type-I), mushroom (type-II) and thin (type-III) types. Individual spines were included in each category based on the specific ratios L/dn and dh/dn, where L is the length of the spine from its base at the dendrite to the tip of its head, dn is the maximum neck diameter and dh is the maximum head diameter, as previously described (Harris et al. 1992; Koh et al. 2002). A schematic of an individual spine indicating the measured parameters is shown in Fig. 1, along with a representative segment of CA1 neurone apical dendrite bearing all three types of spines. Stubby spines have a length similar to the diameter of the neck, which is similar to the diameter of the spine head, all typically ≤1 µm (L≈dn≈dh). Mushroom spines are typically ≤1 µm in length with a neck diameter much smaller than the diameter of the head (dn≪dh). Generally, thin spines are longer than 1 µm, which is much greater than the neck diameter (L≫dn). While the physical dimensions of individual dendritic spines were primarily used to define the morphological categories, they were also quantified and analysed statistically (Table 2). Due to the disparity of spine densities across treatment groups, the quantity of spines in each morphological class were analysed, and are expressed, as the proportion to all spines (total number of spines; T).
Figure 1. Classification of spine types.
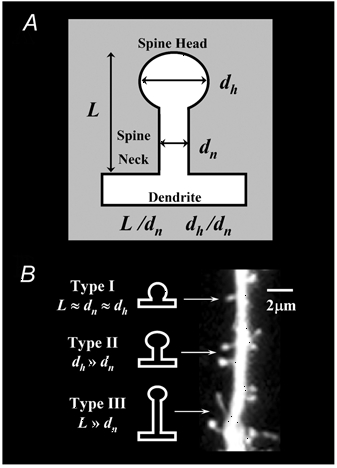
The classification is based on the spine length, the diameter of the head and the diameter of the neck (based on Harris et al. 1992).
Table 2.
Summary of CA1 apical dendritic spine geometry and spine-type proportions by treatment group
| Group | Spine length (μm) | Spine neck diameter (μm) | Spine head diameter (μm) | Type-I proportion | Type-II proportion | Type-III proportion | Slices | Cells |
|---|---|---|---|---|---|---|---|---|
| Serum-free | 0.65 ± 0.02 | 0.25 ± 0.01 | 0.33 ± 0.01 | 0.39 ± 0.04 | 0.38 ± 0.03 | 0.22 ± 0.05 | 4 | 7 |
| BDNF | 0.82 ± 0.03* | 0.43 ± 0.01* | 0.53 ± 0.01* | 0.62 ± 0.03* | 0.23 ± 0.02* | 0.15 ± 0.05 | 4 | 8 |
| TTX | 0.90 ± 0.02* | 0.26 ± 0.01 | 0.37 ± 0.01* | 0.30 ± 0.05 | 0.27 ± 0.02* | 0.43 ± 0.05* | 3 | 6 |
| TTX-BDNF | 0.65 ± 0.02† | 0.31 ± 0.01*† | 0.39 ± 0.01* | 0.58 ± 0.06† | 0.30 ± 0.03 | 0.12 ± 0.03† | 4 | 6 |
| BoNT/C | 0.88 ± 0.03* | 0.49 ± 0.01* | 0.61 ± 0.01* | 0.61 ± 0.07* | 0.28 ± 0.03 | 0.11 ± 0.05 | 3 | 5 |
| BoNT/C-BDNF | 0.91 ± 0.02* | 0.30 ± 0.01*† | 0.40 ± 0.01*† | 0.39 ± 0.04† | 0.25 ± 0.07 | 0.38 ± 0.02*† | 3 | 4 |
| 20% HSM | 0.62 ± 0.02 | 0.25 ± 0.01 | 0.32 ± 0.01 | 0.47 ± 0.03 | 0.33 ± 0.04 | 0.20 ± 0.05 | 3 | 4 |
Values represent the means ±s.e.m. of the geometrical dimensions used to morphologically characterize CA1 apical dendritic spines. The total numbers of slices, cells and spines from which these geometrical data were obtained are also listed for each treatment group.
Significant difference (P < 0.05) compared to serum-free control values
significant difference (P < 0.05) compared to that treatment group's condition control values (i.e. TTX-BDNF vs. TTX).
Statistical analysis
Data were statistically analysed using analysis of variance (ANOVA), Tukey's procedure for multiple comparisons, and independent t tests using SPSS (SPSS Inc., Chicago, IL, USA). Values of P < 0.05 were considered significant. All data shown are presented as means ± standard error of the mean (s.e.m.).
Results
BDNF increases the frequency of mEPSCs, spine density, spine size and the proportion of stubby, type-I spines
Long-term (48–72 h) application of BDNF (250 ng ml−1) to 11–12 days in vitro (div) hippocampal slice cultures maintained in serum-free medium increased the mean frequency of AMPA-mediated mEPSCs recorded from CA1 pyramidal neurones from 0.3 ± 0.09 Hz (six cells from three slices) to 0.69 ± 0.07 Hz (six cells from four slices; P < 0.01; Fig. 2A), without affecting their amplitude (control 11.41 ± 1.5 pA vs. BDNF 12.84 ± 0.8 pA; P = 0.428; Fig. 2A). In the same cells imaged after whole-cell recording, BDNF increased the spine density in secondary and tertiary branches of apical dendrites (BDNF 9.46 ± 0.22 spines per 10 µm of dendrite, eight cells from four cultures vs. control 6.87 ± 0.22 per 10 µm of dendrite, seven cells from four cultures; P < 0.001; Table 1 and Fig. 2B). These results demonstrate that BDNF both enhances quantal synaptic transmission as well as promotes dendritic spine formation in CA1 pyramidal neurones in organotypic cultures of hippocampal slices from 1-week-old rats. These results confirm previous observations following a slightly longer BDNF treatment (2–3 days here vs. 5–9 days in Tyler & Pozzo-Miller, 2001).
