

Abstract
In adult mice, long-term potentiation (LTP) of synaptic transmission at CA3-to-CA1 synapses induced by tetanic stimulation requires l-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors containing GluR-A subunits. Here, we report a GluR-A-independent form of LTP, which is comparable in size to LTP in wild-type mice at postnatal day 14 (P14) but diminishes between P14 and P42 in brain slices of GluR-A-deficient mice. The GluR-A-independent form of LTP is sensitive to d(−)-2-amino-5-phosphonopentanoic acid (d-AP5), but lacks short-term potentiation (STP) and can also be observed in the pairing induction protocol. As judged by unaltered paired-pulse facilitation, this LTP form is postsynaptically expressed despite depleted extrasynaptic AMPA receptor pools with reduced levels of GluR-B, which accumulates in somata and synapses of CA1 pyramidal neurons in GluR-A-deficient mice. Our results show that in the developing hippocampus synaptic plasticity can be expressed by AMPA receptors lacking the GluR-A subunit.
Use-dependent plasticity of synaptic transmission, as manifested by hippocampal LTP (Bliss & Lømo, 1973), is important not only for memory formation in the adult brain, but also for experience-dependent establishment of neuronal connections during brain development, based on the pattern of afferent electrical signals to ‘instruct’ specific neuronal wiring (Katz & Shatz, 1996). Adult mice lacking the AMPA receptor subunit GluR-A (Hollmann et al. 1989; Keinänen et al. 1990) show adequate glutamatergic synaptic transmission, mediated by the remaining AMPA receptor subunits GluR-B and GluR-C (Sommer et al. 1991), but the mice do not express tetanus-induced LTP in hippocampal CA3-to-CA1 connections (Zamanillo et al. 1999; Mack et al. 2001), indicating the importance of GluR-A-containing AMPA receptors in LTP. Studies of virus-infected slice cultures supported this view and provided evidence that synaptic delivery of the GluR-A-containing AMPA receptor channels is dependent on synaptic activity including the activation of the Ca2+-calmodulin-dependent kinase II (CaMKII) which phosphorylates serine residue 831(S831) in the intracellular C-terminal domain of GluR-A (for reviews see: Malinow & Malenka, 2002; Song & Huganir, 2002). Besides the CaMKII activity, the AMPA receptor delivery also requires cAMP-dependent kinase A (PKA)-mediated phosphorylation at serine residue 845 (S845) and the presence of the PDZ interaction domain at the very C-terminus (Shi et al. 1999; Hayashi et al. 2000; Esteban et al. 2003). Notably, mice with substituted serine residues S831 and S845 show reduced LTP (Lee et al. 2003), and similar to adult GluR-A ‘knockout’ mice (Zamanillo et al. 1999), learn normally in the Morris water maze, but show impaired retention of spatial memory (Lee et al. 2003) and of spatial working memory, respectively (Reisel et al. 2002). This indicates that GluR-A-dependent LTP is involved in some aspects of spatial memory, which in its entirety seems to be dependent on NMDA receptor-mediated LTP (e.g. Morris et al. 1986; Tsien et al. 1996). Thus, the NMDA receptor might induce different forms of plasticity. One of these forms is GluR-A dependent and dominates at hippocampal CA3-to-CA1 connections in adult animals. The recent findings that NMDA receptor subtypes use different LTP induction signalling pathways (Köhr et al. 2003), one of which includes the immediate early gene c–fos (Fleischmann et al. 2003), provides further evidence for this speculation. Another report showed recently an additional LTP form which is independent of GluR-A phosphorylation at serine residues S831 and S845 and which is operative in mice under 3 weeks of age (Lee et al. 2003). However, the report left unresolved whether the mutated GluR-A subunits could still participate in the expression of this LTP form.
To understand this ‘juvenile’ form of LTP, it is important to study it in GluR-A-deficient mice. During development, neuronal wiring - a developmental form of neuronal plasticity - seems to be established correctly in GluR-A-deficient mice: LTP at CA3-to-CA1 synapses can be restored when GluR-A expression is switched on at 3 weeks of age (Mack et al. 2001), indicating that other cellular components which are involved in the GluR-A-dependent form of plasticity are present in CA1 pyramidal cells even in absence of GluR-A. We have performed field (Andersen et al. 1977) and cellular (Gustafsson et al. 1987; Chen et al. 1999) recordings and analysed the juvenile development of synaptic plasticity at hippocampal CA1 synapses of wild-type and GluR-A−/− mice between P14 and P42, which we considered as the end of the ontogenic hippocampal development. In parallel, we looked at the expression and distribution of AMPA receptor subunits in the hippocampus with electrophysiological and immunohistochemical methods and visualized AMPA receptors in synapses by electron microscopy. Collectively, our results suggest that at developing CA3-to-CA1 synapses GluR-A-independent LTP can be expressed. This LTP form shows NMDA receptor dependence, is postsynaptic in nature and diminishes with age.
METHODS
GluR-A−/− and wild-type mice
GluR-A−/− mice were crossed with C57BL/6 mice to generate heterozygous GluR-A+/− mice. Homozygous GluR-A−/− and wild-type mice were produced from heterozygous intercrosses and genotyped at P12 or P36 by PCR. Tail-tip biopsy samples were digested overnight with proteinase-K (1 mg ml−1) in 50 mm Tris-HCl, pH 8.0, 100 mm EDTA, 100 mm NaCl, 1 % SDS at 55 °C. DNA was precipitated with 0.8 vol. isopropanol, washed with 1 ml 70 % ethanol and dissolved in sterile H2O-Millipore (300–800 μl). Primer pair 1005 (AAT GCC TAG TAC TAT AGT GCA CG) and 3int3′ (CTG CCT GGG TAA AGT GAC TTG G) amplified fragments of 200 bp from the GluR-A-allele and 1500 bp from the wild-type allele. To confirm the presence of the wild-type allele, primer pair 3int3′ and MH60 (CAC TCA CAG CAA TGA AGC AGG) was used for amplification of a 200 bp fragment. Wild-type littermates were used as controls for most of the experiments. Animal care was in compliance with the institutional guidelines at the animal facilities of the Center of Molecular Biology (ZMBH, INF 282, D-69120 Heidelberg, Germany) and of the Max-Planck-Institute for Medical Research in Heidelberg under the approval of the Animal Care Committee at the Regierungspräsidium Karlsruhe, Germany, licence number 37–9185.81/35/97.
Extracellular field EPSP recordings
EPSPs were recorded in CA1 of transverse hippocampal slices (400 μm) from mice killed with halothane. Experiments were performed blind with respect to mouse genotype. Slices were maintained in an interface chamber as described (Zamanillo et al. 1999). Orthodromic synaptic stimulation (50 μs, < 150 μA, 0.2 Hz) in CA1 was delivered alternately through two tungsten electrodes to activate synapses in apical (stratum radiatum) and basal dendrites (stratum oriens), respectively. Extracellular potentials were monitored by glass electrodes filled with extracellular solution, which were placed in similar positions. After stable synaptic responses in both pathways for at least 15 min, one pathway was tetanized (100 Hz, 1 s), the other served as control. To standardize tetanization strength in different experiments, the tetanic stimulation strength was set in response to a single shock at an intensity just above the threshold for generating a population spike. Synaptic efficacy was assessed measuring the slope of the field EPSP (fEPSP) in the middle third of its rising phase. These measures reduced variability among experiments at the expense of maximal amplitudes. Six consecutive responses (1 min) were averaged and normalized to the mean value recorded 4–7 min prior to tetanic stimulation. In some experiments 50 μmd(−)-2-amino-5-phosphonopentanoic acid (d-AP5) was present during the recordings. In our study we pooled data obtained from the stratum oriens and the stratum radiatum. Statistical evaluation with a linear mixed model revealed no differences in LTP magnitude between the two pathways at the different ages and in the two genotypes (P ranging from 0.08 to 0.79). Data are means ±s.e.m.; the statistical significance of LTP levels between tetanized and non-tetanized inputs were calculated by Student's paired two-tailed t tests. LTP levels between genotypes were statistically evaluated by linear mixed model analysis.
