

Abstract
Muscle inflammation is a common feature in muscle injury and disease. Recently, investigators have speculated that inflammatory cells may increase or decrease muscle damage following modified muscle use, although there are few experimental observations to confirm either possibility. In the present study, a null mutation of gp91phox in neutrophils prevented superoxide production in cytotoxicity assays in which muscle cells were targets, and prevented most neutrophil-mediated cytolysis of muscle cells in comparison to wild-type neutrophils in vitro. We further tested whether deficiency in superoxide production caused a decrease in muscle membrane damage in vivo during modified muscle use. Gp91phox null mutant mice and wild-type mice were subjected to 10 days of muscle hindlimb unloading followed by reloading through return to normal locomotion, which induced muscle membrane lesions and muscle inflammation. Membrane lesions were quantified by measuring the presence of extracellular marker dye in reloaded soleus muscle fibres. There was a 90 % reduction in the number of fibres showing extensive membrane injury in gp91phox null mice compared to controls. Mutation of gp91phox did not change the concentration of neutrophils or macrophages in the reloaded muscle. Furthermore, muscle fibre growth during the reloading period was unaffected by the reduction in membrane injury. Together, these findings show that neutrophils can induce muscle membrane lysis through superoxide-mediated events, and indicate that superoxide-mediated membrane damage in vivo is not required for myeloid cell chemotaxis or muscle growth during muscle reloading.
Muscle membrane lesions that occur in muscle injury or disease are associated with severe disruptions of normal muscle homeostasis. Membrane lesions permit the unregulated transit of solutes in and out of muscle cells, and can lead to loss of ATP and other critical molecules from the muscle cytoplasm. The loss of cytosolic muscle proteins into the extracellular space can be sufficient to raise the content of muscle proteins in the serum to high levels. For example, the serum concentration of muscle creatine kinase can reach 25 000 U l−1 in patients with Duchenne muscular dystrophy (Florence et al. 1985), or 34 000 U l−1 in subjects following muscle exercise (Newham et al. 1983). In addition, the unregulated influx of calcium can activate calcium-dependent proteases (calpains) in the muscle, which can promote further muscle damage (Spencer et al. 1995).
Membrane lysis in injured or diseased muscle is not simply attributable to mechanical damage to the membrane, because indices of muscle membrane lesions, such as elevations of serum creatine kinase levels, can lag behind increased muscle loading by hours to days (e.g. Newman et al. 1983). This delay in the elevation of muscle proteins in serum suggests that some event other than direct mechanical damage causes membrane lesions. More recent experimental findings show that inflammatory cells may play an important role in causing membrane lesions in muscle experiencing modified loading. For example, increases in membrane lesions that are independent of the duration or frequency of muscle loading, but correspond with the invasion of muscle by inflammatory cells, occur in muscle experiencing modified loading (Tidball et al. 1999). Similarly, membrane lesions in the muscles of dystrophic mice that are null mutants for dystrophin (mdx mice) are greatly reduced by macrophage depletions from the mice before the onset of the disease (Wehling et al. 2001).
Myeloid cells, which comprise most of the inflammatory infiltrate in injured or dystrophic muscle are capable of causing muscle membrane lesions in vitro. Macrophages at pathophysiological concentrations in vitro lyse muscle cells through nitric-oxide-dependent processes, while neutrophils lyse through superoxide-dependent processes in vitro (Nguyen & Tidball, 2003a). However, lysis of muscle cells in macrophage-neutrophil co-cultures occurs through superoxide-independent mechanisms (Nguyen & Tidball, 2003a). This latter finding suggests that muscle membrane lysis in vivo in which the inflammatory infiltrate includes neutrophils and macrophages occurs through a superoxide-independent mechanism, although previous investigations have not yet tested this possibility in vivo.
Although inflammatory cells are able to cause muscle injury in vitro, other findings have suggested that inflammatory cells also function to promote muscle growth or the regeneration that follows injury or disease (Malm et al. 2000). In particular, experimental observations indicate that macrophages can promote the proliferation of myogenic cells in vitro (Robertson et al. 1993; Merly et al. 1999) and this function may be a specialized role of a non-phagocytic subpopulation of macrophages (Cantini & Carraro, 1995). Macrophage-derived, soluble factors can also function as chemoattractants for myogenic cells, at least in vitro, which could promote muscle repair following injury (Robertson et al. 1993). Macrophages are also frequently found in the highest concentrations in muscle that is in the process of growth or regeneration that results from modified muscle loading in vivo (St Pierre & Tidball, 1994; Merly et al. 1999) or muscle grafting (Lescaudron et al. 1999). Whether neutrophils also contribute to muscle growth is unknown.
