

Abstract
Exposure of the fetal sheep to moderate to severe hypoxic stress results in both increased cortical blood flow and decreased metabolic rate. Using intravenous infusion of 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), a selective adenosine A1 receptor antagonist that is permeable to the blood brain barrier, we examine the role of adenosine A1 receptors in mediating cortical blood flow and metabolic responses to moderate hypoxia. The effects of DPCPX blockade are compared to controls as well as animals receiving intravenous 8-(p-sulfophenyl)-theophylline) (8-SPT), a non-selective adenosine receptor antagonist which has been found to be blood brain barrier impermeable. Laser Doppler flow probes, tissue PO2, and thermocouples were implanted in the cerebral cortices of near-term fetal sheep. Catheters were placed in the brachial artery and sagittal sinus vein for collection of samples for blood gas analysis. Three to seven days later responses to a 30-min period of fetal hypoxemia (arterial PO2 10–12 mmHg) were studied with administration of 8-SPT, DPCPX, or vehicle. Cerebral metabolic rate was determined by calculation of both brain heat production and oxygen consumption. In response to hypoxia, control experiments demonstrated a 42 ± 7 % decrease in cortical heat production and a 35 ± 10 % reduction in oxygen consumption. In contrast, DPCPX infusion during hypoxia resulted in no significant change in brain heat production or oxygen consumption, suggesting the adenosine A1 receptor is involved in lowering metabolic rate during hypoxia. The decrease in cerebral metabolic rate was not altered by 8-SPT infusion, suggesting that the response is not mediated by adenosine receptors located outside the blood brain barrier. In response to hypoxia, control experiments demonstrated a 35 ± 7 % increase in cortical blood flow. DPCPX infusion did not change this increase in cortical blood flow, however 8-SPT infusion attenuated increases in flow, indicating that hypoxic increases in cerebral blood flow are mediated by adenosine but not via the A1 receptor. In summary, adenosine appears to play a key role in fetal hypoxic defences, acting to increase O2 delivery via adenosine A2 receptors and to decrease metabolic rate via A1 receptors inside the blood brain barrier. These data show for the first time in the mammalian fetus that the adenosine A1 receptor is an important mediator of brain metabolic rate during moderate hypoxia.
Compared to the adult, the brain of the mammalian fetus is remarkably tolerant to hypoxic insult (Duffy et al. 1975; Mallard et al. 1994; Hunter et al. 2003a,c). During acute global hypoxia,fetal blood flow is centralized to the brain, heart, and adrenal glands at the expense of the peripheral tissues (Jensen et al. 1999). This redistribution of flow optimizes oxygen delivery to the vital organs and is a result of increased arterial blood pressure in combination with lowered vascular resistance in these organs (Reuss et al. 1982; Jensen et al. 1999). The increases in arterial blood pressure during hypoxia are in part a result of elevated sympathetic activity and plasma concentrations of catecholamines (Cohen et al. 1982; Iwamoto et al. 1983). In contrast to the increased peripheral resistance to blood flow during hypoxia, cerebral vascular tone is unchanged or decreased, suggesting an important role for one or more mediators of hypoxic cerebral vasodilatation (Jensen et al. 1999; Blood et al. 2002; Bishai et al. 2003; Hunter et al. 2003c). We recently reported evidence suggesting adenosine is of primary importance in the regulation of fetal cortical vascular tone and increased cortical blood flow during hypoxia (Blood et al. 2002).
By increasing cerebral blood flow and oxygen extraction, the fetal brain maintains a normal rate of oxygen consumption during mild to moderate hypoxic stress. However, during severe hypoxic stress (arterial oxyhaemoglobin saturation of ≈35 % or less), cerebral metabolic rate is decreased and becomes directly correlated with oxygen delivery (Parer, 1994). In the fetal sheep, the decrease is associated with low energy-consuming EEG states (Chau & Koos, 1999), depression of evoked brainstem potentials (Gunn et al. 1991), and increased anaerobic cerebral metabolism (Asano et al. 1994). Presently it is not known whether the fall in cerebral oxygen consumption during hypoxia is a result of oxygen starvation or a protective mechanism of adaptive hypometabolism, as has been described in the brain, liver, and whole body of several species (Hochachka et al. 1996; Mortola, 1999; Hochachka & Lutz, 2001).
Growing evidence points to adenosine, an endogenously produced purine nucleoside, as a mediator of both increased cerebral blood flow and decreased cerebral metabolic rate during hypoxic stress (Blood et al. 2002). Plasma and intracerebral adenosine concentrations increase during hypoxia in the fetal sheep (Kjellmer et al. 1989; Koos et al. 1997; Suzuki & Power, 1999), newborn lamb (Laudignon et al. 1991), and adult rat (Winn et al. 1981b; Van Wylen et al. 1986), in association with an inhibition of neuronal activity (Fowler et al. 1999). Intravenous administration of theophylline, a non-specific adenosine receptor antagonist, abolishes increases in cortical blood flow during hypoxia in the fetal sheep (Blood et al. 2002). In adult animals adenosine, acting on the adenosine A1 receptor, suppresses cerebral metabolic rate during hypoxia/asphyxiain hippocampal slices (Croning et al. 1995; Jin & Fredholm, 1997; Fowler et al. 1999) and during severe asphyxia in vivo (Hunter et al. 2003c). In the fetal sheep, breathing movements are inhibited by hypoxia, an adaptation that is abolished by adenosine-receptor blockade at the level of the thalamus (Koos et al. 1994a; Chau & Koos, 1999). Thus, there is considerable evidence to warrant the study of the effect of adenosine on fetal cerebral metabolic rate during moderate hypoxia.
The present study was designed to test the hypothesis that adenosine A1 receptor activation mediates both decreased cortical metabolic rate and increased cortical blood flow during moderate hypoxia in the near-term fetal sheep. In order to test this hypothesis late-term fetal sheep were chronically instrumented for measurement of cortical blood flow by laser Doppler flowmetry, cortical heat production, and cortical oxygen consumption. Fetal sheep were subjected to severe hypoxia during intravenous infusion of either DPCPX, a selective adenosine A1 receptor antagonist that crosses the blood brain barrier (Marston et al. 1998), or 8-SPT, a non-selective adenosine receptor antagonist that does not cross the blood brain barrier (Baumgold et al. 1992; Coney & Marshall, 1998; Meno et al. 2001).
METHODS
Surgical preparation
All study procedures were approved by the Loma Linda University Animal Care Research Committee. Western ewes carrying singleton or twin fetuses were used for the study; in the case of twins only one fetus was studied. Surgical instrumentation was performed between 122 and 128 days gestation (term = 47 days gestation) as previously described (Lan et al. 2000). Anaesthesia was induced with 0.5 g thiopental and maintained with 1.5-2.5 % inhaled halothane. A polyvinyl catheter was placed in the femoral vein of the ewe for administration of intravenous fluids during the surgery, as well as administration of a euthanasia solution (Euthasol) at the termination of the experiments. Through a midline maternal incision in the abdomen, the fetal head and neck were exposed. Polyvinyl catheters were placed in a brachial artery for blood gas sampling and arterial blood pressure measurement, in the subclavian vein for drug administration, and in the amniotic sac for measurement of amniotic fluid pressure and administration of antibiotics to the fetus (Blood et al. 2002). Then 0.5 mm burr holes were drilled bilaterally in the fetal skull for insertion of a laser Doppler flowmetry probe in the right parietal cortex and a thermocouple (IT-18 thermometer, Physitemp, Clifton, NJ, USA) in the left cortex as described previously (Lan et al. 2000; Bishai et al. 2003). The laser Doppler flow probe (Oxford Optronix, Oxford, UK) consisted of a four-part composite probe (diameter ≈400 μm) containing emitting and receiving laser Doppler channels, a PO2 electrode, and thermocouple. A second thermocouple was inserted into the brachial artery of the fetus together with the polyvinyl catheter, for measurement of arterial temperature. A polyvinyl catheter was also inserted into the sagittal sinus for collection of cerebral venous blood samples. The uterus and abdomen of the ewe were sutured shut in layers and the catheters and probes were exteriorized through the maternal flank and held in a protective pouch between experiments. The ewe was given 900 000 u of intramuscular penicillin for 3 days. The fetus received 500 mg of ampicillin and 40 mg of gentamicin daily in the amniotic fluid, until the termination of the experiment.