Figure 2. Under conditions of normal neuronal activity, BDNF increases the frequency of AMPA-mEPSCs and dendritic spine density while promoting the formation of stubby spines (type-I).
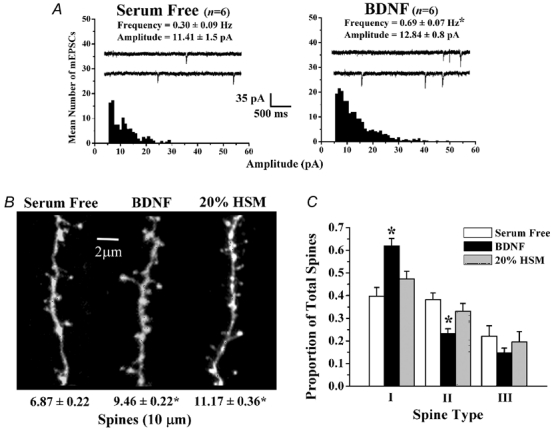
A, probability distributions of the mean amplitudes of AMPA-mediated mEPSCs recorded from CA1 pyramidal neurones in serum-free control (left) and brain-derived neurotrophic factor (BDNF)-treated (right) hippocampal slice cultures. Mean frequency, amplitude and representative continuous records of spontaneous mEPSCs are also shown for each group (inset). B, maximum-intensity z-projection confocal images of representative apical dendritic segments of CA1 pyramidal neurones filled with Alexa-594 in serum-free control (left), BDNF (centre) and 20 % horse serum (HSM) (right) slice cultures. C, histogram plots of the proportion of each morphologically distinct spine type (I, II and III) in control, BDNF and 20 % HSM-treated slice cultures. In this and all remaining figures, asterisks indicate statistically significant differences (P < 0.05).
Table 1.
Summary of total CA1 apical dendritic spine densities
| Group | Dendritic segments | Dendritic length (μm) | Spine density per 10 μm | Slices | Cells |
|---|---|---|---|---|---|
| Serum-free | 54 | 2200.75 | 6.87 ± 0.22 | 4 | 7 |
| BDNF | 76 | 2072.46 | 9.46 ± 0.22* | 4 | 8 |
| TTX | 77 | 2344.57 | 6.46 ± 0.1 7 | 3 | 6 |
| TTX-BDNF | 66 | 1844.64 | 8.97 ± 0.20*† | 4 | 6 |
| BoNT/C | 49 | 1650.65 | 5.89 ± 0.21* | 3 | 5 |
| BoNT/C-BDNF | 40 | 1292.77 | 8.33 ± 0.33*† | 3 | 4 |
| 20% HSM | 23 | 885.97 | 11.17 ± 0.36* | 3 | 4 |
BDNF, brain-derived neurotrophic factor; BoNT/C, botulinum neurotoxin C; HSM, horse serum. Total spine density is expressed as mean ±s.e.m. normalized to 10 μm dendritic length.
Significant difference (P < 0.05) compared to serum-free control values
significant difference (P < 0.05) compared to that treatment group's condition control values (i.e. TTX-BDNF vs. TTX).
Since variations in synaptic activity are thought to affect the morphology of individual spines (Segal et al. 2000; Sorra & Harris, 2000; Yuste & Bonhoeffer 2001; Nimchinsky et al. 2002), and since BDNF enhanced synaptic activity (i.e. the frequency of mEPSCs, see Fig. 2A), we further characterized the morphology of individual spines after BDNF exposure. Dendritic spines were classified as type-I (stubby), type-II (mushroom) or type-III (thin) using the length (L), neck diameter (dn) and head diameter (dh) of individual spines, as previously described (Harris et al. 1992; Koh et al. 2002; see Methods and Fig. 1). Shorter and stubbier spines (type-I) are thought to promote more coordinated and widespread Ca2+ transients in the parent dendrite, as well as to coordinate Ca2+ signalling among adjacent spines, whereas long and thin spines (type-III) spines may isolate Ca2+ transients from the parent dendrite and other spines (Segal et al. 2000; Yuste et al. 2000; Nimchinsky et al. 2002). Due to the disparity of spine densities across treatment groups, the quantity of spines in each morphological class was analysed, and is expressed, as a proportion of total spines (T; see Fig. 5B for raw density of specific spine types per 10 µm of apical dendrite). Compared to serum-free controls, BDNF increased the proportional density of stubby, type-I spines (control I/T 0.39 ± 0.04, 303 spines, seven cells from four slices vs. BDNF I/T 0.62 ± 0.03, 304 spines, eight cells from four slices; P < 0.01; Table 2 and Fig. 2C). BDNF also reduced the proportion of mushroom, type-II spines (control II/T 0.38 ± 0.03 vs. BDNF II/T 0.23 ± 0.02; P = 0.002; Table 2 and Fig. 2C). BDNF treatment did not significantly affect the proportion of thin, type-III spines (BDNF III/T 0.15 ± 0.05 vs. control III/T 0.22 ± 0.05; P = 0.646; Table 2 and Fig. 2C). Compared to serum-free controls, BDNF also induced a hypertrophy of spines by eliciting an increase in the length, neck diameter and head diameter of individual dendritic spines (P < 0.05; Table 2). Due to the consequence of spine form on Ca2+ signals (Segal et al. 2000; Yuste et al. 2000; Nimchinsky et al. 2002), these observations suggest that BDNF exerts its effects on synaptic plasticity, as well as in learning and memory, by facilitating synchronous widespread Ca2+ elevations within spines and their corresponding dendritic compartments.