Paired-pulse facilitation
Paired-pulse responses (mean of five trials at 10 different stimulation strengths at 50 ms interstimulus interval) in both the test and control pathways were monitored 15 min before and 50–65 min after LTP induction. The maximal slope of the rising phase of the fEPSP was measured and the facilitation ratio was calculated as fEPSP2 slope/fEPSP1 slope. In order to pool data from different experiments we selected responses to a stimulation strength just below threshold for eliciting a population spike on the second fEPSP.
Evoked whole-cell EPSCs and pairing-induced cellular LTP
Whole-cell voltage clamp recordings were made from CA1 pyramidal cells in 250 μm thick slices under visual control (Stuart et al. 1993). EPSCs were evoked from two independent inputs, one of which was paired and the other one was used as control, with two patch pipettes as stimulating electrodes. In contrast with a previous report (Mack et al. 2001) the control pathway was measured by stimulating synapses of the basal dendrites and the paired pathway was an input to synapses of the apical dendrite. The two stimulus pipettes were > 200 μm apart, located below and above the soma of a CA1 pyramidal cell. All measurements were at −70 mV membrane potential. LTP was evoked and recorded according to Chen et al. (1999) by voltage clamping the membrane potential of the postsynaptic pyramidal cell to 0 mV for 3 min while stimulating the paired pathway every 1.5 s. Recording pipettes were filled with caesium-based solutions. The measured amplitudes were normalized to the mean EPSCs before pairing. The NMDA dependence was tested in the presence of 100 μmd-AP5. Data are means ±s.e.m.; the statistical significance levels are from a Student's paired two-tailed t test for comparing responses from paired vs. unpaired input. LTP between genotypes was compared by Student's unpaired two-tailed t test.
Soma patch currents and synaptic currents
Recordings from ‘nucleated’ patches isolated from the soma of CA1 pyramidal neurons from brain slices were obtained with pipettes of 3–5 MΩ access resistance. Soma patch currents were evoked by brief (2 ms) application of glutamate (1 mm). EPSCs were recorded at −70 mV and analysed as described (Zamanillo et al. 1999). To block inhibitory activity, the GABA-A blocker bicuculline (20 μm) was added to the bath solution.
Immunohistochemical staining
Mice were anaesthetized with halothane (Hoechst, Frankfurt, Germany) and perfused intracardially with 0.1 m sodium phosphate buffer, pH 7.4, at room temperature followed by ice-cold 4 % paraformaldehyde in phosphate-buffered saline (PBS). Brains were removed and postfixed for 1 h in 4 % paraformaldehyde at 4 °C. The brains were rinsed in PBS, embedded in 2 % agarose in PBS as supportive material for sectioning and were cut sagittally into 50–70 μm sections on a vibratome (Leica VT 1000S, Leica Microsystems, Nussloch, Germany). Sections were incubated with PBS and 0.5 % H2O2 in PBS for 10 min at room temperature, washed twice with PBS, blocked with 5 % normal goat serum (NGS) in PBS, 1.0 % BSA, 0.3 % Triton X-100 for 30 min and incubated at room temperature overnight in same buffer with primary polyclonal anti-glutamate receptor antibodies (anti-glutamate receptor 1 (1 μg ml−1), 2 (4 μg ml−1), 2/3 (1.5 μg ml−1) and 4 (4 μg ml−1); all from Chemicon, Temecula, CA, USA). The following day sections were rinsed three times for 10 min with PBS, 0.3 % BSA, 0.1 % Triton X-100 and incubated with horseradish peroxidase-coupled secondary antibody (1:600, anti-rabbit; Vector Laboratories, Burlingame, CA, USA) in PBS, 0.3 % BSA, 0.1 % Triton X-100 for 2 h. Finally, sections were washed two times with PBS, 0.3 % BSA, 0.1 % Triton X-100 and twice with PBS and developed for peroxidase with 0.05 % 3,3′-diaminobenzidine hydrochloride (Sigma-Aldrich, Taufkirchen, Germany) and 0.01 % H2O2. After two washes in PBS and 20 mm Tris-HCl pH 7.6, sections were mounted on glass slides, dried overnight, dipped into xylol and coverslipped in Eukitt (Kindler GmbH, Freiburg, Germany).
Immunoblots
Dissected hippocampi were homogenized in ice-cold 25 mm Hepes, pH 7.4 including protease inhibitor Cocktail CompleteTM (Roche Diagnostics, Mannheim, Germany) with 10–12 strokes at 900 r.p.m. in a glass teflon homogeniser and centrifuged for 5 min at 2000 r.p.m., 4 °C in a table centrifuge to remove debris and nuclei. For each sample, 8 μg protein (Protein Assay Kit, Pierce, Rockford, USA) was resolved on a 7 % SDS-polyacrylamide (Laemmli 1970) gel and transferred to nitrocellulose membrane. Blots were probed with polyclonal anti-glutamate receptor 1 (0.1 μg ml−1, Chemicon, Temecula, CA, USA), 2 (0.2 μg ml−1 Chemicon, Temecula, CA, USA), 3 (1:600, monoclonal; Zymed Laboratories, San Francisco, CA, USA), 4 (0.25 μg ml−1, Oncogene, Boston, MA, USA), anti-NMDAR1 (0.2 μg ml−1, Chemicon, Temecula, CA, USA), anti-α-CaMKII (clone 6G9, 1:25000, Chemicon, Temecula, CA, USA) and anti-β-actin (clone AC-15 ascites, 1:40000, Sigma, St Louis, MO, USA), followed by horseradish peroxidase-linked anti-rabbit or anti-mouse secondary antibodies (1:15000, Jackson ImmunoResearch, West Grove, PA, USA). Proteins were visualized by enhanced chemiluminescence (ECL-Plus, Amersham Pharmacia Biotech, Freiburg, Germany).
Immunofluorescence for confocal microscopy
Immediately after cutting, brain sections were transferred into 50 mm Tris-HCl buffer containing 1.5 % NaCl, pH 7.4 (TBS) and permeabilized in TBS, 0.4 % Triton X-100 for 30 min. Blocking was in TBS, 4 % NGS, 0.2 % Triton X-100 for 30 min at room temperature. Sections were then incubated in TBS, 4 % NGS, 0.1 % Triton X-100 at 4 °C overnight with anti-glutamate receptor 2 antibody (4 μg ml−1; Chemicon, Temecula, CA, USA). The following day, sections were washed three times for 10 min each with cold TBS, 1 % NGS and incubated for 2.5 h with goat anti-rabbit fluorescein-isothiocyanate (FITC)-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories-Dianova, Hamburg, Germany) in TBS, 1.5 % NGS, in the dark at room temperature. Afterwards, sections were washed twice in TBS, 1 % NGS and twice in TBS. After a brief rinse in 10 mm Tris-HCl, pH 7.5, sections were mounted in Vectashield Mounting Medium for Fluorescence (Vector Laboratories, Burlingame, CA, USA) and visualized by confocal microscope under epifluorescence illumination (TCS-NT laser scanning microscope, Leica Microsystems, Heidelberg, Germany).
EM immunocytochemistry
Five GluR-A-deficient and four wild-type mice were deeply anaesthetized with an overdose of Narkodorm-n (250 mg kg−1 body weight) and transcardially perfused with 0.9 % saline followed by a fixative containing 4 % paraformaldehyde, 0.1 % glutaraldehyde and 0.2 % picric acid in 0.1 m phosphate buffer (pH 7.4). Brains were removed and postfixed in the same fixative overnight, cut on a vibratome into 300 μm sections, and the CA1 area of the hippocampus was excised. The tissue blocks were cryoprotected in glycerol, cryofixed in nitrogen-cooled propane, substituted in methanol containing 1.5 % uranylacetate and embedded in Lowicryl HM20 (Chemische Werke Lowi, Waldkraiburg, Germany). Ultrathin sections were processed for postembedding immunocytochemistry, employing anti-GluR1 and anti-GluR2 antibodies (5 μg ml−1 protein; Chemicon, Temecula, CA, USA), respectively. Immunolabelling was visualized by 10 nm gold-coupled secondary antibodies (1:50; Amersham, Arlington Heights, IL, USA). Data analysis was performed on 12 grids per animal (six incubated with anti-GluR1, six incubated with anti-GluR2) of wild-type and GluR-A-deficient mice, respectively. Direct comparisons between synapses of wild-type and GluR-A-deficient mice were made on sections processed simultaneously. On each grid, 300 randomly selected synapses were counted in CA1 stratum radiatum, and the number of labelled postsynaptic densities (PSDs) among these contacts as well as the number of gold grains per individual PSD were evaluated. To exclude false positives only synapses labelled with at least two gold particles were counted. The data from GluR-B-labelled material of wild-type and GluR-A-deficient mice were fitted by Poisson distributions, and numbers of labelled PSDs and the mean numbers of gold grains per PSD were determined from the fitted distributions. Differences were assessed by Student's t test for paired samples.