Experiments were carried out 5 ± 2 days following the operation, with a minimum of 48 h between the control and experimental protocols conducted on each animal. Experiments were conducted while the ewe was standing in a metabolic cart at room temperature with minimal distractions; alfalfa pellets and water were available to the ewe as desired.
Experimental protocols
The control protocol consisted of a 60 min baseline period followed by a 30 min period of hypoxia induced by breathing the ewe with 10–12 % O2 in a balance of nitrogen. This was administered into a bag placed over the head of the ewe with a flow rate of 30 l min−1. The hypoxic period was followed by a 60 min recovery period during which the ewe was allowed to breathe room air.
The time course was identical in all experimental protocols except that an adenosine receptor blockade was initiated by intravenous infusion of either 8-(p-sulphophenyl)-theophylline (8-SPT) or 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) beginning 30 min prior to the initiation of hypoxia and ending 30 min after completion of hypoxia. 8-SPT is a universal adenosine receptor antagonist that is relatively membrane impermeable (Evoniuk et al. 1987; Meno et al. 2001). It was diluted in saline to 10 mg ml−1 and pH was adjusted to 7.4 with sodium hydroxide. The solution was infused intravenously as a 15 mg bolus followed by an infusion of 1.8 mg min−1 for the remainder of the infusion period. This dose was selected as the greater of two administered in separate studies by Koos et al. (1995) and Giussani et al. (2001). both of which were reported to attenuate cardiovascular responses to hypoxia in the fetal sheep.
DPCPX is a selective adenosine A1 receptor antagonist that is membrane permeable (Marston et al. 1998). It was prepared in 0.1 m NaOH to a concentration of 2.5 mg ml−1 (final pH ≈10) and infused at a rate of 2.5 mg min−1 for the first 10 min of the infusion period followed by 0.75 mg min−1 for the remainder of the infusion period. This dose of DPCPX is equivalent to that given to fetal sheep by Koos & Maeda (2001) where it was found to antagonize the effects of the selective adenosine A1 receptor agonist N6-cyclopentyl-adenosine. Both 8-SPT and DPCPX were obtained from Sigma Chemical Co, St Louis, MO, USA.
The total volumes infused were 17.7 ml for the 8-SPT group, 34 ml for the DPCPX group, and 30 ml of saline for the comparable period in the control protocol.
Seven sheep were studied under the control protocol. Because we have found previously that the cortical blood flow responses to hypoxia are consistent when repeated at least 48 h apart (Lan et al. 2000), two of these sheep were subsequently (48–72 h later) studied under the 8-SPT protocol and four were studied under the DPCPX protocol. An additional five sheep were studied under the 8-SPT protocol only, and three more were studied under the DPCPX protocol only. The mean fetal gestational age at the time of experiment was 129 ± 1 days (0.89 gestation).
Upon completion of the experiments, the ewe and fetus were killed using Euthasol (17 ml, Western Medical Supply, Arcadia, CA, USA). The location of the probes, thermocouple, and catheters was verified and fetal body weights were recorded.
Blood sampling
Blood samples (0.5 ml each) were collected into heparinized syringes from the arterial and sagittal sinus catheters during each experiment at 0, 15, 30, 40, 50, and 60 min during the baseline period, 5, 10, 20, and 30 min during the hypoxic period, and 10, 20, 30, 45, and 60 min during the recovery period. The samples were analysed immediately for measurement of blood gases (ABL3, Radiometer, Copenhagen, Denmark), haemoglobin content and oxyhaemoglobin saturation (OSM2 Hemoximeter, Radiometer), and glucose and lactate concentrations (YSI 2700 analyser, Yellow Springs Instruments, Dayton, OH, USA). A total of 30 blood samples (30 ml) were collected making the total amount of blood drawn during each experiment amount to ≈8 % of the total estimated fetal blood volume of ≈400 ml.
Electronic data acquisition and handling
Cortical blood flow was measured by use of a laser Doppler flowmeter (Oxford Optronics, Oxford, UK). The perfusion signal provided an index of blood perfusion in a 1–2 mm3 hemisphere of tissue around the tip of the laser Doppler probe. Cortical blood flow, mean arterial blood pressure (BP), cortical tissue PO2 and arterial and cortical temperatures were recorded continuously. Analogue outputs were digitized (sampling rate 100 Hz) and stored using an analogue-to-digital converter (Powerlab 16/SP, ADInstruments, Colorado Springs, CO, USA) and data acquisition software (Chart v.4, ADInstruments). Heart rate was calculated from the pressure wave of the arterial pressure measurements. Cortical vascular resistance was calculated as (mean brachial arterial blood pressure - mean sagittal sinus pressure)/(cortical blood flow). Due to loss of sagittal sinus catheter patency, arterial blood pressure was assumed to be equivalent to cerebral perfusion pressure in three of the sheep studied under each of the three protocols.
Because laser Doppler flowmetry provides a relative, not absolute, measure of flow, flow signals from the baseline period of each experiment were averaged and considered as 100 %, with subsequent measurements expressed as a percentage of the baseline values.
Cortical heat production and oxygen consumption were calculated using the Fick principle. Heat production was calculated by multiplying the temperature difference between the artery and the brain by cortical blood flow. For a detailed description of the measurement of fetal brain heat production and metabolism refer to Hunter et al. (2003b). Cortical oxygen consumption was calculated by multiplying the arterial-venous difference in oxygen content by cortical blood flow. Both parameters were normalized to a percentage of the baseline period.
Statistical analyses
Data are presented as means ±s.e.m. For statistical analysis of continuously measured parameters, mean values for the final 10 min of each study period were used. The significance of changes over time was evaluated using a one-way ANOVA with repeated measures and Dunnett's post hoc test when a significant difference was detected by ANOVA. The significance of differences between the control and 8-SPT or between the control and DPCPX protocols was evaluated using a two-way ANOVA with a Bonferroni post hoc test. The statistical software Graphpad Prism 3.0 (Graphpad Software Inc., San Diego, CA, USA) was used for all statistical analyses.
RESULTS
Fetal haemodynamics, blood gases, pH, oxyhaemoglobin saturation, glucose, and lactate values
Fetal blood pressure and heart rate values for the final 10 min of each study period are shown in Table 1. Arterial blood pressure increased significantly in response to hypoxia in all experiments, and then returned to baseline values during recovery. In addition, DPCPX infusion resulted in a significant increase in blood pressure. Heart rate decreased significantly during hypoxia in the control and 8-SPT experiments, but was not significantly altered during the DPCPX experiments. As demonstrated by the mean values for arterial PO2, oxyhaemoglobin saturation, and oxygen content (Table 2), each of the three study groups was subjected to a comparable hypoxic insult. This was characterized by a decrease in arterial PO2 from ≈22 to 12 mmHg, PCO2 from 50–45 mmHg, mild reductions in pH, and subsequent recovery of all variables over the next hour.
Table 1.