Figure 5. BDNF induces dendritic spine formation, and selectively affects the morphological maturation of spines across several distinct levels of synaptic transmission.
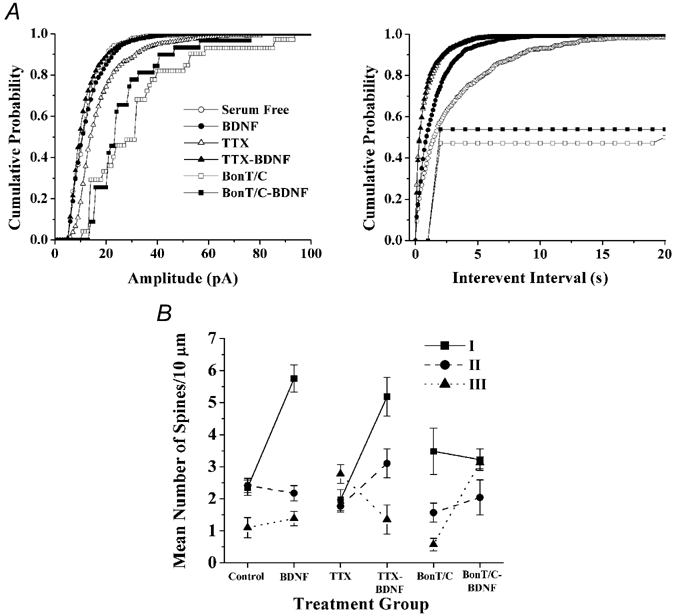
A, cumulative probability distributions of AMPA-mEPSC amplitudes (left) and frequencies (right) for all recordings obtained from CA1 pyramidal neurones in serum-free control (^), BDNF (•), TTX (▵), TTX-BDNF (▾), BoNT/C (□) and BoNT/C-BDNF-treated (▪) hippocampal slice cultures. B, scatter plot illustrating the mean number of type-I (▪, solid line), type-II (•, dashed line) and type-III (▾, dotted line) dendritic spines per 10 µm of apical dendrite in each treatment group, and presented as control vs. BDNF, TTX vs. TTX-BDNF and BoNT/C vs. BoNT/C-BDNF comparisons. Summation of the densities for each spine type yields overall spine density (T), expressed in mean number of spines per 10 µm of secondary and tertiary apical dendrites.
Are the effects of BDNF on spine morphology specific, or is BDNF simply exerting a non-specific, trophic effect on spine growth and form? CA1 pyramidal neurones maintained in culture media containing 20 % HSM had the highest spine density compared to serum-free controls and BDNF-treated groups (20 % HSM 11.17 ± 0.36 spines per 10 µm of dendrite, four cells from three cultures; P < 0.001; Table 1 and Fig. 2B), but had proportions of spine types reminiscent to serum-free controls (Table 2 and Fig. 2C). In addition, the sizes (L, dn and dh) of individual dendritic spines did not differ between the 20 % HSM treatment group and the serum-free controls (P > 0.05; Table 2). These results indicate that the effects of BDNF of dendritic spine growth and form are not merely due to a generalized trophic action, suggesting that BDNF specifically promotes the growth of individual spines, as well as the formation of stubby, type-I spines.
Spontaneous, action potential-independent synaptic transmission provides sufficient activity for BDNF to promote spine formation and to induce changes in spine form
Because the effect of BDNF on spines could be secondary to enhanced synaptic transmission and neuronal activity throughout the slice (Levine et al. 1995; Li et al. 1998), we characterized its actions on spine growth and form after blocking action potentials with TTX. As observed in cultured hippocampal neurones (Burrone et al. 2002; Thiagarajan et al. 2002), chronic (48–72 h) TTX application to hippocampal slice cultures increased the frequency and amplitude of mEPSCs in CA1 neurones, compared to serum-free controls (P < 0.05; Fig. 5A). Under conditions of chronic TTX, BDNF reduced the mean mEPSC amplitude (TTX 17.92 ± 1.9 pA vs. TTX-BDNF 11.82 ± 0.8 pA; P < 0.05; Fig. 3A), without affecting their frequency (TTX-BDNF 1.78 ± 0.6 Hz, six cells from five slices vs. TTX 1.37 ± 0.2 Hz, six cells from four slices; P > 0.05; Fig. 3A). These results indicate that homeostatic scaling of quantal amplitudes, as well as their modulation by BDNF, are also expressed by CA1 neurones in organotypic slice cultures from postnatal rats, similar to that observed in embryonic cortical neurons in vitro (Rutherford et al. 1998; Desai et al. 1999). The ensuing visualization of dendritic spines in the same recorded cells revealed that BDNF also increased spine density when applied in the presence of TTX (TTX-BDNF 8.97 ± 0.20 spines per 10 µm of dendrite, six cells from four cultures vs. TTX 6.46 ± 0.17 spines per 10 µm of dendrite, six cells from three cultures; P < 0.001; Table 1 and Fig. 3B). The spine density in TTX was not significantly different than that of serum-free controls (P > 0.05; Table 1).