RESULTS
Tetanus-induced LTP can be GluR-A-independent in juvenile mice
We have previously shown that synaptic plasticity at CA3-to-CA1 synapses of hippocampal CA1 pyramidal cells, as measured by tetanus-induced LTP, is lacking in adult GluR-A−/− mice (> P60; Zamanillo et al. 1999). Here, we compare field EPSPs (fEPSPs) before and after tetanic stimulation of Schaffer collaterals (SC) in juvenile mice. Experiments were performed on transverse hippocampal slices from mice at P14, P28, P35 and P42, respectively. In wild-type mice, tetanic activation of CA3-to-CA1 fibres in stratum radiatum or stratum oriens induced a homosynaptic increase of the fEPSP slope of a similar magnitude at all ages tested (Fig. 1, Table 1A). In GluR-A−/− mice the homosynaptic fEPSP slope increase was only observed in brain slices of young mice and the magnitude of LTP gradually declined between P14 and P42 (Fig. 1, Table 1A). Although the mean LTP amplitude, measured between 40 and 45 min after tetanization, was smaller in P14 GluR-A−/− mice than in wild-type mice the values did not differ significantly (P = 0.242; Fig. 1B, Table 1A). At P42, there was no significant difference between the tetanized and control pathway (P = 0.06), as in older GluR-A ‘knockout’ mice (> P60) (Zamanillo et al. 1999). LTP in juvenile GluR-A-deficient mice at P14 was NMDA receptor dependent as it was fully blocked by 50 μmd-AP5, with values not significantly different from those of the non-tetanized control pathway (P = 0.94, Table 1A).
Figure 1. Disappearance of tetanization-induced field LTP between P14 and P42 in GluR-A-deficient mice.
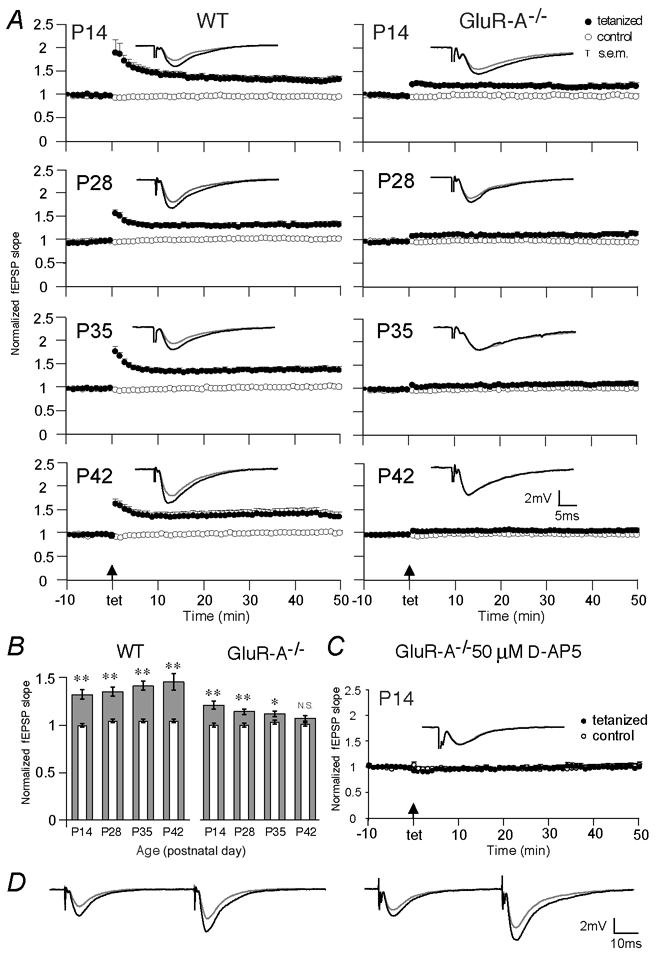
A, normalized extracellular field EPSP (fEPSP) slopes evoked at CA3-to-CA1 synapses in brain slices of wild-type (left) and GluR-A−/− mice (right) of the tetanized (filled circles) and non-tetanized control (open circles) pathway from 10 min before to 50 min after tetanic stimulation are displayed for mice at postnatal day 14 (P14), P28, P35 and P42. The insets show the means of six consecutive synaptic responses in the tetanized pathway before and 40 min after tetanization (grey and black traces, respectively) superimposed at different ages. B, mean LTP values, given as normalized extracellular fEPSP slopes, obtained 40–45 min after tetanization (broad grey columns) in wild-type and GluR-A−/− mice as a function of age. The thin open columns give the corresponding values for the non-tetanized control pathway. Vertical bars: s.e.m. Asterisks indicate degree of significance between LTP values in the tetanized and control pathways (two-tailed paired t test; *P = 0.02, **P < 0.002; N.S.: not significant). C, normalized extracellular fEPSP slopes evoked at CA3-to-CA1 synapses in brain slices of GluR-A−/− at P14 in the tetanized (filled circles) and non-tetanized control (open circles) pathway from 10 min before to 50 min after tetanic stimulation in the presence of 50 μmd-AP5. The inset shows the means of six consecutive synaptic responses before and 40 min after tetanization (grey and black traces, respectively) in the tetanized pathway. For A and C, only the last 10 min of a > 15 min stable baseline are shown. Vertical bars: s.e.m., arrows: time points of tetanic stimulation. Numbers of mice, see Table 1. D, paired-pulses prior to and 60 min after LTP induction (grey and black traces, respectively) in slices from wild-type (left) and GluR-A−/− mice (right) at P14 (see Methods and Table 1B).
Table 1.
LTP expression during development in wild-type and GluR-A-deficient mice
| Wild-type | GluR-A−/− | |||||
|---|---|---|---|---|---|---|
| Potentiation | Control | e/a | Potentiation | Control | e/a | |
| A. LTP values in tetanic stimulation experiments | ||||||
| P14 | 1.32 ± 0.05 | 0.99 ± 0.02 | 29/7 | 1.21 ± 0.04 | 1.00 ± 0.02 | 22/5 |
| P28 | 1.35 ± 0.05 | 1.04 ± 0.02 | 25/5 | 1.14 ± 0.03 | 1.00 ± 0.02 | 23/5 |
| P35 | 1.41 ± 0.05 | 1.04 ± 0.02 | 25/5 | 1.12 ± 0.03 | 1.03 ± 0.02 | 21/4 |
| P42 | 1.45 ± 0.09 | 1.04 ± 0.02 | 16/3 | 1.07 ± 0.03 | 1.01 ± 0.02 | 21/5 |
| P14 with 50 μmd-AP5 | — | — | — | 1.00 ± 0.05 | 1.01 ± 0.04 | 13/2 |
| B. LTP and paired-pulse facilitation in tetanic stimulation experimentsτ | ||||||
| P14–19 | 1.29 ± 0.06 | 1.00 ± 0.03 | 14/4 | 1.11 ± 0.04 | 0.98 ± 0.02 | 13/4 |
| PPF before | 1.74 ± 0.07 | 1.70 ± 0.09 | — | 1.84 ± 0.11 | 1.72 ± 0.06 | — |
| PPF after | 1.85 ± 0.07 | 1.68 ± 0.08 | — | 1.73 ± 0.08 | 1.89 ± 0.12 | — |
| C. LTP values in pre- and postsynaptic pairing experiments | ||||||
| P14 | 1.61 ± 0.29 | 0.75 ± 0.10 | 17/5 | 1.67 ± 0.16 | 0.96 ± 0.11 | 17/5 |
| P42 | 3.12 ± 0.34 | 1.39 ± 0.16 | 14/4 | 1.43 ± 0.11 | 1.07 ± 0.13 | 11/3 |
Long-term potentiation (LTP) values are given as normalized extracellular field EPSP (fEPSP) slopes measured 40–45 min after tetanization (A and B) or as normalized EPSC amplitudes recorded 45–50 min after the ‘pairing’ protocol (C); potentiation, test pathway; control, control pathway; e, number of experiments; a, number of mice; PPF before, paired-pulse facilitation 15 min before tetanization; PPF after, paired-pulse facilitation 50–65 min after tetanization.