In the present investigation, we tested in vivo whether muscle membrane lysis occurs through superoxide-mediated events during modified muscle use. Hindlimb unloading followed by reloading through normal weight bearing in mice was used as a model system (the hindlimb unloading/reloading model) because the treatment produces a well-characterized sequence of muscle invasion by myeloid cells, and extensive muscle membrane damage (Krippendorf & Riley, 1993; 1994; Kasper, 1995; Tidball et al. 1999). Muscle membrane damage and muscle inflammation in soleus muscles from wild-type mice and mice that are null mutants for gp91phox were compared because gp91phox is an essential subunit of NADPH oxidase in phagocytes, which is required for superoxide production (Pollock et al. 1995). Our findings show that the null mutation of gp91phox causes a significant reduction of muscle fibre injury during muscle reloading, as shown by the reduced presence of large lesions in the membranes of muscle cells. Null mutation of gp91phox did not cause changes in the concentrations of neutrophils or macrophages in the reloaded muscles. In addition, the reduction in membrane lesions in the muscles of gp91phox mutant mice did not affect the rate of muscle growth during muscle reloading, which indicates that membrane lesions are not required for muscle repair in this injury model.
METHODS
All experimental protocols involving the use of animals were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the University of California, Los Angeles Institutional Animal Care and Use Committee.
Isolation of neutrophils
Adult female mice received an intraperitoneal injection of sterile 12 % sodium caseinate solution and were killed by overdose of sodium pentobarbital 16 h later. The peritoneal exudate containing leukocytes was then collected. Cells were resuspended in Hanks' balanced salt solution (HBSS) overlaid on Histopaque 1077 (Sigma, St Louis, USA), and then centrifuged at 400 g for 45 min at room temperature. Neutrophils were pelleted and separated from other leukocytes, which partitioned at the HBSS-Histopaque interface. Neutrophils were then resuspended in HBSS and some cells were adhered to slides by centrifugation at 14 g for 3 min using a Cytospin (Shandon, Pittsburgh, USA), and stained with haematoxylin to confirm by nuclear morphology that they were neutrophils.
Assays of superoxide concentration
Superoxide concentration was determined by superoxide-dismutase-inhibitable reduction of ferricytochrome C (Talpain et al. 1995).
Cytotoxicity assays
C2C12 muscle cells were plated in 96-well plates in 10 % fetal bovine serum (FBS) in Dulbecco's modified Eagle's medium (DMEM) for 7 days, at which time they were a confluent monolayer. Myotubes were then placed in serum-free media overnight to induce fusion, and then returned to DMEM containing 10 % FBS for 2 days before use in cytotoxicity assays.
Myotubes were incubated in HBSS containing chromium-51 (51Cr) for 2 h and washed twice with HBSS before addition of neutrophils to the cultures. The concentrations of neutrophils used in co-cultures are expressed relative to the area (mm2) of myotube-covered substratum. Calculation of effector:target ratios in cytotoxicity assays was not possible because variability in myoblast proliferation and fusion prior to the assay resulted in an unknown number of target myotubes. Neutrophils were co-cultured with myotubes in HBSS containing 0.25 % FBS and 400 μm arginine for 24 h before the medium was assayed for 51Cr release by scintillation counting. Neutrophils in some co-cultures were activated by the addition of 0.6 μm phorbol 12-myristate 13-acetate (PMA), or 9.2 μmN-formyl methionine-leucine-phenylalanine (FMLP) added to the culture media. Cytotoxicity is expressed as a percentage of total myotube lysis, where 100 % lysis was determined by incubating myotubes with 20 % Triton X-100 in HBSS for 1 h to obtain complete myotube release of 51Cr, and 0 % lysis was set at the spontaneous release of 51Cr in the absence of neutrophils. All conditions were duplicated at least five times for each experiment, and each experiment was performed at least three times. Each experiment under a given set of conditions yielded similar results, although the data for only one representative experiment for each set of conditions are presented. Each value is expressed as the mean ±s.e.m. Values are compared using Student's t test, with P < 0.05 taken to indicate statistical significance.
Western analysis
The presence of gp91phox in neutrophils was assessed by Western analysis. Isolated neutrophils from gp91−/− or wild-type mice were homogenized in 80 mm Tris, pH 6.8 containing 0.1 m dithiothreitol and 70 mm SDS. The extract was boiled and then centrifuged at 12 000 g for 10 min to remove particulates. Protein concentration was measured (Minamide & Bamburg, 1990) and 30 μg of each sample was loaded on 10 % polyacrylamide gels for electrophoresis (Laemmli, 1970). Protein was then transferred electrophoretically to nitrocellulose membranes (Burnette, 1981), which were then overlaid with mouse anti-gp91phox (BD Pharmingen, San Diego, USA) diluted to 1:1000 in Tris-buffered saline (TBS; 50 mm Tris HCl, pH 7.6 containing 150 mm NaCl and 0.1 % NaN3) containing 3 % powdered dry milk and 0.05 % Tween 20. After extensive washing with TBS, the membranes were incubated with horseradish-peroxidase-conjugated second antibody, and the bound antibody was detected by enhanced chemiluminescence (Amersham, Piscataway, USA).