Figure 3. Action potential-independent neurotransmitter release (minis) provides sufficient activity for BDNF to induce spine formation, and preferentially increase the proportion of stubby spines (type-I).
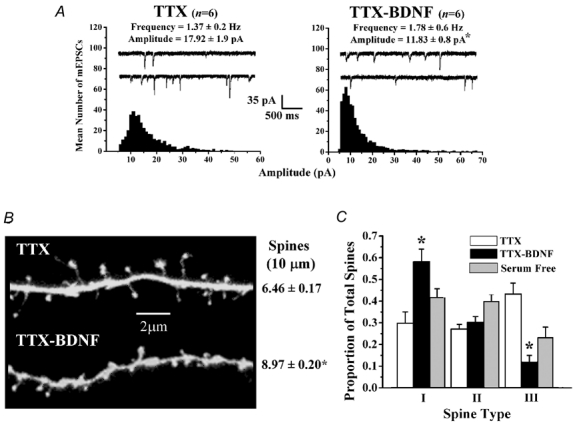
A, probability distributions of the mean amplitudes of AMPA-mediated mEPSCs in slice cultures treated with TTX (left) and TTX-BDNF (right) slice cultures. B, representative dendritic segments of CA1 pyramidal neurones in slice cultures treated with TTX (top) and TTX-BDNF (bottom). C, histogram plots of the proportion of each morphologically distinct spine type in slice cultures treated with TTX and TTX-BDNF. Data from serum-free controls (from Fig. 2) are presented here to facilitate visual comparisons.
Blockade of neuronal activity also affected the morphology of individual spines. Chronic TTX treatment increased the proportion of type-III spines (P < 0.05; Table 2), and reduced the proportion of type-II spines (P < 0.05; Table 2), without affecting type-I spines (P > 0.05; Table 2), suggesting that reduced levels of neuronal activity promoted spine elongation. In fact, spines in TTX-treated slices were significantly longer and had a significantly larger spine head diameter compared to serum-free controls (P < 0.05; Table 2). Under conditions of chronic TTX treatment, BDNF increased the proportion of stubby, type-I spines (TTX-BDNF I/T 0.58 ± 0.06, 319 spines, six cells from four slices vs. TTX I/T 0.30 ± 0.05, 299 spines, six cells from three slices; P < 0.001; Table 2 and Fig. 3C), and reduced the proportion of thin, type-III spines (TTX- BDNF III/T 0.12 ± 0.03 vs. TTX III/T 0.43 ± 0.05; P < 0.001; Table 2 and Fig. 3C), without significantly affecting the proportion of mushroom, type-II spines (TTX-BDNF II/T 0.30 ± 0.03 vs. TTX II/T 0.27 ± 0.02; P > 0.05; Table 2 and Fig. 3C). Compared to TTX treatment alone, BDNF treatment in the presence of TTX elicited a reduction in the length of spines and an increase in the diameter of spine necks (P < 0.05; Table 2). These results are similar to those observed under conditions of normal synaptic transmission, and thus demonstrate that spontaneous action potential-independent synaptic transmission provides sufficient activity for BDNF to increase the proportion of those stubby-type spines, which are potentially responsible for widespread and coordinated Ca2+ transients thought to mediate enduring changes in synaptic strength.
Even in the absence of SNARE-mediated neurotransmitter release, BDNF promotes spine formation while increasing the proportion of thin, type-III spines
To completely rule out the contribution of presynaptic transmitter release on spine growth and form, especially considering that BDNF is one of the most potent modulators of presynaptic function (Poo, 2001; Tyler et al. 2002b), we used botulinum neurotoxin C (BoNT/C) to cleave syntaxin-1A (Blasi et al. 1993), an essential SNARE protein for synaptic vesicle fusion (Südhof, 2000). Chronic (48–72 h) incubation of slice cultures with BoNT/C caused a 30-fold reduction in the frequency of mEPSCs, compared to serum-free controls (P < 0.05; Fig. 5A). Reminiscent of homeostatic quantal scaling (Turrigiano & Nelson, 2000), the amplitude of the few detected mEPSCs was significantly larger than those detected in control cells (P < 0.05; Fig. 5A). Under these conditions, BDNF did not rescue the severe inhibition in the frequency of mEPSCs (BoNT/C 0.01 ± 0.003 Hz, six cells from three slices vs. BoNT/C-BDNF 0.01 ± 0.003 Hz, six cells from three slices; P = 0.45; Fig. 4A). On the other hand, BDNF did cause a trend towards smaller mEPSCs, albeit not significant (BoNT/C 30.36 ± 6.0 pA vs. BoNT/C-BDNF 26.41 ± 3.4 pA; P = 0.58; Fig. 4A); the small number of events detected in BoNT/C-treated slice cultures is likely to have reduced the power of our statistical analyses. Consistent with the findings of McKinney and colleagues (1999). BoNT/C-treated CA1 neurones had a significantly lower spine density compared to controls (P < 0.05; Table 1). Despite the near complete blockade of vesicular fusion by BoNT/C, and thus of synaptic transmission, BDNF was still capable of increasing spine density in CA1 pyramidal neurones (BoNT/C-BDNF 8.33 ± 0.33 spines per 10 µm of dendrite, four cells from three cultures vs. BoNT/C 5.89 ± 0.21 spines per 10 µm of dendrite, five cells from three cultures; P < 0.001; Table 1 and Fig. 4C). These observations indicate that BDNF triggers a postsynaptic spinogenic programme, independent of presynaptic, SNARE-dependent vesicular neurotransmitter release.