Data for fetal arterial blood pressure and heart rate during the last 10 min of each of the five protocol periods
| Baseline | Pre-hypoxia infusion | Hypoxia | Recovery with infusion | Post-infusion recovery | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | ||
| Blood pressure (mmHg) | Control | 44 | 1 | 44 | 2 | 53 | 4* | 48 | 3 | 47 | 3 |
| 8-SPT | 45 | 2 | 46 | 2 | 54 | 3* | 49 | 2 | 48 | 2 | |
| DPCPX | 45 | 2 | 51 | 3† | 56 | 2† | 55 | 3 | 52 | 4 | |
| Heart rate (bpm) | Control | 167 | 5 | 166 | 5 | 151 | 8* | 176 | 7 | 185 | 4 |
| 8-SPT | 170 | 7 | 158 | 8 | 142 | 14* | 171 | 8 | 180 | 7 | |
| DPCPX | 168 | 5 | 185 | 14 | 172 | 12 | 189 | 11 | 178 | 8 | |
Significant difference from baseline value (P < 0.05);
significant difference from baseline value (P < 0.01).
Table 2.
Data for arterial blood and plasma parameters during each of the five protocol periods
| Pre-hypoxia | Recovery | Post-infusion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | infusion | Hypoxia | with infusion | recovery | |||||||
| Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | ||
| PO2 (mmHg) | Control | 22.4 | 1.1 | 22.3 | 1.2 | 12.1 | 0.4† | 22.0 | 1.2 | 20.4 | 0.9 |
| 8-SPT | 21.4 | 1.0 | 22.1 | 1.4 | 11.0 | 0.6† | 22.6 | 0.9 | 22.8 | 1.4 | |
| DPCPX | 21.7 | 1.0 | 18.3 | 1.9 | 10.9 | 1.0† | 19.6 | 2.6 | 23.1 | 1.4 | |
| Hb (mg dl−1) | Control | 8.9 | 0.5 | 9.1 | 0.5 | 9.3 | 0.5 | 8.9 | 0.5 | 8.8 | 0.5 |
| 8-SPT | 8.9 | 0.2 | 8.5 | 0.3 | 9.2 | 0.2 | 8.7 | 0.2 | 8.5 | 0.3 | |
| DPCPX | 8.8 | 0.3 | 9.1 | 0.3 | 9.7 | 0.3 | 9.6 | 0.4 | 8.7 | 0.4 | |
| O2 content (mm) | Control | 3.6 | 0.3 | 3.4 | 0.2 | 1.7 | 0.1* | 3.3 | 0.2 | 3.3 | 0.2 |
| 8-SPT | 3.5 | 0.2 | 3.3 | 0.3 | 1.4 | 0.1† | 3.2 | 0.3 | 3.3 | 0.3 | |
| DPCPX | 3.5 | 0.2 | 3.3 | 0.3 | 2.0 | 0.2† | 3.4 | 0.4 | 3.5 | 0.3 | |
| PCO2 (mmHg) | Control | 50.1 | 1.8 | 50.1 | 2.2 | 44.0 | 1.9* | 48.0 | 2.6 | 48.0 | 2.6 |
| 8-SPT | 48.6 | 1.4 | 49.2 | 2.0 | 44.3 | 2.0* | 48.9 | 1.5 | 42.0 | 1.5 | |
| DPCPX | 47.3 | 1.2 | 46.7 | 2.1 | 46.6 | 3.2 | 47.4 | 2.0 | 54.0 | 2.0 | |
| pH | Control | 7.36 | 0.01 | 7.34 | 0.02 | 7.34 | 0.03 | 7.28 | 0.03* | 7.31 | 0.03 |
| 8-SPT | 7.36 | 0.01 | 7.35 | 0.01 | 7.37 | 0.02 | 7.26 | 0.02† | 7.31 | 0.02* | |
| DPCPX | 7.37 | 0.01 | 7.36 | 0.02 | 7.32 | 0.03* | 7.26 | 0.02† | 7.26 | 0.03† | |
| Glucose (mm) | Control | 1.04 | 0.14 | 1.13 | 0.16 | 1.19 | 0.11 | 1.34 | 0.12† | 1.26 | 0.13† |
| 8-SPT | 0.97 | 0.22 | 1.02 | 0.25 | 1.20 | 0.20 | 1.52 | 0.21† | 1.38 | 0.25† | |
| DPCPX | 0.98 | 0.07 | 1.17 | 0.09 | 1.51 | 0.08† | 1.63 | 0.11† | 1.53 | 0.16† | |
| Lactate (mm) | Control | 1.39 | 0.25 | 1.79 | 0.45 | 3.74 | 0.93 | 5.40 | 1.13† | 4.26 | 1.20* |
| 8-SPT | 1.53 | 0.22 | 1.56 | 0.22 | 3.54 | 0.53 | 6.11 | 1.06† | 4.52 | 0.97* | |
| DPCPX | 1.06 | 0.05 | 2.03 | 0.32 | 4.74 | 0.62 | 7.10 | 0.73† | 6.61 | 0.64† | |
Significant difference from baseline value (P < 0.05);
significant difference from baseline value (P < 0.01).
Cortical blood flow and cortical vascular resistance
Cortical blood flow responses are shown in Fig. 1 for all three protocols. In the control experiments hypoxia resulted in a significant increase in cortical blood flow, with measurements rising steadily during the first 10 min of hypoxia to reach a mean value of 135 ± 7 % of baseline during the last 10 min of the hypoxic insult. Cortical blood flow then returned to baseline during the recovery period, and remained unchanged during the remainder of the experiment.
Figure 1. Time course of changes in cortical blood flow during hypoxia.
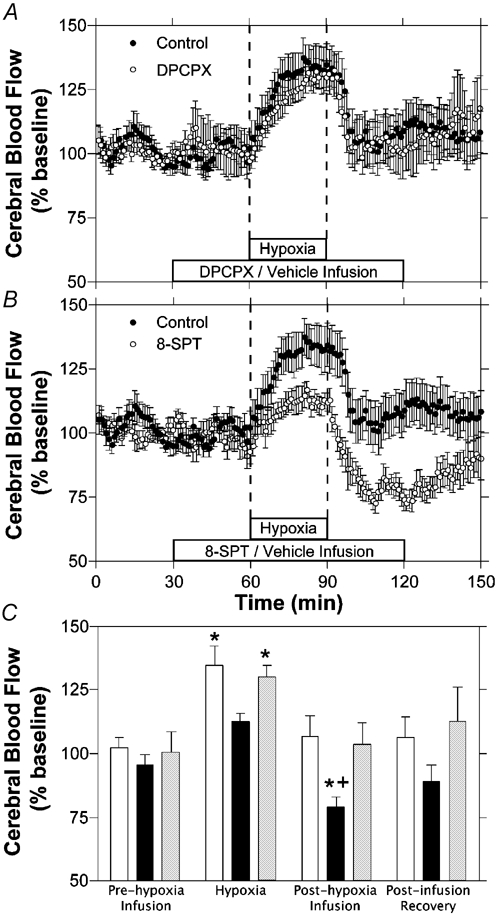
Results are shown for hypoxia with intravenous infusion of vehicle (A and B, n = 7), DPCPX (A, n = 7) or 8-SPT (B, n = 7). C, comparison of cortical blood flow changes from baseline measurements for control (open bars), 8-SPT (black bars) and DPCPX (grey bars) experiments. Bars represent mean values for each of the three experimental groups during the last 10 min of each of the four experimental periods labelled. Error bars indicate s.e.m.* Significant change from baseline period. + Significantly different from control experiments at that time period.
In the DPCPX experiments, there was no significant change in cortical blood flow during the first 30 min of DPCPX infusion. During hypoxia, cortical blood flow rose significantly to a mean of 130 ± 5 % of baseline levels, a response similar to that observed in control experiments. In the recovery period, cortical blood flow returned to baseline levels within 10 min and remained at baseline levels for the remainder of the DPCPX infusion and recovery period. DPCPX infusion had no significant effect on cortical blood flow compared to control experiments.