The GluR-A-independent LTP form is postsynaptically expressed, as paired-pulse facilitation (PPF) experiments did not substantiate a presynaptic locus of enhancement (Fig. 1D; Table 1B). There was no difference in PPF before and after LTP induction (P > 0.10), nor any correlation between the degree of changes in PPF and the amount of LTP obtained, regardless of genotype (Pearson's correlation coefficient, P > 0.10).
A striking feature of the field response pattern to tetanization in GluR-A−/− mice was the lack of the large responses which are normally seen for minutes after tetanization (Fig. 1A) and comprise post-tetanic potentiation (PTP) and short-term potentiation (STP) (Bliss & Lømo, 1973).
Pairing-induced LTP in GluR-A−/− mice decreases during postnatal development
A development-dependent decline of synaptic potentiation in GluR-A−/− mice was also observed in whole-cell recordings of EPSCs in CA1 pyramidal neurons after ‘pairing’ low frequency presynaptic stimulation of CA3-to-CA1 afferents with postsynaptic depolarization (Chen et al. 1999). The amplitude of evoked EPSCs was enhanced in young (P14–P16) wild-type and GluR-A-deficient mice when measured after ‘pairing’ (Fig. 2A and B). The time course of the potentiation was slightly different in mutant and wild-type mice (Fig. 2A), but the amplitude was comparable for both genotypes 50 min after pairing (P = 0.51, Fig. 2B) with enhancement specific for the paired pathways (Fig. 2B, Table 1C). At P42, the pairing-induced LTP of EPSCs was increased about twofold in wild-type compared to P14 (P = 0.014), but was much smaller in GluR-A-deficient mice (P = 0.001; Fig. 2, Table 1C). Thus, as for tetanus-induced potentiation, in juvenile mice enhanced transmission across CA3-to-CA1 synapses can be induced by pairing of evoked EPSCs with postsynaptic depolarization, whether GluR-A subunits are present or absent. Similarly, the remaining pairing-induced LTP at P14 in GluR-A−/− mice seemed to be dependent on NMDA receptors as well, and under our conditions its formation could be blocked by d-AP5. Between P14 and P42, pairing-induced LTP decreased when GluR-A was not present, indicating a requirement for GluR-A in adult mice. At P42, the size of synaptic enhancement differed fourfold between wild-type and GluR-A-deficient mice (Fig. 2B, Table 1C), which reflects a combination of reduced LTP amplitude in GluR-A−/− mice and a large increase in LTP in wild-type mice during development.
Figure 2. Changes of pairing-induced cellular LTP during development.
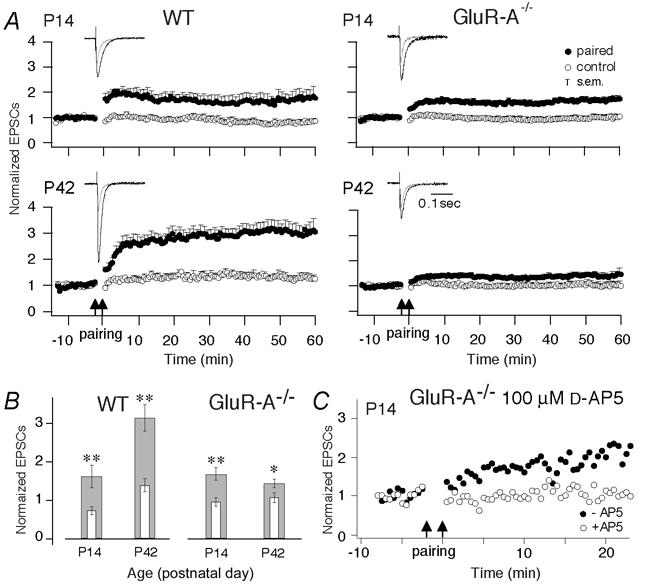
A, normalized EPSC amplitudes evoked at CA3-to-CA1 synapses in brain slices of wild-type (left) and GluR-A−/− mice (right) in the paired (filled circles) and unpaired pathways (open circles) from 10 min before to 60 min after pairing at P14 and P42. Cellular LTP was evoked by pairing low-frequency presynaptic stimulation (0.67 Hz) with postsynaptic depolarization to 0 mV for 3 min indicated by the two arrows. The insets show the means of five consecutive synaptic responses in the paired pathway before and 40 min after pairing (grey and black traces, respectively) superimposed at different ages. Vertical bars: s.e.m.; number of mice see Table 1C. B, mean LTP values, given as normalized EPSC amplitudes, averaged from recordings from 45 to 50 min after pairing (broad grey columns) in wild-type and GluR-A−/− mice as a function of age. The thin open columns give the corresponding values for unpaired control pathways. Vertical bars: s.e.m. Asterisks indicate degree of significance between LTP values in the paired and control pathways (two-tailed paired t test; *P = 0.01, *P < 0.001). C, normalized EPSC amplitudes evoked at CA3-to-CA1 synapses in brain slices of GluR-A−/− mice in the paired pathways from 10 min before to 20 min after pairing in the absence (filled circles) and presence (open circles) of 100 μmd-AP5.
The developmental increase of AMPA EPSCs is lacking in GluR-A-deficient mice
The loss of LTP at adult CA3-to-CA1 synapses of GluR-A−/− mice was not paralleled by a developmental change in EPSCs. We compared AMPA and NMDA receptor-mediated components of evoked EPSCs in brain slices of mice of different ages (Fig. 3A). For wild-type mice the ratio of AMPAR/NMDAR-mediated EPSCs was small at P14 and increased about twofold until P42 (P = 0.001 for P14 and P42, Fig. 3B). In GluR-A-deficient mice AMPAR/NMDAR-mediated EPSCs started, as in wild-type, small at P14 and remained small at P28 and P42 (P = 0.63 comparing P14 and P42; Fig. 3B).
Figure 3. Developmental changes in synaptically evoked EPSCs and glutamate-evoked whole-soma currents.
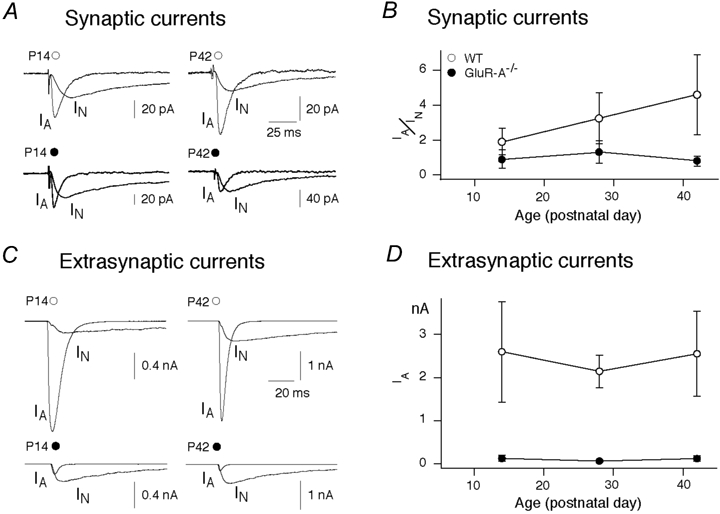
A, recordings of synaptically evoked AMPA and NMDA EPSCs at P14 and P42 from CA1 pyramidal cells in slices from wild-type (open circle) and GluR-A-deficient (filled circle) mice. Traces are averages of 50–100 sweeps. B, increase of postsynaptic AMPA receptor-mediated EPSCs of CA1 pyramidal cells measured at P14, P28 and P42 in wild-type mice (open circles; n = 12, 7 and 10, respectively; P = 0.001 for P14 versus P42) and developmentally unaffected AMPA receptor-mediated EPSCs in GluR-A-deficient mice at P14, P28 and P42 (filled circles; n = 8, 5 and 3; P = 0.63 for P14 versus P42). AMPA receptor-mediated EPSCs are normalized to NMDA receptor-mediated EPSCs (IA/IN). Vertical bars: s.e.m.C, recordings of glutamate-activated AMPA and NMDA receptor currents from nucleated patches from CA1 pyramidal cells of acute brain slices obtained from P14 and P42 wild-type (open circles) and GluR-A-deficient (filled circles) mice. Nucleated patches were exposed to 1 mm glutamate for 2 ms. IA = AMPA receptor-mediated current, IN = NMDA receptor-mediated current. Traces are averages of 5–10 sweeps. D, unchanged extrasynaptic AMPA receptor-mediated currents of CA1 pyramidal cells (IA) at P14, P28 and P42 in wild-type mice (open circles; n = 5, 7 and 5, respectively) and also unaffected, but low AMPA receptor-mediated currents in GluR-A-deficient mice (filled circles; n = 8 at all ages).