Figure 4. In the near absence of SNARE-dependent neurotransmitter release, BDNF induces spine formation, while increasing the proportion of thin spines (type-III).
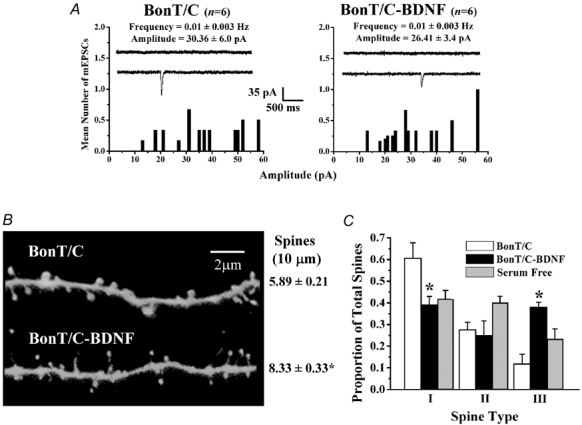
A, probability distributions of the mean amplitudes of AMPA-mediated mEPSCs in slice cultures treated with botulinum neurotoxin C (BoNT/C) (left) and BoNT/C-BDNF (right). B, representative dendritic segments of CA1 pyramidal neurones in BoNT/C (top) and BoNT/C-BDNF (bottom) slice cultures. C, histogram plots of the proportion of each morphologically distinct spine type in slice cultures treated with BoNT/C and BoNT/C-BDNF. Data from serum-free controls (from Fig. 2) are presented here to facilitate visual comparisons.
We next determined to what extent vesicular neurotransmitter release is required for the morphological sculpting of individual spines by BDNF. BoNT/C treatment alone increased the proportion of stubby, type-I spines (P < 0.05; Table 2), and reduced the proportion of mushroom, type-II spines (P < 0.05; Table 2), without affecting the proportion of thin, type-III spines (P > 0.05; Table 2), when compared to serum-free controls. BoNT/C treatment also induced an increase in the overall size of individual dendritic spines compared to serum-free controls (P < 0.05; Table 2). When applied in the presence of BoNT/C, BDNF increased the proportion of thin, type-III spines (BoNT/C III/T 0.11 ± 0.05 vs. BoNT/C-BDNF III/T 0.38 ± 0.02; P < 0.05; Table 2 and Fig. 4C), while decreasing the proportion of stubby, type-I spines (BoNT/C I/T 0.61 ± 0.07, 222 spines, five cells from three cultures vs. BoNT/C-BDNF I/T 0.39 ± 0.04, 363 spines, four cells from three cultures; P < 0.001; Table 2 and Fig. 4C). There were no differences in the proportions of mushroom, type-II spines between the BoNT/C and BoNT/C-BDNF groups (BoNT/C II/T 0.28 ± 0.03 vs. BoNT/C-BDNF II/T 0.25 ± 0.07; P > 0.05; Table 2 and Fig. 4C). Further, BDNF treatment in the presence of BoNT/C reduced both spine neck and head diameters (P < 0.05; Table 2), without affecting spine length (P > 0.05; Table 2), when compared to BoNT/C treatment alone. These data illustrate that in the almost complete absence of SNARE-mediated synaptic transmission, the majority of dendritic spines observed in BDNF-treated CA1 neurones are of the thin, type-III class.
Taken altogether, these results suggest that the combined actions of BDNF and spontaneous miniature synaptic transmission enhance the morphological sculpting of dendritic spines of CA1 pyramidal neurones. The prevalent spine form under those conditions, the stubby ones, may play a fundamental role in Ca2+-dependent integrative synaptic plasticity, such as long-term potentiation, by virtue of their geometry, which is amenable for coordinated, widespread Ca2+ transients in the parent dendrite and neighbouring spines (Segal et al. 2000; Yuste et al. 2000; Nimchinsky et al. 2002; Kasai et al. 2003).
Discussion
Due to their dynamic and plastic properties, dendritic spines have been the major focus for exploring activity-dependent structural modifications in the CNS (Harris, 1999; Segal & Andersen, 2000; Sorra & Harris, 2000; Yuste & Bonhoeffer, 2001; Nimchinsky et al. 2002). Here, we present evidence that long-term BDNF application enhanced dendritic spine formation, as well as the proportion of stubby spines (type-I) under conditions of both action potential-dependent and independent synaptic transmission. Even when SNARE-dependent vesicular synaptic transmission was inhibited with BoNT/C, BDNF was still capable of inducing spine formation, although the majority of spines were of the thin class (type-III). Interestingly, blockade of BDNF signalling with a scavenger of endogenous BDNF, TrkB-IgG, increased spine length in cerebellar Purkinje cells (Shimada et al. 1998), making spines similar to the thin class of spines described here (type-III). Consistent with an activity-dependent process of morphological differentiation of synapses, and reminiscent of the process of ocular dominance column formation in the visual cortex (McAllister et al. 1999), our data suggest that BDNF interacts with spontaneous miniature synaptic activity to sculpt fine dendritic structure in CA1 pyramidal neurones of the postnatal hippocampus.