In 8-SPT experiments, baseline cortical blood flow was not changed during the drug infusion administered before hypoxia. During hypoxia, cortical blood flow rose to 113 ± 3 % of baseline the last 10 min of hypoxia, a value that was not significantly different from baseline and measurably less than in control fetuses. Following hypoxia, cortical blood flow decreased significantly to 79 ± 4 % of baseline levels in the last 10 min of the 8-SPT infusion and rose gradually towards baseline levels during the final 30 min of the experiment. Cortical blood flow was significantly reduced during the final 10 min of 8-SPT infusion compared to that of controls from the same time period.
Cerebrovascular resistance to flow did not change significantly relative to baseline values during hypoxia in the control experiments, as shown in Fig. 2. During DPCPX experiments, mean resistance tended to increase upon initiation of DPCPX infusion and to fall during hypoxia, although neither change was determined to be significantly different from baseline. Following hypoxia, resistance rose significantly to a mean of 130 ± 14 % during the final 10 min of DPCPX infusion and then returned to baseline values during the final 30 min of the experiment. During 8-SPT experiments, resistance remained unchanged during the hypoxic period, and then rose significantly to a mean of 145 ± 13 % of baseline during the final 10 min of 8-SPT infusion and remained elevated at 132 ± 12 % during the final 10 min of the experiment. Compared to control experiments, 8-SPT infusion resulted in significantly increased resistance during the post-hypoxic period of the study, while DPCPX infusion resulted in significantly increased cerebrovascular resistance only during the initial 30 min of the infusion period.
Figure 2. Time course of changes in cortical vascular resistance during hypoxia.
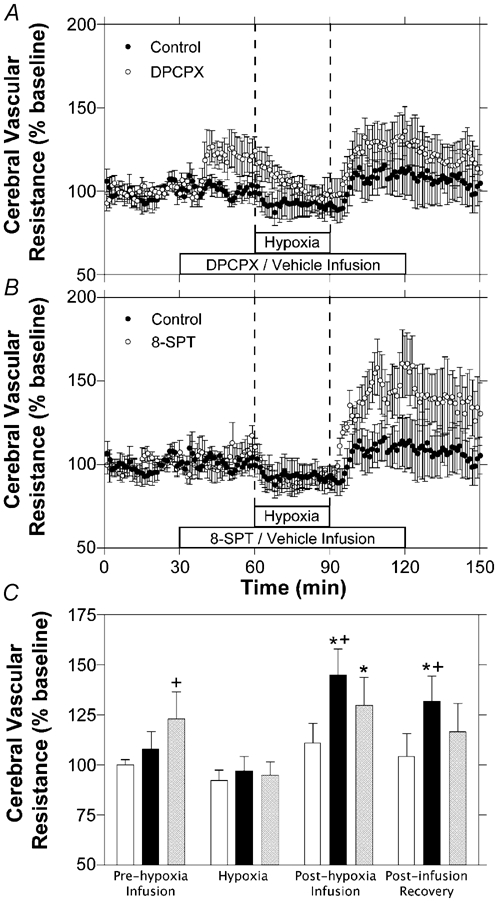
Results are shown for hypoxia with intravenous infusion of saline (A and B, n = 7), DPCPX (A, n = 6) or 8-SPT (B, n = 6). C, comparison of changes from baseline in cortical vascular resistance calculations for control (open bars), 8-SPT (black bars) and DPCPX (grey bars) experiments. Bars represent mean values for each of the three experimental groups during the last 10 min of each of the four experimental periods labelled. Error bars indicate s.e.m.* Significant change from baseline period. + significantly different from control experiments at that time period.
Cerebral cortical PO2
Cortical tissue PO2 (tPO2) is presented in Fig. 3 as a percentage of baseline. During the baseline period mean tPO2 for the control (n = 6), DPCPX (n = 6), and 8-SPT (n = 4) groups was 9 ± 1, 7 ± 2, and 6 ± 2 mmHg, respectively. During hypoxia, tPO2 fell rapidly to a nadir of 1–2 mmHg for each of the three study groups and there were no significant differences between responses of the 8-SPT, DPCPX, or control experiments. Finally, during the recovery period, tPO2 rapidly returned to baseline values in all three groups for the remainder of the experiment.
Figure 3. Time course of changes in cortical tissue PO2 during hypoxia.
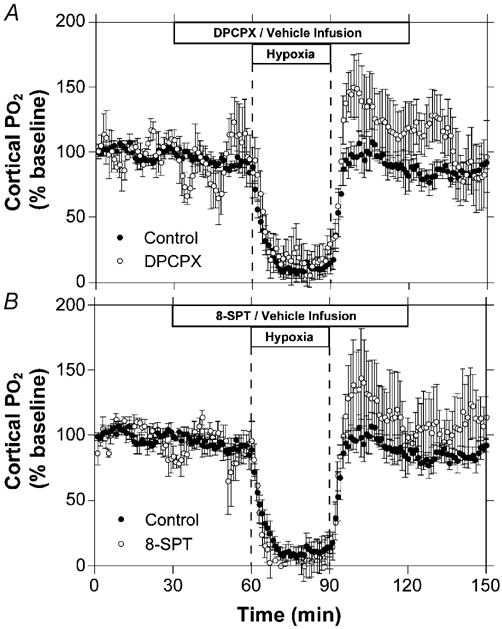
Results are shown for hypoxia with intravenous infusion of saline (A and B, n = 6), DPCPX (A, n = 5) or 8-SPT (B, n = 5). Error bars indicate s.e.m.
Cortical heat production and oxygen and glucose consumption
Cortical heat production data are presented in Fig. 4. Vehicle, DPCPX, and 8-SPT infusions had no effect on heat production during the initial 30 min of infusion. During control experiments, mean cortical heat production decreased significantly to 58 ±7 % of baseline values during the hypoxic insult, rapidly returned to baseline values following termination of hypoxia, and remained unchanged for the remainder of the experiment. During the DPCPX experiments, mean cortical heat production did not vary significantly from baseline values at any point throughout the experiments. During 8-SPT experiments, mean cortical heat production decreased significantly to 65 ± 10 % of baseline values during the hypoxic insult, rapidly returned to baseline values following hypoxia, and remained unchanged for the remainder of the experiment. 8-SPT infusion had no significant effect on cortical heat production compared to the control experiments. In contrast, DPCPX infusion prevented the fall in cortical heat production during the hypoxic insult compared to the control experiments.
Figure 4. Time course of changes in cortical heat production during hypoxia.

Results are shown for hypoxia with intravenous infusion of saline (A and B, n = 7), DPCPX (A, n = 7) or 8-SPT (B, n = 7). C, comparison of cortical heat production changes from baseline measurements for control (open bars), 8-SPT (black bars) and DPCPX (grey bars) experiments. Bars represent mean values for each of the three experimental groups during the last 10 min of each of the four experimental periods labelled. Error bars indicate s.e.m.* Significant change from baseline period. + Significantly different from control experiments at that time period.
Relative changes in cortical oxygen uptake and glucose consumption, calculated by the Fick principle, are shown in Table 3. Hypoxic insult resulted in a significant decrease in cortical oxygen consumption in the control and 8-SPT experiments. In contrast, there was no significant change in cortical oxygen consumption during hypoxia in the DPCPX experiments.
Table 3.