We further studied extrasynaptic AMPA receptor currents measured at the soma membrane of CA1 pyramidal cells by using nucleated patches and fast glutamate application. Assuming similar patch sizes, the AMPA receptor currents did not change in wild-type and GluR-A-deficient mice between P14 and P42 (Fig. 3C). In GluR-A-deficient mice however, at all ages investigated (P14, P28 and P42), the somatic AMPA currents were only a small fraction, about one-tenth, of those in wild-type (Fig. 3C and D). Therefore, throughout development, lack of GluR-A leads to the selective loss of AMPA currents in extra-synaptic membranes.
Absence of developmental redistribution of AMPA receptor subunits in GluR-A-deficient mice
In wild-type mice, the developmental increase of synaptic AMPA/NMDAreceptor EPSC ratios is accompanied by a shift of GluR-A- and GluR-B-containing AMPA receptors from somatic to dendritic sites, as revealed by immunolabelling (Fig. 4). In the hippocampal layer CA1 of wild-type mice both the somatic layer of stratum pyramidale and the dendritic layers of strata radiatum and oriens - where CA3-to-CA1 synapses are located - show immunosignal for GluR-A and GluR-B subunits at P14 (Fig. 4, left). At P42 the somatic layers show a reduced immunosignal, whereas the dendritic layers still exhibit a strong signal. For the GluR-B subunit this is obvious in Fig. 4 and less recognizable for the GluR-A subunit, which was described before to show this age-dependent redistribution (Mack et al. 2001). For the GluR-C subunit the situation is less clear since its distribution can only be monitored with an antibody recognizing both the GluR-B and the GluR-C subunits. Using this antibody we obtained a stronger relative immunosignal in the somatic layer of stratum pyramidale at P42 compared to P14 which might indicate that the GluR-C-containing AMPA receptors follow a different distribution pattern than those without GluR-C. The availability of GluR-C specific antibodies needs to clarify this point. The immunosignal for GluR-D was selectively detected in interneurons where it co-localized with interneuron markers (see Supplementary Material Fig. 2) with no changes in developmental profile (Fig. 4).
Figure 4. Developmental changes in AMPA receptor subunit distribution.
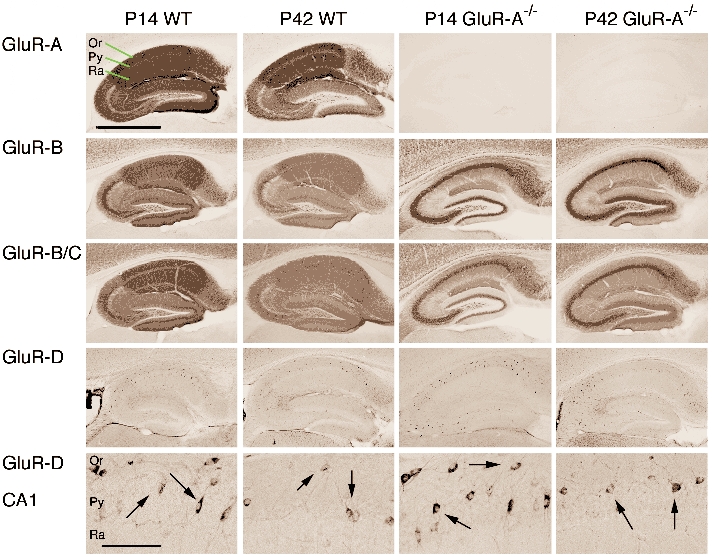
GluR-A to -D subunit distribution in hippocampi of wild-type and GluR-A-deficient mice at P14 and P42. Sagittal vibratome brain sections were immunostained with specific antibodies directed against GluR-A, -B, -B/C and -D subunits. Micrograph pictures of the hippocampal subunit distribution visualized by diaminobenzidine (DAB)-labelled secondary antibodies are given (scale bar for 4 upper rows: 1 mm). In the bottom row higher magnification shows putative interneurons positive for the GluR-D subunit (arrows) in the CA1 region. (Scale bar for lower row: 0.13 mm).
Brains from GluR-A-deficient mice showed aberrant distribution of other AMPA receptor subunits, most notably of GluR-B (Fig. 4; right, Supplementary Material Fig. 1), which accumulated in the cell layer of hippocampal principal neurons (Zamanillo et al. 1999). The intense GluR-B-specific staining of the stratum pyramidale in GluR-A−/− mice was equally prominent at P14 and P42 (Fig. 4, right second row), and might represent unassembled GluR-B subunits and/orhomomeric GluR-B receptors with very small single-channel conductance (Swanson et al. 1997). The anti-GluR-B/-C antibody provided a similar pattern with a slightly higher immunosignal in the dendritic layers (Fig. 4, right third row). GluR-D immunostaining remained restricted to hippocampal interneurons at both P14 and P42, and was not different from wild-type, as clearly seen with high magnification (Fig. 4, bottom row). Therefore, a hypothetical age-dependent difference in the expression pattern of the AMPA receptor subunits GluR-B, -C and -D can be excluded as a mechanism underlying the postnatal decline of the GluR-A-independent LTP in GluR-A-deficient mice.
In summary, GluR-A-deficient mice show similar AMPA receptor expression profiles in synapses and somata at P14 and P42, whereas in wild-type mice the AMPA receptors shift during this time from perisomatic to synaptic and dendritic sites. In spite of the low immunohistochemical signal of GluR-B and GluR-C subunits in dendritic layers of GluR-A-deficient mice, the synaptic transmission remained robust, which suggests that in the hippocampus of wild-type mice, most dendritic GluR-A immunolabelling is mediated by extrasynaptic AMPA receptors and/orunassembled GluR-A.
Glutamate receptor expression is delayed and the levels of GluR-B and GluR-D subunits are reduced in GluR-A-deficient mice
The redistribution from somatic to dendritic sites of AMPA receptors in wild-type mice between P14 and P42 is not a result of increased AMPA or NMDA receptor expression. We measured the expression levels of the AMPA receptor subunits GluR-A to -D and the NMDA receptor subunit NMDAR1 (Moriyoshi et al. 1991) between P2 and P180 in immunoblots of hippocampal proteins (Fig. 5). We found low expression of all subunits at P2. The expression increased, reaching a maximum between P14 and P28, and was then maintained to adulthood. Thus, the redistribution of the AMPA receptors becomes prominent after the levels of all four AMPA receptor subunits have reached their maximum.
Figure 5. Developmental changes in AMPA receptor subunit expression.