BDNF application under conditions of normal action potential-dependent synaptic transmission increased the proportion of both stubby (type-I) and mushroom (type-II) spine categories. In addition, spontaneous action potential-independent synaptic transmission (minis) provides sufficient synaptic activity for BDNF to exert its structural effects on dendritic spines. Lastly, in the absence of action potentials (TTX-treated slices), BDNF increased both the total spine density and the proportion of stubby spines (type-I), while reducing the proportion of thin spines (type-III). Since long and thin spines may isolate spine Ca2+ transients from the parent dendrite and neighbouring spines, and considering that coordinated spine Ca2+ transients are thought to be necessary for synaptic plasticity (Yuste & Majewska, 2001; Kasai et al. 2003), we propose that BDNF enhances synaptic plasticity not only by increasing spine density, but also by specifically promoting the formation of those spines more amenable for widespread Ca2+ signalling.
The values of spine density in our 11–12 div slice cultures maintained in media containing 20 % HSM are close to those observed at 21 div slice cultures and P21 rats in a recent study of the development of rat CA1 pyramidal neurones in organotypic slice cultures versus acute slices of matching developmental ages (De Simoni et al. 2003). On the other hand, the proportions of spine classes in our 11–12 div 20 % HSM cultures resembled those found in 7 div cultured slices and P14 rats in the same study (De Simoni et al. 2003). These discrepancies might arise from the slightly older postnatal rat pups used for slice culture in our studies (P7 vs. P5), and by the fact that penicillin was used in De Simoni et al. (2003) as an antibiotic; we purposely avoided the use of penicillin – or any other antibiotic or antimycotic – due to its actions on GABAA receptor channels (Twyman et al. 1992), and the resulting elevated neuronal activity that may confound our observations. For reasons of potential neurotoxicity, we also avoided the use of cell proliferation inhibitors in our culture media.
How does spine geometry relate to synaptic activity levels? Similar to the bell-shaped relationship between intracellular Ca2+ levels and growth cone behaviour (Zheng et al. 1996), a correlation between synaptic activity – and the corresponding Ca2+ levels – with spine formation and elongation has been recently proposed. In this model, very low and very high Ca2+ levels inhibit spine growth (and sometimes induce spine retraction or ‘spine pruning’), whereas intermediate Ca2+ levels promote spine formation and elongation (Segal et al. 2000). Consistent with this model of activity-dependent remodelling of spines, chronic TTX treatment had no effect on spine density by itself, but it increased the proportion of thin spines (type-III). These observations appear to differ from the increased spine density observed after TTX application to acute hippocampal slices (Kirov & Harris, 1999). However, these authors described an increase in the number of long and thin spines in response to TTX, similar to the increased proportion of thin spines described here (type-III). Since those studies used acute slices and short TTX exposures (Kirov & Harris, 1999), the discrepancy with the present results is likely to be due to the difference in slice preparation (acute slices vs. organotypic cultures) and/or treatment duration (minutes to hours vs. 48–72 h). Our results are in good agreement with the observation that chronic TTX treatment of hippocampal slice cultures did not affect spine density in apical dendrites of CA1 neurones (McKinney et al. 1999). McKinney and colleagues (1999) described a trend, albeit not significant, favouring their type-3 spines, similar to the type-III described here. Similarly, TTX increased the relative number of long and thin spines on dentate granule cell dendrites, without affecting overall spine density (Drakew et al. 1999). Taken together, these observations indicate that thin spines (type-III) predominate under conditions where only miniature synaptic transmission is present, due to intermediate dendritic Ca2+ levels. However, when only miniature synaptic transmission is present in conjunction with BDNF signalling (TTX-BDNF), miniature synaptic transmission provides sufficient activity for BDNF to mediate the activity-dependent morphological sculpting of dendritic spines from thin to stubby spines in CA1 pyramidal neurones.
Another fascinating observation is that BDNF increases spine density even in the near absence of SNARE-mediated synaptic transmission. Under these conditions, however, BDNF selectively increased the proportion of thin spines (type-III), while decreasing the proportion of stubby spines (type-I), suggesting that in the absence of presynaptic vesicular fusion, BDNF induced spine elongation. Considering that Ca2+ release from intracellular stores increases spine length (Korkotian & Segal, 1999), this last effect of BDNF on spine form may arise from intermediate Ca2+ levels brought up by Ca2+ mobilization from inositol 1,4,5-trisphosphate (IP3) stores evoked by TrkB-induced phospholipase C (PLC) activation (Segal & Greenberg, 1996). It appears that synapse formation does not require SNARE-mediated neurotransmitter release, as observed in the developing brains of mutant mice lacking munc-18, a protein required for synaptic vesicle fusion (Verhage et al. 2000). Although a dendritic spine alone does not indicate the presence of a presynaptic terminal, and thus a synapse, our data suggest that BDNF promotes synapse formation, or at least the formation of the postsynaptic compartment, in the absence of vesicular neurotransmitter release. In this context, the release of BDNF during development may serve as a chemotropic signal to guide synapse formation by enhancing dendritic spine formation independently of neurotransmitter release.