Data for cerebral oxygen and glucose uptake ( ) during each of the five protocol periods
) during each of the five protocol periods
| Baseline | Pre-hypoxia infusion | Hypoxia | Post-hypoxia infusion | Post-infusion recovery | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | Mean | s.e.m. | ||
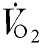 (% baseline) (% baseline) |
Control | 100 | – | 110 | 14 | 64 | 10† | 137 | 24 | 132 | 17 |
| 8-SPT | 100 | – | 125 | 18 | 69 | 7† | 131 | 27 | 9 | ||
| DPCPX | 100 | – | 106 | 10 | 87 | 19 | 137 | 37 | 107 | 30 | |
 glucose (% baseline)* glucose (% baseline)*
|
Control | 100 | – | 109 | 4 | 93 | 8 | 107 | 7 | 127 | 19 |
| 8-SPT | 100 | – | 120 | 5 | 108 | 13 | 115 | 6 | 172 | 10† | |
| DPCPX | 100 | – | 117 | 3 | 111 | 6 | 79 | 8‡ | 122 | 11 | |
Cortical uptake of glucose was calculated as the product of cortical flow and the arteriovenous difference.
Significant difference from baseline value (P < 0.05);
significant difference from control for the same time period (P < 0.05).
Cortical glucose consumption was unchanged by infusion of either 8-SPT or DPCPX during the baseline period, and did not change significantly during the hypoxic insult for any of the three experimental groups. Glucose consumption remained unchanged from baseline values throughout the recovery for the control and DPCPX groups, although consumption was significantly lower in the DPCPX group compared to controls during the recovery period while DPCPX infusion was continued. Glucose consumption was significantly increased during the recovery period in the 8-SPT group.
DISCUSSION
The principal findings of this study are that adenosine is an important mediator of decreased cerebral metabolic rate during moderate hypoxia and that this effect on metabolism is mediated by the adenosine A1 receptors located within the brain. The fact that hypoxic decreases in metabolism are abolished by infusion of DPCPX, a selective adenosine A1 antagonist, is consistent with the idea of adaptive hypometabolism of the fetal brain in response to hypoxic stress. The A1 receptors mediating this adaptive hypometabolism appear to be located inside the blood brain barrier, since intravenous infusion of 8-SPT, a non-selective adenosine receptor antagonist with poor blood brain barrier permeability, resulted in no significant change in the fall in fetal cortical oxygen consumption or heat production during hypoxia. In addition, adenosine appears to mediate increased cerebral blood flow by its actions on A2 but not A1 receptors during hypoxia because cerebral blood flow increases were markedly attenuated by infusion of 8-SPT but not DPCPX.
Adenosine is a purine nucleoside produced principally from dephosphorylation of AMP and to a lesser extent from hydrolysis of S-adenosyl-homocysteine. In the fetal sheep, both hypoxia and asphyxia result in increased plasma concentrations of adenosine (Koos & Doany, 1991; Kubonoya & Power, 1997). Microdialysis measurements also show increased adenosine concentrations in the interstitial fluid of the fetal sheep brain with hypoxia (Koos et al. 1997) and cord occlusion (Watson et al. 2002).
Adenosine mediates various physiological processes acting via at least four different adenosine receptor types named A1, A2A, A2B, and A3. The receptors are all coupled to G-proteins and have been found in a wide range of species and types of tissues (for review, see Fredholm et al. 2001)). A1 receptor expression is notably high in the brain, adrenal gland, atria, skeletal muscle, and liver. In the brain, the activation of the A1 receptor results in neuronal membrane stabilization and decreased neuronal firing (Croning et al. 1995; Fowler et al. 1999). The A2A and A2B receptors can be found in cerebral vascular smooth muscle where both mediate vasodilatation in response to hypoxia (Coney & Marshall, 1998) and neuronal stimulation (Meno et al. 2001).
The exact mechanisms by which adenosine concentrations increase during hypoxia are not fully understood. In the guinea-pig heart (Decking et al. 1997) as well as the rat brain (Lynch et al. 1998), it appears that hypoxia-stimulated increases in adenosine concentrations result from decreased phosphorylation of adenosine by adenosine kinase (Decking et al. 1997), the enzyme that facilitates the major pathway of adenosine metabolism in the rat brain (Lloyd & Fredholm, 1995). In contrast, the breakdown of ATP does not appear to be a major contributing factor to increases in adenosine concentrations during hypoxic stress (Doolette, 1997). In the brain of the fetal sheep, however, evidence exists that the primary hypoxic increases in extracellular adenosine concentration are a result of increased degradation of extracellular 5′-AMP (Koos et al. 1997).
Thus, there is evidence that adenosine concentrations increase in response to hypoxia and may be involved in regulating both increased cerebral blood flow and decreased neuronal activity to optimize both oxygen delivery and usage.
Effect of adenosine on cerebral metabolic rate during hypoxia
It has been nearly 30 years since adenosine was first reported to inhibit neuronal firing in isolated brain slices (Kuroda et al. 1976; Phillis & Edstrom, 1976). Since then, there have been numerous reports supporting a neuroinhibitory role for adenosine (for review see Dunwiddie & Masino, 2001). In the fetal sheep brain, hypoxic/anoxicstress results in a marked suppression of electrocortical activity (Bocking & Harding, 1986; Gunn et al. 1991), an effect that is attenuated in the presence of DPCPX (Hunter et al. 2003b). Likewise, adenosine has been implicated in the inhibition of fetal breathing movements during hypoxic stress by its action on receptors in the thalamus (Koos et al. 1994b; Chau & Koos, 1999). Significantly, recent work in the near-term fetal sheep has shown that adenosine A1 receptor activation during anoxia is a major mediator of the pronounced hypoxic tolerance in the fetal sheep. This work by Hunter et al. demonstrated that inhibition of the A1 receptor during a 10 min period of complete cord occlusion resulted in significantly increased neuronal cell death compared to controls, delayed the suppression of EEG activity and resulted in increased cerebral metabolism as evidenced by continued heat production (Hunter et al. 2003a).
In the present study, cortical oxygen consumption and heat production decreased as fetal arterial PO2 fell below 11–12 mmHg during both control and 8-SPT experiments. In contrast, cerebral metabolic rate was not reduced during hypoxia with DPCPX infusion, providing strong evidence that the adenosine A1 receptor mediates the reduction in cerebral metabolic rate in response to hypoxic stress. These data, taken together with previous work, support the presence of a mechanism of adaptive hypometabolism in the brain of the fetal sheep such that oxygen consumption is actively reduced during hypoxic stress, rather than being reduced by oxygen starvation.
Since 8-SPT is a relatively non-selective receptor antagonist that inhibits the same adenosine A1 receptors as DPCPX, it is of interest that cerebral metabolic rate was not altered by 8-SPT administration as with DPCPX. One explanation for this finding may be due to the relatively plasma membrane-impermeable properties of 8-SPT resulting in its inability to cross the blood brain barrier. 8-SPT has been shown to not cross the blood brain barrier of the adult mouse (Baumgold et al. 1992) and rat (Coney & Marshall, 1998; Meno et al. 2001), as evidenced by a lack of significant cerebrospinal fluid (CSF) concentrations with significant plasma concentrations following intraperitoneal injection (Baumgold et al. 1992; Meno et al. 2001). Although cerebrospinal concentrations of 8-SPT were not determined in the present study, the blood brain barrier of the fetal sheep is intact as early as 0.6 gestation (Stonestreet et al. 2000; Harris et al. 2001). Exclusion of 8-SPT by the blood brain barrier in the present experiments would indicate that the effects of DPCPX on metabolic rate are due to blockade of adenosine receptors located inside the blood brain barrier. Alternatively, it is possible that the hypometabolism observed during 8-SPT infusion is due to the lack of increased cerebral blood flow compared to controls, such that the observed decrease in metabolic rate may be due to oxygen starvation rather than hypometabolism.
The finding of hypoxic adaptive hypometabolism in the brain is not new, as it has also been reported as a protective mechanism in the brains of other animals. For example, exposure of the freshwater turtle to anoxia results in a five-fold decrease in cerebral energy turnover with no significant decrease in cerebral ATP stores (Hochachka & Lutz, 2001). Likewise, in the rat, hypoxia results in a strong inhibition of neuronal firing (Croning et al. 1995; Fowler et al. 1999). In both instances, the investigators provided strong evidence suggesting these responses are mediated by activation of the adenosine A1 receptor. In the adult mouse, knockout of the adenosine A1 receptor results in significantly decreased neuronal tolerance to hypoxia (Johansson et al. 2001).