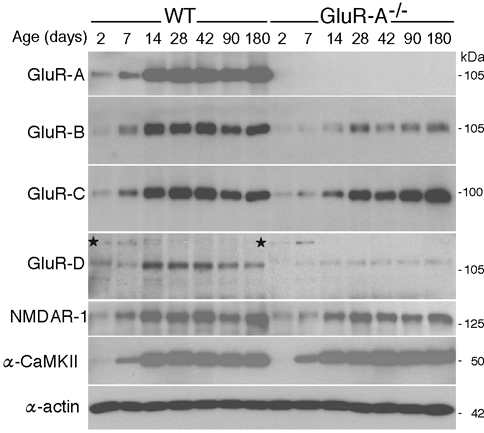
Expression of ionotropic glutamate receptor subunits in hippocampi of wild-type (WT) and GluR-A-deficient (GluR-A−/−) mice from P2 to P180. Total hippocampal proteins were isolated, and for each sample 8 μg was loaded on a 7 % SDS-polyacrylamide gel. Separated proteins were transferred to nitrocellulose, and the glutamate receptor subunits were visualized by selective antibodies and monitored by autoradiography. For each subunit the 14 samples (7 time points for each genotype) were analysed in one gel run. The α-subunit of the Ca2+-calmodulin-dependent protein kinase ll (α-CaMKII) was used as a positive control and β-actin was used as a control for lane loading. The experiment was repeated with at least three sets of mice. One representative example for each subunit is given. In wild-type mice, the expression of all glutamate receptor subunits reached its maximum between P14 and P28. In GluR-A-deficient mice the expression maximum was delayed until P28. In the GluR-D panel a non-specific protein recognized by a CHEMICON anti-GluR-D antibody is indicated by an asterix. This protein shows a transient expression in the first postnatal days in hippocampi of wild-type and GluR-A-deficient mice and is also present in GluR-D ‘knockout’ mice (not shown).
In GluR-A−/− mice we observed a delay in the upregulation of AMPA receptor subunits and of the NMDA receptor subunit NMDAR1, with maximum expression first reached around P28 (Fig. 5, right). Because the α-subunit of the Ca2+-calmodulin-dependent protein kinase II showed its expected expression profile (Fig. 5, α-CaMKII), with peak expression at P14 (Kelly & Vernon, 1985), the delayed upregulation of the glutamate receptor subunits does not reflect a generalized delay in development. In addition, we found that the number of GluR-B and GluR-D subunits was reduced in GluR-A-deficient mice older than P14 (Fig. 5, rows 2 and 4). GluR-B and GluR-D are the major GluR-A partners of hippocampal AMPA receptors in principal neurons and interneurons, respectively. Thus, absence of GluR-A leaves much of GluR-B and GluR-D unassembled and the half-life of unassembled GluR-B and GluR-D subunits is expected to be shorter than the half-life of assembled subunits
Adult GluR-A-deficient mice show increased numbers of GluR-B-containing CA1 synapses
In CA1 pyramidal cells of GluR-A−/− mice the remaining functional AMPA receptors are mainly localized in synapses as indicated by the very low AMPAR/NMDARcurrent ratios in soma patches and much higher AMPAR/NMDARcurrent ratios in synaptic measurements. Therefore, the strong somatically immunolabelled GluR-B in stratum pyramidale of GluR-A−/− brain slices (Fig. 4 and Fig. 6A; Supplementary Material Fig. 3) must reflect unassembled or non-functional (low conductance) homomeric GluR-B channels. The functional GluR-B-containing channels might be represented by the speckled GluR-B signal on proximal dendritic shafts of the CA1 cells in slices from GluR-A-deficient mice. In slices from wild-type mice, dendritic shafts cannot be resolved due the intense GluR-B expression in strata radiatum and oriens, which is absent in GluR-A−/− mice (Fig. 4 and Fig. 6A; Supplementary Material Fig. 3).
Figure 6. Accumulation of the GluR-B subunits in somata and at synapses of GluR-A-deficient mice.
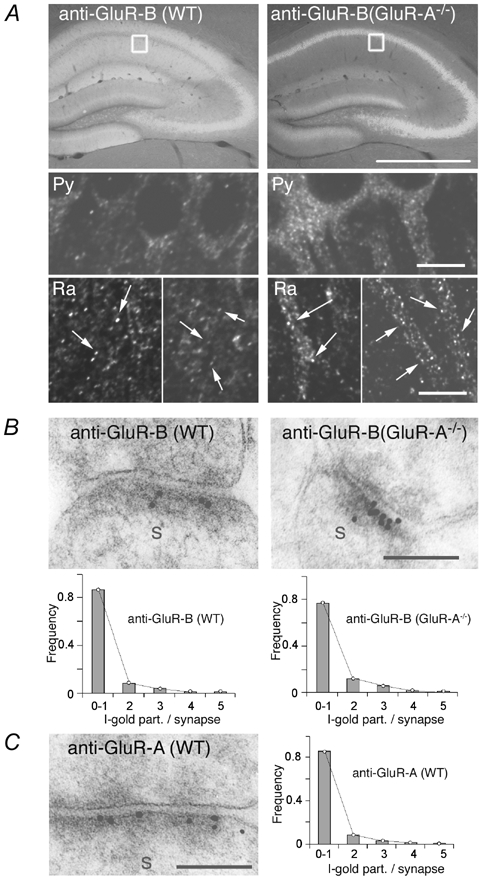
A, immunofluorescence-labelled GluR-B (white signal) in hippocampus of wild-type and GluR-A-deficient mice indicating somatic accumulation of the GluR-B subunit in GluR-A-deficient mice. Scale bar: 1 mm. Insets display the approximate position of the confocal images of labelled GluR-B in the stratum pyramidale (Py) and radiatum (Ra) in the CA1 region. Pyramidal CA1 cell nuclei are black, without fluorescent signal. In GluR-A−/− mice cell somata and the most proximal part of the dendrite show strong fluorescent staining (right). In the stratum radiatum of wild-type mice the GluR-B-specific fluorescent signal is evenly distributed in tiny spots (arrows, less then 1 μm) all over the stratum radiatum. The overall fluorescence is high and cellular structures cannot be resolved. In GluR-A-deficient mice, total fluorescence in the stratum radiatum is weaker and the proximal part of dendrites can now be seen. The tiny GluR-B immunospots (arrows) are also present. They are sitting on the shafts of the dendrites and might represent large GluR-B-containing spine heads. Scale bar: 10 μm. B, example of an increased number of immunogold-labelled GluR-B-containing receptors at spine synapses (s) in the stratum radiatum of GluR-A-deficient (right) compared to wild-type mice (left). Scale bar: 0.1 μm. C, immunogold labelling for GluR-A at a spine synapse(s) in the stratum radiatum of the CA1 region in wild-type mice. Scale bar: 0.1 μm. B and C, graphs show the quantitative analysis of GluR-A- and GluR-B-containing AMPA receptors in CA1 synapses. The grey bars give the frequency of gold grains per synapse (I-gold part./synapse) detected for GluR-B and GluR-A, respectively, in wild-type (WT) and GluR-A-deficient (GluR-A−/−) mice. To account for possible background staining, synapses showing only one gold grain were counted as unlabelled. Experimental data were then fitted by Poisson distribution (lines) to estimate the total number of AMPA receptor-containing synapses (see Table 2).
To study the detailed ultrastructural GluR-B localization, we visualized GluR-A and GluR-B subunits in electron microscopic sections using cryosubstitution and immunogold techniques (Frotscher et al. 1999). In adult (> P60) hippocampus of wild-type mice the percentage of synapses labelled with gold particles specific for GluR-A (Fig. 6C) and GluR-B (Fig. 6B) was comparable (Table 2). In contrast, the number of anti-GluR-B labelled postsynaptic densities (PSDs) was about 1.5-fold increased compared to wild-type (Table 2). The average number of gold grains per synapse was also increased slightly in GluR-A-deficient mice (Fig. 6B, Table 2), suggesting increased numbers of synaptic GluR-B-containing AMPA receptors.
Table 2.