Although some spines are still present, chronic inhibition of vesicular neurotransmitter release with BoNT/C reduced spine density in apical dendrites of CA1 neurones, without affecting the proportion of any of the morphologically distinct classes of spines. Under these conditions of almost complete absence of presynaptic function, BDNF still increased spine density, suggesting that spine formation is an intrinsic property of postsynaptic neurons, whereas spine maintenance requires presynaptic input. The first indication of this inherent property was provided by the observation that Purkinje cells formed new dendritic spines in response to removal of their climbing fibre input (Sotelo et al. 1975). In addition, cultured neurones form dendritic filipodia, the precursors of spines (Fiala et al. 1998), before receiving presynaptic inputs (Papa et al. 1995). We interpret our observations of BDNF-induced spine formation in the absence of neurotransmitter release to indicate that BDNF facilitates spine formation by enhancing an intrinsic property of the postsynaptic cell. However, most of the spines formed under these conditions were morphologically different to those formed in response to BDNF in combination with spontaneous miniature synaptic transmission. Then, to what extent does synaptic activity play a role in the BDNF-induced effects presented here?
Since BDNF is capable of increasing spine density in the absence of SNARE-mediated release, and without affecting the frequency of mEPSCs under the same conditions, it seems that BDNF promotes spine formation independently of neurotransmitter release; those spines are, however, of the thin class (type-III). Neurotransmitter release does, however, play a critical role in the structural refinement of individual spines induced by BDNF. In the presence of spontaneous action potential-dependent and action potential-independent synaptic transmission, BDNF increased the proportion of stubby spines (type-I). From these morphological observations alone, one might posit that, during BDNF exposure, there are no fundamental differences in the levels of synaptic activation at individual synapses between the normal (action potential-dependent) and TTX (action potential-independent) treatment conditions; this is likely to be due to the fact that most hippocampal synapses release, at most, only one vesicle – per active zone – per action potential (Stevens & Wang, 1995).
Chronic blockade of neuronal activity with TTX increased the frequency of mEPSCs in cultured hippocampal neurones (Burrone et al. 2002; Thiagarajan et al. 2002), and increased the amplitude of AMPA-mediated mEPSC in cultured cortical (Rutherford et al. 1998; Turrigiano et al. 1998; Desai et al. 1999) and hippocampal (Burrone et al. 2002; Thiagarajan et al. 2002) pyramidal-like neurones. Similarly, chronic TTX increased the frequency and amplitude of AMPA mEPSCs in CA1 pyramidal neurons in our slice cultures. These effects are considered to be forms of ‘synaptic scaling’ or homeostatic synaptic plasticity, maintaining the integrated synaptic strength of an individual neurone, while still permitting relative changes in the strengths at individual synapses (Turrigiano & Nelson, 2000). Interestingly, BDNF/TrkB signalling has been shown to mediate this type of homeostatic synaptic plasticity. As previously shown in cultured neocortical neurones (Rutherford et al. 1998; Desai et al. 1999), we show here that BDNF blocked the increase in mEPSC amplitude in CA1 pyramidal neurons from TTX-treated hippocampal slice cultures. Chronic inhibition of SNARE-dependent vesicular neurotransmitter release also seemed to increase the amplitude of the few detected mEPSCs. Under these conditions, BDNF produced a trend towards smaller mEPSCs, albeit not significant, probably due to the small number of events detected in BoNT/C-exposed cultures.
Despite the difficulty of establishing a causal relationship, several neurological disorders have correlated dendritic spine pathologies, most notably those associated with mental retardation (Fiala et al. 2002). In this context, it is noteworthy that those spines induced by BDNF in the absence of transmitter release (BDNF-BoNT/C; Fig. 4) are reminiscent to the tortuous and thin spines observed in patients with Down and Fragile X syndromes (Fiala et al. 2002). These observations suggest that the interplay between BDNF signalling and spontaneous miniature transmitter release not only promote the formation of those spines more amenable for Ca2+-dependent synaptic plasticity, but it may also be fundamental for the structural differentiation of cortical networks relevant to human cognition.
Conclusions
Our combined physiological and morphological observations in CA1 pyramidal neurones demonstrate that BDNF increases spine density irrespective of the levels of synaptic transmission, and that miniature synaptic transmission provides sufficient activity for the functional translation of this BDNF-triggered spinogenesis into clearly defined morphological spine types. Thus, we conclude that BDNF acts as a homeostatic regulator of both synaptic structure and function in order to maintain optimal neuronal communication among hippocampal excitatory synapses. These actions at synapses may underlie not only the actions of BDNF on synaptic plasticity at the cellular level, but also the role of BDNF signalling in hippocampal-dependent learning and memory (Tyler et al. 2002a).
Acknowledgments
We thank Drs K Harris (Medical College of Georgia), R Lester (UAB), R Llinás (NYU) and M Segal (Weizmann Institute) for critical reading of the manuscript and insightful discussions; T Inoue (Tokyo University) for data acquisition software; and the High Resolution Imaging Facility (UAB) for the use of the laser scanning confocal microscope. Supported by NIH grants RO1-NS40593 (L.P.-M.), P30-HD38985 and PO1-HD38760. We also thank Amgen for the generous supply of BDNF.