Regulation of cerebral blood flow during hypoxia
Intravenous infusion of theophylline, a non-selective adenosine receptor antagonist, results in a lack of increased cortical blood flow during hypoxia in the fetal sheep (Blood et al. 2002). Theophylline crosses cell membranes freely, resulting in blockade of adenosine receptors on either side of the blood brain barrier (McPhee & Maxwell, 1987; Morii et al. 1987). However, as mentioned previously, 8-SPT is relatively impermeable to the plasma membrane and has been shown to not cross the blood brain barrier (Baumgold et al. 1992; Coney & Marshall, 1998; Meno et al. 2001). In the present study, 8-SPT infusion resulted in an inhibition of much of the characteristic increase of cortical blood flow provoked by hypoxia, thus supporting previous work indicating the importance of adenosine receptor activation in mediating increased cortical blood flow during hypoxia and suggesting that these receptors are located outside the blood brain barrier. Such findings are in agreement with studies in the adult rat, in which topical application of 8-SPT to the exposed cortex had no effect on hypoxic cerebral vasodilatation, also suggesting that the receptors mediating cerebral vasodilatation are located outside the blood brain barrier (Coney & Marshall, 1998).
Unlike 8-SPT, DPCPX infusion had no effect on the hypoxic increases in cortical blood flow, suggesting that adenosine A1 receptors are not involved in the cortical blood flow response. These findings are in agreement with work by others studying changes in arteriole diameter following application of selective adenosine receptor agonists to the exposed cortex of the preterm fetal sheep. These previous studies found that the adenosine A2 receptor plays a greater role in cerebral vasodilatation than does the A1 receptor (Kurth & Wagerle, 1992). Similar results have also been observed in the adult rat where adenosine A2 agonists resulted in greater cerebral vasodilatation than A1 agonists (Meno et al. 1993). Likewise, selective adenosine A2A antagonists have been shown to effectively block increases in cerebral arteriole diameter, while A1 antagonists had little or no effect (Coney & Marshall, 1998).
Although there is growing evidence that adenosine is a strong mediator of cerebral blood flow during hypoxia in the fetus, the possible mechanisms by which it may act are many and complex. Adenosine may lower cerebrovascular resistance during hypoxia by direct activation of adenosine receptors on the cerebrovascular smooth muscle, resulting in cerebrovascular vasodilatation. It is also possible that adenosine acts by the regulation of other mediators of cerebrovascular tone. For example, vasopressin decreases cerebral vascular resistance and infusion of vasopressin receptor antagonists to the fetus during hypoxia attenuates increases in cerebral blood flow (Eisenach et al. 1992). During hypoxia, fetal plasma vasopressin concentrations increase, a response that is blocked by infusion of 8-SPT (Koos et al. 1994b). Adenosine's vasodilatory actions may also be exerted by stimulation of prostaglandin and nitric oxide production in the cerebral vasculature, pathways that have been observed in skeletal muscle of the adult rat (Marshall, 2002). However, in contrast to the findings of the present study, hypoxic vasodilatation in skeletal muscle appears to be mediated primarily by the A1 receptor, indicating that considerable tissue-specific differences may exist (Marshall, 2002). We have recently observed a partial attenuation of hypoxic increases in cortical blood flow during nitric oxide synthase blockade with l-NAME, indicating that nitric oxide may indeed play a role in addition to or as a result of adenosine receptor activation (Hunter et al. 2003c). Clearly, the cascade of events leading to vasodilatation is multifactorial and further work is necessary to determine which of several possible pathways are involved in the adenosine-mediated hypoxic increases in fetal cerebral blood flow.
The control of adenosine on cerebral blood flow during hypoxia may also include systemic regulation of arterial blood pressure. Autoregulation of cerebral blood flow is intact in the fetal sheep by 0.6 gestation, such that cerebral perfusion remains relatively constant over a range of perfusion pressures from 45 to 85 mmHg (Papile et al. 1985). However, during hypoxic stress, fetal autoregulation is lost and cerebral blood flow becomes pressure passive (Tweed et al. 1983). The findings of the present study support this previous work, since fetal arterial blood pressure increased gradually over the first 20 min to a plateau of 10–15 mmHg above baseline, while cerebral vascular resistance remained unchanged. Therefore, at least part of the hypoxic increase in cerebral blood flow is due to the combined increase in driving pressure and decreased autoregulatory response. It is possible that the decrease in arterial PCO2 noted for the control and 8-SPT groups may have played a role in preventing the fall in cerebral vascular resistance during hypoxia. However, this possibility seems unlikely since PCO2 is a relatively poor regulator of fetal cerebral blood flow compared to the adult (Ashwal et al. 1984; Yamashita et al. 1991). The fact that there was no significant attenuation of the hypoxic hypertension during 8-SPT or DPCPX infusion, and no clear difference between the hypoxic cerebral vascular resistance of control and 8-SPT experiments in the present study makes it impossible to discern the relative contribution of systemic and local effects of adenosine. It seems likely that the hypoxic increase in cerebral blood flow is due to a combination of both increased perfusion pressure and local vasodilatation, both of which have been shown to be mediated, at least in part, by adenosine (Winn et al. 1981a; Koos et al. 1995; Giussani et al. 2001)
Following restoration of normoxia, cerebral blood flow decreased to 75–80 % of baseline values in those fetuses receiving 8-SPT, a response that was associated with pronounced increases of cerebral vascular resistance persisting for an hour or more. In contrast, blood flow did not decline below initial baseline values in control experiments. These results are consistent with previous studies in the fetal sheep using theophylline (Blood et al. 2002) and may indicate a role for adenosine in inhibiting the effect of vasoconstricting factors. As these post-hypoxic increases of resistance were not observed in fetuses receiving DPCPX, they are probably attributable to the action of A2 receptors.
Summary
The present findings add to a growing body of evidence that adenosine is an important modulator of adaptive responses to hypoxic stress in the fetal brain. Cortical blood flow is modulated during hypoxia by activation of adenosine receptors that are probably located outside the blood brain barrier. Adenosine also maintains cortical blood flow in the post-hypoxia period. Finally, the data suggest an active suppression of cortical metabolic rate, mediated by adenosine A1 receptors located on the inside of the blood brain barrier. Together these mechanisms are likely to be an important part of the remarkable tolerance of the fetal brain to hypoxic stress and provide strong evidence for adaptive hypometabolism mediated by adenosine.
Acknowledgments
The authors gratefully acknowledge the skillful technical assistance of Shannon Bragg in surgically instrumenting the animals and conducting the experiments. This study was supported in part by USPHS award HL R01-812-654-941.