Quantitative analysis of GluR-A- and GluR-B-containing AMPA receptors in CA1 synapses
| Wild-type | GluR-A−/− | |||
|---|---|---|---|---|
| Experimental data | Fitted Poisson distribution | Experimental data | Fitted Poisson distribution | |
| GluR-A-specific Ab | ||||
| Percentage of positive PSDs | 13.6 ± 1.3 | 42.0 ± 2.0 | 0.2 ± 0.06 | † |
| No. of grains per individual synapse | 2.47 ± 0.04 | 1.17 ± 0.08 | n.a. | — |
| GluR-B-specific Ab | ||||
| Percentage of positive PSDs | 12.4 ± 0.8 | 35.9 ± 2.4* | 22.5 ± 1.3 | 51.4 ± 0.7* |
| No. of grains per individual synapse | 2.51 ± 0.04 | 1.24 ± 0.08** | 2.63 ± 0.04 | 1.49 ± 0.08** |
Ab, antibody; PSDs, postsynaptic densities;
P = 0.0008,
P = 0.0051;
† fitting of Poisson distribution was not possible; n.a., not analysed. To account for possible background staining, synapses showing only one gold grain were not counted. Experimental data were then fitted by Poisson distribution to estimate the total number of AMPA receptor-containing synapses. On the basis of this distribution the mean number of gold grains per individual synapse was calculated.
DISCUSSION
Two forms of LTP in young mice
Using GluR-A ‘knockout’ mice we could demonstrate that - despite the lack of GluR-A - significant hippocampal CA3-to-CA1 LTP can be induced in young mice (P14–P28), either by brief presynaptic tetanic stimulation or by pairing low frequency presynaptic stimulation with postsynaptic depolarization. During the following weeks, the ability of these synapses to express LTP diminishes and becomes very low at P42 in the pairing-induced and undetectable in the tetanus-induced LTP, as reported earlier for older (> P60) GluR-A−/− mice (Zamanillo et al. 1999).
Our study, currently, cannot exclude that this GluR-A-independent LTP is one of the proposed ‘reserve mechanisms’ (Zhu & Malinow, 2002) which is recruited only when GluR-A-containing AMPA receptors are not available. However, according to the principles of Darwin's gene selection theory, it is difficult to explain how such ‘reserve mechanisms’ are maintained in evolution, and how they are restricted in expression to a short phase of postnatal development. No natural scenarios are known when AMPA or NMDA receptor subtypes become transiently not available during development and a ‘reserve mechanism’ comes to the rescue of the affected progeny. It is more likely that also in wild-type the GluR-A-independent LTP form coexists with the GluR-A-dependent form in CA1 pyramidal cell synapses, especially in young mice. During maturation of hippocampal connections between P15 and P42, the GluR-A-independent form slowly decreases while the GluR-A-dependent form increases to become dominant in the adult brain.
Mechanisms for GluR-A-independent LTP
The GluR-A-dependent and independent forms of LTP are both input specific, long-lasting, d-AP5-sensitive, postsynaptically expressed and mechanistically induced by the same protocols. Despite these similarities both forms of LTP are likely to rely on different mechanisms. This is strongly supported by slice recordings from rats showing that PKA is essential for NMDA receptor-induced LTP of CA1 pyramidal cells in young animals whereas in adults, NMDA receptor-activated CaMKII is responsible for the formation of LTP (Yasuda et al. 2003). Further support that in young mice the GluR-A-independent LTP, which might need PKA for its expression, does not operate through GluR-A-containing AMPA receptors, is provided by mutant mice lacking - in addition to the CaMKII phosphorylation site in position S831 - the PKA phosphorylation site in position S845 of the GluR-A subunit. These mice show LTP at P14 which is strongly reduced at adult ages, indicating that the PKA phosphorylation of GluR-A at S845 is not important for LTP in young mice (Lee et al. 2003). However, in short-term plasticity, GluR-A phosphorylation might be involved since the hallmark of the LTP in juvenile GluR-A-deficient mice is the striking lack of STP in field recordings. Similar to the adult LTP the GluR-A-independent form is expressed postsynaptically since we did not detect an improved glutamate release in paired pulse facilitation experiments before and after LTP expression.
It is striking that the presence of GluR-A-independent LTP correlates with the activation of putative ‘silent’ synapses (Stricker et al. 1996; Malenka & Nicoll, 1997), which are made operative during a similar developmental time period (Isaac et al. 1995; Durand et al. 1996; Liao & Malinow, 1996) and which is accompanied by pre- and postsynaptic structural changes and alterations in the number of contacts (Engert & Bonhoeffer, 1999; Maletic-Savatic et al. 1999). Whether the remaining AMPA receptor subunits GluR-B, -C, -D contribute to these processes and to the expression of LTP is not resolved yet. The dependence on NMDA receptor activation of the GluR-A-independent LTP might favour speculations that activity-dependent AMPA receptor insertion participates also in GluR-A-independent LTP. In recent reports a contribution of GluR-D, instead of GluR-A, in postnatal LTP was suggested from gene transfer studies in slice cultures (Zhu et al. 2000). According to our results this seems unlikely since we were not able to detect specific GluR-D immunosignals in CA1 pyramidal cells in young as well as in adult mice, neither in wild-type nor in GluR-A-deficient mice. Thus, similar to CA3 pyramidal cells (Geiger et al. 1995), CA1 pyramidal neurons probably do not express this apparently interneuron specific GluR-D subunit. Also, homomeric GluR-B channels are not good candidates for expressing LTP given that these channels mediate only small currents (Burnashev et al. 1992; Swanson et al. 1997). Better candidates are heteromeric GluR-B/-C channels containing the GluR-B(long) splice variant (Kohler et al. 1994). The recent findings that GluR-B(long) expression declines between P14 and P42 and that recombinant expression of the C-terminal domain of GluR-B(long) in hippocampal slices from juvenile GluR-A-deficient mice reduce LTP expression (Kolleker et al. 2003) would make GluR-B(long) an attractive alternative to GluR-A in mediating activity dependent synaptic AMPA receptor insertion for LTP expression.
Is GluR-A-independent LTP related to developmental redistribution of AMPA receptors?
Between P14 and P42, AMPA receptors redistribute in the CA1 region of the hippocampus, from a predominantly somatic to a dendritic location. This redistribution resulting in dendritic accumulation of AMPA receptors may be required for the adult form of LTP in CA3-to-CA1 synapses but, perhaps, could also be a normal aspect of the connectivity build-up that takes place during the same time in development (Petralia et al. 1999).
It is interesting to note that the majority of AMPA receptors in mature CA1 pyramidal cells are extrasynaptic and therefore they are not involved in synaptic transmission. These extrasynaptic AMPA receptors are readily detected in GluR-A- and GluR-B-specific immunohistochemical stainings of the dendritic fields and in dendritic (Andrásfalvy & Magee, 2001) and in nucleated patch recordings. Extrasynaptic AMPA receptors can be removed, as in GluR-A−/− mice, without loss of synaptic AMPA receptor currents. Thus, GluR-A seems to be necessary for the dendritic localization in a ‘facultative transport pathway’ that operates by accumulation of extrasynaptic GluR-A-containing receptors and regulates the number of GluR-A-containing receptors which reach synaptic localization (Andrásfalvy et al. 2003). After LTP induction, GluR-A-containing receptors of the extrasynaptic pool may be incorporated into activated synapses, thereby enhancing synaptic transmission.
When GluR-A subunits are lacking, GluR-B/-C receptors might translocate via a ‘constitutive pathway’ directly into spine heads (Osten et al. 2000; Shi et al. 2001), as revealed in our immunogold experiments by increased synaptic GluR-B and an increased number of synapses containing GluR-B. Homomeric GluR-B receptors might not be translocated to synaptic sites, but may recycle or remain in perisomatic compartments (Greger et al. 2002), before being degraded. Evidence for this may be seen from the decreased GluR-B levels and the somatic accumulation of GluR-B in hippocampus of GluR-A-deficient mice. As a consequence, in absence of GluR-A, the extrasynaptic dendritic sites are depleted in AMPA receptors which is best documented in brain slices by the strong reduction of GluR-B immunosignal in the stratum radiatum and stratum oriens and the lack of somatic currents in GluR-A-deficient mice. The remaining GluR-B signal in the dendritic field of the hippocampus seems to be exclusively synaptically localized as visualized by confocal and immunogold immunohistochemistry. Experiments with virus-infected slice cultures from rat brains at P5-P7 proposed this function for the GluR-A subunit in developing synapses (Shi et al. 1999; Hayashi et al. 2000), supporting the contribution of GluR-A-dependent LTP to total LTP in the juvenile rodent brain.