References
- Bi G, Poo M. Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Yamasaki S, Binz T, Niemann H, Jahn R. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993;12:4821–4828. doi: 10.1002/j.1460-2075.1993.tb06171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Cajal SR. Histologie du Systeme Nerveux de l'Homme et des Vertebres. Vol. 1. Paris: 1909. [Google Scholar]
- Collin C, Miyaguchi K, Segal M. Dendritic spine density and LTP induction in cultured hippocampal slices. J Neurophysiol. 1997;77:1614–1623. doi: 10.1152/jn.1997.77.3.1614. [DOI] [PubMed] [Google Scholar]
- Desai NJ, Rutherford LC, Turrigiano GC. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakew A, Frotscher M, Heimrich B. Blockade of neuronal activity alters spine maturation of dentate granule cells but not their dendritic arborization. Neuroscience. 1999;94:767–774. doi: 10.1016/s0306-4522(99)00378-4. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5:1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- Horch HW, Kruttgen A, Protbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–364. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Hubel D, Wiesel T. Ferrier lecture. Functional architecture of macaque monkey visual cortex. Proc R Soc Lond B Biol Sci. 1977;198:1–59. doi: 10.1098/rspb.1977.0085. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasamatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Katz L, Shatz C. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- Koh IY, Lindquist WB, Zito K, Nimchinsky EA, Svoboda K. An image analysis algorithm for dendritic spines. Neural Comput. 2002;14:1283–1310. doi: 10.1162/089976602753712945. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Release of calcium from stores alters the morphology of dendritic spines in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1999;96:12068–12072. doi: 10.1073/pnas.96.21.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci U S A. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Boden P, Hill RG. Rat hippocampal slices ‘in vitro’ display spontaneous epileptiform activity following long-term organotypic culture. J Neurosci Methods. 1989;27:35–49. doi: 10.1016/0165-0270(89)90051-4. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller D, Buchs PA, Stoppini L. Time course of synaptic development in hippocampal organotypic cultures. Dev Brain Res. 1993;71:93–100. doi: 10.1016/0165-3806(93)90109-n. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The synapses upon dendritic spines. Z Zellforsch Mikrosk Anat. 1969;100:487–506. doi: 10.1007/BF00344370. [DOI] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof I. The small pyramidal neuron of the rat cerebral cortex. The perikarion, dendrites and spines. J Anat. 1970;127:321–356. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Inoue T, Murphy DD. Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J Neurophysiol. 1999;81:1404–1411. doi: 10.1152/jn.1999.81.3.1404. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Mahanty NK, Connor JA, Landis DM. Spontaneous pyramidal cell death in organotypic slice cultures from rat hippocampus is prevented by glutamate receptor antagonists. Neuroscience. 1994;63:471–487. doi: 10.1016/0306-4522(94)90544-4. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Petrozzino JJ, Connor JA. G protein-coupled receptors mediate a fast excitatory postsynaptic current in CA3 pyramidal neurons in hippocampal slices. J Neurosci. 1995;15:8320–8330. doi: 10.1523/JNEUROSCI.15-12-08320.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GC. BDNF has opposite effects on the quantal amplitude of pyramidal and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Poo M-M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- Segal I, Korkotian I, Murphy DD. Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci. 2000;23:53–57. doi: 10.1016/s0166-2236(99)01499-x. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Shapiro ML, Eichenbaum H. Hippocampus as a memory map: synaptic plasticity and memory encoding by hippocampal neurons. Hippocampus. 1999;9:365–384. doi: 10.1002/(SICI)1098-1063(1999)9:4<365::AID-HIPO4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J Neurosci. 1998;18:8559–8570. doi: 10.1523/JNEUROSCI.18-21-08559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorra KE, Harris KM. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus. 2000;10:501–511. doi: 10.1002/1098-1063(2000)10:5<501::AID-HIPO1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Hillman DE, Zamora AJ, Llinas R. Climbing fiber deafferentation: its action on Purkinje cell dendritic spines. Brain Res. 1975;98:574–581. doi: 10.1016/0006-8993(75)90374-1. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle revisited. Neuron. 2000;28:317–320. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Tanzi E. I fatti e le induzioni nell'odierna isologia del sistema nervosa. Revista Sperimentale di Freniatria e di Medicina Legale delle Alienazione Mentali. 1893;19:419–472. [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, Leslie K, Desai N, Rutherford L, Nelson S. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, Nelson S. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Twyman RE, Green RM, MacDonald RL. Kinetics of open channel block by penicillin of single GABAA receptor channels from mouse spinal cord neurones in culture. J Physiol. 1992;445:97–127. doi: 10.1113/jphysiol.1992.sp018914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: On the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem. 2002a;9:227–234. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Perrett S, Pozzo-Miller LD. The role of neurotrophins in neurotransmitter release. Neuroscientist. 2002b;8:524–531. doi: 10.1177/1073858402238511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia A, Plomp J, Brussaard A, Heeroma J, Vermeer H, Toonen R, Hammer R, Berg TVD, Missler M, Geuze H, Sudhof T. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nat Neurosci Rev. 2002;3:965–974. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A. On the function of dendritic spines. Neuroscientist. 2001;7:387–395. doi: 10.1177/107385840100700508. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Holthoff K. From form to function: calcium compartmentalization in dendritic spines. Nat Neurosci. 2000;3:653–659. doi: 10.1038/76609. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Poo MM, Connor JA. Calcium and chemotropic turning of nerve growth cones. Perspect Dev Neurobiol. 1996;4:205–213. [PubMed] [Google Scholar]
- Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- Segal M, Andersen P. Dendritic spines shaped by synaptic activity. Curr Opin Neurobiol. 2000;10:582–586. doi: 10.1016/s0959-4388(00)00123-9. [DOI] [PubMed] [Google Scholar]