REFERENCES
- Asano H, Homan J, Carmichael L, Korkola S, Richardson B. Cerebral metabolism during sustained hypoxemia in preterm fetal sheep. Am J Obstet Gynecol. 1994;170:939–944. doi: 10.1016/s0002-9378(94)70310-8. [DOI] [PubMed] [Google Scholar]
- Ashwal S, Dale PS, Longo LD. Regional cerebral blood flow: studies in the fetal lamb during hypoxia, hypercapnia, acidosis, and hypotension. Pediatr Res. 1984;18:1309–1316. doi: 10.1203/00006450-198412000-00018. [DOI] [PubMed] [Google Scholar]
- Baumgold J, Nikodijevic O, Jacobson KA. Penetration of adenosine antagonists into mouse brain as determined by ex vivo binding. Biochem Pharmacol. 1992;43:889–894. doi: 10.1016/0006-2952(92)90257-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishai JM, Blood AB, Hunter CJ, Longo LD, Power GG. Fetal lamb cerebral blood flow (CBF). and oxygen tensions during hypoxia: a comparison of laser Doppler and microsphere measurements of CBF. J Physiol. 2003;546:869–878. doi: 10.1113/jphysiol.2002.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood AB, Hunter CJ, Power GG. The role of adenosine in regulation of cerebral blood flow during hypoxia in the near-term fetal sheep. J Physiol. 2002;543:1015–1023. doi: 10.1113/jphysiol.2002.023077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocking AD, Harding R. Effects of reduced uterine blood flow on electrocortical activity, breathing, and skeletal muscle activity in fetal sheep. Am J Obstet Gynecol. 1986;154:655–662. doi: 10.1016/0002-9378(86)90625-3. [DOI] [PubMed] [Google Scholar]
- Chau A, Koos BJ. Metabolic and cardiorespiratory responses to hypoxia in fetal sheep: adenosine receptor blockade. Am J Physiol. 1999;276:R1805–1811. doi: 10.1152/ajpregu.1999.276.6.R1805. [DOI] [PubMed] [Google Scholar]
- Cohen WR, Piasecki GJ, Jackson BT. Plasma catecholamines during hypoxemia in fetal lamb. Am J Physiol. 1982;243:R520–525. doi: 10.1152/ajpregu.1982.243.5.R520. [DOI] [PubMed] [Google Scholar]
- Coney AM, Marshall JM. Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. J Physiol. 1998;509:507–518. doi: 10.1111/j.1469-7793.1998.507bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croning MD, Zetterstrom TS, Grahame-Smith DG, Newberry NR. Action of adenosine receptor antagonists on hypoxia-induced effects in the rat hippocampus in vitro. Br J Pharmacol. 1995;116:2113–2119. doi: 10.1111/j.1476-5381.1995.tb16419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- Doolette DJ. Mechanism of adenosine accumulation in the hippocampal slice during energy deprivation. Neurochem Int. 1997;30:211–223. doi: 10.1016/s0197-0186(96)00055-1. [DOI] [PubMed] [Google Scholar]
- Duffy TE, Kohle SJ, Vannucci RC. Carbohydrate and energy metabolism in perinatal rat brain: relation to survival in anoxia. J Neurochem. 1975;24:271–276. doi: 10.1111/j.1471-4159.1975.tb11875.x. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Eisenach JC, Tong C, Stump DA, Block SM. Vasopressin and fetal cerebrovascular regulation. Am J Physiol. 1992;263:R376–381. doi: 10.1152/ajpregu.1992.263.2.R376. [DOI] [PubMed] [Google Scholar]
- Evoniuk G, Von Borstel RW, Wurtman RJ. Antagonism of the cardiovascular effects of adenosine by caffeine or 8-(p-sulfophenyl)theophylline. J Pharmacol Exp Ther. 1987;240:428–432. [PubMed] [Google Scholar]
- Fowler JC, Partridge LD, Gervitz L. Hydroxylamine blocks adenosine A1 receptor-mediated inhibition of synaptic transmission in rat hippocampus. Brain Res. 1999;815:414–418. doi: 10.1016/s0006-8993(98)01114-7. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Ap IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Gardner DS, Cox DT, Fletcher AJ. Purinergic contribution to circulatory, metabolic, and adrenergic responses to acute hypoxemia in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;280:R678–685. doi: 10.1152/ajpregu.2001.280.3.R678. [DOI] [PubMed] [Google Scholar]
- Gunn AJ, Cook CJ, Williams C E, Johnston BM, Gluckman PD. Electrophysiological responses of the fetus to hypoxia and asphyxia. J Dev Physiol. 1991;16:147–153. [PubMed] [Google Scholar]
- Harris AP, Robinson R, Koehler RC, Traystman RJ, Gleason CA. Blood-brain barrier permeability during dopamine-induced hypertension in fetal sheep. J Appl Physiol. 2001;91:123–129. doi: 10.1152/jappl.2001.91.1.123. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Lutz PL. Mechanism, origin, and evolution of anoxia tolerance in animals. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:435–459. doi: 10.1016/s1096-4959(01)00408-0. [DOI] [PubMed] [Google Scholar]
- Hunter CJ, Bennet L, Power GG, Roelfsema V, Blood AB, Quaedackers JS, George S, Guan J, Gunn AJ. Key neuroprotective role for endogenous adenosine A1 receptor activation during asphyxia in the fetal sheep. Stroke. 2003b;34:2240–2245. doi: 10.1161/01.STR.0000083623.77327.CE. [DOI] [PubMed] [Google Scholar]
- Hunter CJ, Blood AB, Power GG. Cerebral metabolism during cord occlusion and hypoxia in the fetal sheep: a novel method of continuous measurement based on heat production. J Physiol. 2003c;552:241–251. doi: 10.1113/jphysiol.2003.048082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CJ, Blood AB, White CR, Pearce WJ, Power GG. Role of nitric oxide in hypoxic cerebral vasodilatation in the ovine fetus. J Physiol. 2003a;549:625–633. doi: 10.1113/jphysiol.2002.038034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto HS, Rudolph AM, Mirkin BL, Keil LC. Circulatory and humoral responses of sympathectomized fetal sheep to hypoxemia. Am J Physiol. 1983;245:H767–772. doi: 10.1152/ajpheart.1983.245.5.H767. [DOI] [PubMed] [Google Scholar]
- Jensen A, Garnier Y, Berger R. Dynamics of fetal circulatory responses to hypoxia and asphyxia. Eur J Obstet Gynecol Reprod Biol. 1999;84:155–172. doi: 10.1016/s0301-2115(98)00325-x. [DOI] [PubMed] [Google Scholar]
- Jin S, Fredholm BB. Adenosine A1 receptors mediate hypoxia-induced inhibition of electrically evoked transmitter release from rat striatal slices. Eur J Pharmacol. 1997;329:107–113. [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie T V, Masino S A, Poelchen W, Gimenez-Llort L, Escorihuela R M, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu X J, Hardemark A, Betsholtz C, Herlenius E, Fredholm B B. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellmer I, Andine P, Hagberg H, Thiringer K. Extracellular increase of hypoxanthine and xanthine in the cortex and basal ganglia of fetal lambs during hypoxia-ischemia. Brain Res. 1989;478:241–247. doi: 10.1016/0006-8993(89)91504-7. [DOI] [PubMed] [Google Scholar]
- Koos BJ, Chau A, Ogunyemi D. Adenosine mediates metabolic and cardiovascular responses to hypoxia in fetal sheep. J Physiol. 1995;488:761–766. doi: 10.1113/jphysiol.1995.sp021007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos BJ, Doany W. Role of plasma adenosine in breathing responses to hypoxia in fetal sheep. J Dev Physiol. 1991;16:81–85. [PubMed] [Google Scholar]