Physiological significance of GluR-A-independent LTP
GluR-A-deficient mice are further proof that the GluR-A-independent LTP form is sufficient for the establishment of functional hippocampal connections, since (i) spine number, spine structure and excitatory synapse function are not affected, (ii) they are comparable to wild-type mice in several hippocampus-mediated behavioural tasks, including the Morris water maze (Zamanillo et al. 1999; Reisel et al. 2002) and (iii) the GluR-A-dependent LTP form can be restored in GluR-A-deficient adult mice by transgenic GluR-A expression (Mack et al. 2001). Therefore, during development the GluR-A independent LTP is, conceivably, related to the establishment of proper hippocampal synaptic connectivity and/orweighting, which occurs before the hippocampus becomes a functionally important integration centre.
In contrast, the GluR-A-mediated LTP seems to be of special importance for the function of the hippocampus in the adult brain. This is indicated by very high hippocampal GluR-A expression, which is stronger than in most other brain regions, in particular in the neocortex (Molnar et al. 1993; Zamanillo et al. 1999), and by the high ratio of extrasynaptic versus synaptic AMPA receptors. AMPA receptor levels increase during the first 3–4 weeks of postnatal development in mice and, by P42, GluR-A-dependent LTP is the dominant form at CA3-to-CA1 connections. Due to the strong dependence of LTP expression on GluR-A in adult mice, it came as a surprise that spatial reference memory in the water maze was not impaired in adult GluR-A ‘knockouts’ (Zamanillo et al. 1999; Reisel et al. 2002). However, Reisel et al. (2002) have recently shown that spatial memory does require GluR-A-dependent LTP. In contrast to the spared reference memory performance in the water maze, GluR-A−/− mice exhibited a robust and enduring spatial working memory impairment during non-matching to place testing on the elevated T-maze (Reisel et al. 2002).
In conclusion, our comparative studies on GluR-A-deficient and wild-type mice suggest that NMDA receptors can induce different LTP forms with their own molecular mechanisms at the very same synaptic hippocampal connections (CA3-to-CA1). This view is supported by the finding of operative, delayed theta-burst pairing potentiation in hippocampi of adult GluR-A-deficient mice (Hoffman et al. 2002). Future work will need to identify those LTP mechanisms that have physiological relevance on computational abilities in the brain.
Supplementary material
The online version of this paper can be found at:DOI: 10.1113/jphysiol.2003.053637 and contains material entitled:Somatic GluR-B accumulation and GluR-D expresssion in GluR-A deficient miceV. Jensen, K. M. M. Kaiser and T. Borchardt contributed equally to this work
Supplementary Material
Acknowledgments
This work was supported by the European Union (EU-Grant QLRT-1999-01022 LTP expression), by the Norwegian Research Council, by the Deutsche Forschungsgemeinschaft (SFB 505, C 6) and by the Volkswagen-Stiftung. The authors thank Dr Frank Single for help in immunohistochemical analysis, Dr Jürgen Schulte-Mönting for help with statistical analysis of immunogold-labelled sections, Margerita Pfeffer, Annette Herold, Karin Mews for technical assistance, Marianne Winter for photographic assistance, Professor Petter Laake for help with statistical analysis and Dr Pavel Osten for discussion.
REFERENCES
- Andersen P, Sundberg SH, Sveen O, Wigstrom H. Specific long-lasting potentiation of synaptic transmission in hippocampal slices. Nature. 1977;266:736–737. doi: 10.1038/266736a0. [DOI] [PubMed] [Google Scholar]
- Andrásfalvy B, Magee JC. Distance-dependent increase in AMPA receptor number in the dendrites of adult hippocampal CA1 pyramidal neurons. J Neurosci. 2001;21:9151–9159. doi: 10.1523/JNEUROSCI.21-23-09151.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrásfalvy B, Smith MA, Borchardt T, Sprengel R, Magee JC. Impaired regulation of synaptic strength in hippocampal neurons from GluR-1-deficient mice. J Physiol. 2003;552:35–45. doi: 10.1113/jphysiol.2003.045575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Lisman J. Requirements for LTP induction by pairing in hippocampal CA1 pyramidal cells. J Neurophysiol. 1999;82:526–532. doi: 10.1152/jn.1999.82.2.526. [DOI] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fleischmann A, Hvalby Ø, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Spanagel R, Sprengel R, Wagner EF, Gass P. Impaired long-term memory and NR2A-type NMDA receptor dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci. 2003;8:9116–9122. doi: 10.1523/JNEUROSCI.23-27-09116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Mews K, Adelmann G. Morphological characteristics of glutamatergic synapses in the hippocampus. In: Jonas P, Monyer H, editors. Handbook of Experimental Pharmacology. Berlin, Heidelberg, New York: Blackwell Science Inc; 1999. pp. 343–362. [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Gustafsson B, Wigström H, Abraham WC, Huang YY. Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J Neurosci. 1987;7:774–780. doi: 10.1523/JNEUROSCI.07-03-00774.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Sprengel R, Sakmann B. Molecular dissection of hippocampal theta-burst pairing potentiation. Proc Natl Acad Sci U S A. 2002;99:7740–7745. doi: 10.1073/pnas.092157999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, O'Shea-Greenfield A, Rogers SW, Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature. 1989;342:643–648. doi: 10.1038/342643a0. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- Kelly PT, Vernon P. Changes in the subcellular distribution of calmodulin-kinase II during brain development. Brain Res. 1985;350:211–224. doi: 10.1016/0165-3806(85)90265-2. [DOI] [PubMed] [Google Scholar]
- Kohler M, Kornau HC, Seeburg PH. The organization of the gene for the functionally dominant alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluR-B. J Biol Chem. 1994;269:17367–17370. [PubMed] [Google Scholar]
- Köhr G, Jensen V, Koester HJ, Mihaljevic ALA, Utvik JK, Kvello A, Ottersen OP, Sprengel R, Seeburg PH, Hvalby Ø. Intracellular domains of NMDA receptor subtypes are determinants for LTP induction. J Neurosci. 2003;23 doi: 10.1523/JNEUROSCI.23-34-10791.2003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolleker A, Zhu JJ, Schupp BJ, Qin Y, Mack V, Borchardt T, Köhr G, Malinow R, Seeburg P, Osten P. The GluR-B(long) splice variant mediates synaptic AMPA receptor insertion triggered by spontaneous synaptic activity. Neuron. 2003 doi: 10.1016/s0896-6273(03)00722-0. in press. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Liao D, Malinow R. Deficiency in induction but not expression of LTP in hippocampal slices from young rats. Learn Mem. 1996;3:138–149. doi: 10.1101/lm.3.2-3.138. [DOI] [PubMed] [Google Scholar]
- Mack V, Burnashev N, Kaiser KM, Rozov A, Jensen V, Hvalby O, Seeburg PH, Sakmann B, Sprengel R. Conditional restoration of hippocampal synaptic potentiation in GluR-A-deficient mice. Science. 2001;292:2501–2504. doi: 10.1126/science.1059365. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Silent synapses speak up. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Molnar E, Baude A, Richmond SA, Patel PB, Somogyi P, McIlhinney RA. Biochemical and immunocytochemical characterization of antipeptide antibodies to a cloned GluR1 glutamate receptor subunit: cellular and subcellular distribution in the rat forebrain. Neuroscience. 1993;53:307–326. doi: 10.1016/0306-4522(93)90198-o. [DOI] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Kohr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27:313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Esteban JA, Wang YX, Partridge JG, Zhao HM, Wenthold RJ, Malinow R. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nat Neurosci. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JN. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5:868–873. doi: 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Sommer B, Kohler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Stricker C, Field AC, Redman SJ. Changes in quantal parameters of EPSCs in rat CA1 neurones in vitro after the induction of long-term potentiation. J Physiol. 1996;490:443–454. doi: 10.1113/jphysiol.1996.sp021156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby Ø, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, Lubke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Malinow R. Acute versus chronic NMDA receptor blockade and synaptic AMPA receptor delivery. Nat Neurosci. 2002;5:513–514. doi: 10.1038/nn0602-850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.