- Koos BJ, Kruger L, Murray TF. Source of extracellular brain adenosine during hypoxia in fetal sheep. Brain Res. 1997;778:439–442. doi: 10.1016/s0006-8993(97)01207-9. [DOI] [PubMed] [Google Scholar]
- Koos BJ, Maeda T. Adenosine A(2A) receptors mediate cardiovascular responses to hypoxia in fetal sheep. Am J Physiol Heart Circ Physiol. 2001;280:H83–89. doi: 10.1152/ajpheart.2001.280.1.H83. [DOI] [PubMed] [Google Scholar]
- Koos BJ, Mason BA, Ervin MG. Adenosine mediates hypoxic release of arginine vasopressin in fetal sheep. Am J Physiol. 1994a;266:R215–220. doi: 10.1152/ajpregu.1994.266.1.R215. [DOI] [PubMed] [Google Scholar]
- Koos BJ, Mason BA, Punla O, Adinolfi AM. Hypoxic inhibition of breathing in fetal sheep: relationship to brain adenosine concentrations. J Appl Physiol. 1994b;77:2734–2739. doi: 10.1152/jappl.1994.77.6.2734. [DOI] [PubMed] [Google Scholar]
- Kubonoya K, Power GG. Plasma adenosine responses during repeated episodes of umbilical cord occlusion. Am J Obstet Gynecol. 1997;177:395–401. doi: 10.1016/s0002-9378(97)70204-7. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Saito M, Kobayashi K. Concomitant changes in cyclic AMP level and postsynaptic potentials of olfactory cortex slices induced by adenosine derivatives. Brain Res. 1976;109:196–201. doi: 10.1016/0006-8993(76)90393-0. [DOI] [PubMed] [Google Scholar]
- Kurth CD, Wagerle LC. Cerebrovascular reactivity to adenosine analogues in 0. 6–0.7 gestation and near-term fetal sheep. Am J Physiol. 1992;262:H1338–1342. doi: 10.1152/ajpheart.1992.262.5.H1338. [DOI] [PubMed] [Google Scholar]
- Lan J, Hunter CJ, Murata T, Power GG. Adaptation of laser-Doppler flowmetry to measure cerebral blood flow in the fetal sheep. J Appl Physiol. 2000;89:1065–1071. doi: 10.1152/jappl.2000.89.3.1065. [DOI] [PubMed] [Google Scholar]
- Laudignon N, Farri E, Beharry K, Aranda JV. Rapid effects of hypoxia on the cerebrospinal fluid levels of adenosine and related metabolites in newborn and one-month-old piglets. Biol Neonate. 1991;59:54–59. doi: 10.1159/000243322. [DOI] [PubMed] [Google Scholar]
- Lloyd HG, Fredholm BB. Involvement of adenosine deaminase and adenosine kinase in regulating extracellular adenosine concentration in rat hippocampal slices. Neurochem Int. 1995;26:387–395. doi: 10.1016/0197-0186(94)00144-j. [DOI] [PubMed] [Google Scholar]
- Lynch JJ, 3rd, Alexander KM, Jarvis MF, Kowaluk EA. Inhibition of adenosine kinase during oxygen-glucose deprivation in rat cortical neuronal cultures. Neurosci Lett. 1998;252:207–210. doi: 10.1016/s0304-3940(98)00376-0. [DOI] [PubMed] [Google Scholar]
- McPhee AJ, Maxwell GM. The effect of theophylline on regional cerebral blood flow responses to hypoxia in newborn piglets. Pediatr Res. 1987;21:573–578. doi: 10.1203/00006450-198706000-00014. [DOI] [PubMed] [Google Scholar]
- Mallard EC, Williams CE, Johnston BM, Gluckman PD. Increased vulnerability to neuronal damage after umbilical cord occlusion in fetal sheep with advancing gestation. Am J Obstet Gynecol. 1994;170:206–214. doi: 10.1016/s0002-9378(94)70409-0. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Roles of adenosine in skeletal muscle during systemic hypoxia. Clin Exp Pharmacol Physiol. 2002;29:843–849. doi: 10.1046/j.1440-1681.2002.03734.x. [DOI] [PubMed] [Google Scholar]
- Marston HM, Finlayson K, Maemoto T, Olverman HJ, Akahane A, Sharkey J, Butcher SP. Pharmacological characterization of a simple behavioral response mediated selectively by central adenosine A1 receptors, using in vivo and in vitro techniques. J Pharmacol Exp Ther. 1998;285:1023–1030. [PubMed] [Google Scholar]
- Meno JR, Crum AV, Winn HR. Effect of adenosine receptor blockade on pial arteriolar dilation during sciatic nerve stimulation. Am J Physiol Heart Circ Physiol. 2001;281:H2018–2027. doi: 10.1152/ajpheart.2001.281.5.H2018. [DOI] [PubMed] [Google Scholar]
- Meno JR, Ngai AC, Winn HR. Changes in pial arteriolar diameter and CSF adenosine concentrations during hypoxia. J Cereb Blood Flow Metab. 1993;13:214–220. doi: 10.1038/jcbfm.1993.26. [DOI] [PubMed] [Google Scholar]
- Morii S, Ngai AC, Ko KR, Winn HR. Role of adenosine in regulation of cerebral blood flow: effects of theophylline during normoxia and hypoxia. Am J Physiol. 1987;253:H165–175. doi: 10.1152/ajpheart.1987.253.1.H165. [DOI] [PubMed] [Google Scholar]
- Mortola JP. How newborn mammals cope with hypoxia. Respir Physiol. 1999;116:95–103. doi: 10.1016/s0034-5687(99)00038-9. [DOI] [PubMed] [Google Scholar]
- Papile LA, Rudolph AM, Heymann MA. Autoregulation of cerebral blood flow in the preterm fetal lamb. Pediatr Res. 1985;19:159–161. doi: 10.1203/00006450-198502000-00001. [DOI] [PubMed] [Google Scholar]
- Parer JT. Fetal cerebral metabolism: the influence of asphyxia and other factors. J Perinatol. 1994;14:376–385. [PubMed] [Google Scholar]
- Phillis JW, Edstrom JP. Effects of adenosine analogs on rat cerebral cortical neurons. Life Sci. 1976;19:1041–1053. doi: 10.1016/0024-3205(76)90296-4. [DOI] [PubMed] [Google Scholar]
- Reuss ML, Parer JT, Harris JL, Krueger TR. Hemodynamic effects of alpha-adrenergic blockade during hypoxia in fetal sheep. Am J Obstet Gynecol. 1982;142:410–415. doi: 10.1016/s0002-9378(16)32381-x. [DOI] [PubMed] [Google Scholar]
- Stonestreet BS, Sadowska GB, McKnight AJ, Patlak C, Petersson KH. Exogenous and endogenous corticosteroids modulate blood-brain barrier development in the ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2000;279:R468–477. doi: 10.1152/ajpregu.2000.279.2.R468. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Power GG. Role of adenosine in regulation of brain temperature in fetal sheep. Am J Obstet Gynecol. 1999;181:681–687. doi: 10.1016/s0002-9378(99)70513-2. [DOI] [PubMed] [Google Scholar]
- Tweed WA, Cote J, Pash M, Lou H. Arterial oxygenation determines autoregulation of cerebral blood flow in the fetal lamb. Pediatr Res. 1983;17:246–249. doi: 10.1203/00006450-198304000-00002. [DOI] [PubMed] [Google Scholar]
- Van Wylen DG, Park TS, Rubio R, Berne RM. Increases in cerebral interstitial fluid adenosine concentration during hypoxia, local potassium infusion, and ischemia. J Cereb Blood Flow Metab. 1986;6:522–528. doi: 10.1038/jcbfm.1986.97. [DOI] [PubMed] [Google Scholar]
- Watson CS, Schaefer R, White SE, Homan JH, Fraher L, Harding R, Bocking AD. Effect of intermittent umbilical cord occlusion on fetal respiratory activity and brain adenosine in late-gestation sheep. Reprod Fertil Dev. 2002;14:35–42. doi: 10.1071/rd01013. [DOI] [PubMed] [Google Scholar]
- Winn HR, Rubio GR, Berne RM. The role of adenosine in the regulation of cerebral blood flow. J Cereb Blood Flow Metab. 1981a;1:239–244. doi: 10.1038/jcbfm.1981.29. [DOI] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. Am J Physiol. 1981b;241:H235–242. doi: 10.1152/ajpheart.1981.241.2.H235. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Kamiya K, Nagai H. CO2 reactivity and autoregulation in fetal brain. Childs Nerv Syst. 1991;7:327–331. doi: 10.1007/BF00304831. [DOI] [PubMed] [Google Scholar]