Hindlimb unloading/reloading model
Male C57BL/6J and gp91phox null mutant mice (C57BL/6-Cybbtm1) were obtained from Jackson Laboratory (Bar Harbor, USA) and subjected to hindlimb unloading for 10 days by using a previously described suspension apparatus (Morey-Holton & Globus, 2002). After 10 days of hindlimb unloading, mice were removed from the suspension apparatus and either immediately killed (six C57 and six gp91 phox−/− mice) or allowed to locomote under normal cage activity for 6 h (six C57 and six gp91phox−/− mice) or 24 h of reloading (six C57 and six gp91phox−/− mice). In addition, six C57 and six gp91phox−/− mice that were not subjected to hindlimb unloading were used as ambulatory controls. All mice were 3 months of age at the time of experimentation. Mice were monitored daily throughout the experimentation, and they exhibited no signs of distress, such as loss of appetite, failure to groom or vocalization. At the end of experimentation, mice were killed by an overdose of sodium pentobarbital.
Assessment of muscle membrane injury
Muscle fibre membrane injury was assessed quantitatively by measuring the presence of the extracellular marker dye, procion orange, within muscle fibres. Membrane lesions were assayed by influx of extracellular marker dye rather than measuring the concentration of muscle proteins in the serum, because in this experimental model there is little muscle damage to muscles other than the soleus, and since the soleus muscle is only approximately 10 mg in mass, the release of cytosolic proteins from the soleus into the serum is difficult to resolve reliably. Procion orange was selected as a marker of membrane lesions because it is a vital dye that is not actively transported across cell membranes, and it has a small mass (631 Da), which makes it a sensitive marker of membrane lesions.
After sacrifice, the left soleus of each mouse was dissected and incubated in 0.5 % procion orange dye (Sigma) in Krebs' Ringer solution for 1 h followed by two 5 min washes with Krebs' Ringer. The muscles were then rapidly frozen in isopentane that had been cooled with liquid nitrogen. Cross-sections 10 μm thick were taken from the mid-belly of each soleus. Sections were viewed by fluorescence microscopy and images were captured using a digital imaging system (Bioquant, Nashville, TN, USA). All sections were prepared and viewed under identical conditions.
Muscle fibre membrane injury was assessed by determining the percentage of fibres in entire cross-sections of the muscles that were brightly fluorescent. Bright fluorescence indicated that the procion orange, an extracellular marker dye, had entered the fibres through membrane lesions. This index of injury has been used frequently in previous investigations of muscle membrane injury (e.g. Greelish et al. 1999; Hack et al. 2000; Wehling et al. 2001), although no threshold for fluorescence intensity that is minimally necessary to indicate membrane injury has been established. Our recent findings have shown that muscle injury can cause increases in the influx of intracellular marker dye in fibres that are not obviously more fluorescent than neighbouring fibres, yet have experienced an increase in membrane damage (Nguyen & Tidball, 2003b). We tested for the presence of fibres possessing membrane damage that were not obviously more fluorescent than neighbouring fibres by measuring the intracellular fluorescence intensity of all fibres in complete cross-sections of entire soleus muscles. Intracellular fluorescence was measured in 8 μm diameter sampling circles placed at the centre of every fibre present in each cross-section of every muscle sampled. Background fluorescence measurements were made at a site adjacent to the muscle that contained no tissue, and that value was set to 0 for each section analysed. Thus, the index of muscle injury in this second injury assay was expressed as the fluorescence intensity of all fibres sampled in a muscle, after correction for background fluorescence. In this assay, the fluorescence intensities of approximately 760 fibres were measured for each muscle, and are expressed as arbitrary units. For each experimental condition, data are expressed as the mean fluorescence intensity and the differences between the mean values assessed by the Mann-Whitney unpaired test (P < 0.05).
Measurement of neutrophil and macrophage concentrations
The right soleus of each animal was analysed quantitatively by immunohistochemistry. Muscles were frozen in isopentane immediately upon dissection. Mid-belly cross-sections (10 μm thick) were stained with antibodies specific for neutrophils (rat anti-Ly6G; Pharmingen) or macrophages (rat anti-F4/80). Anti-F4/80 was obtained by affinity chromatography using agarose-bound mouse anti-rat IgG (Sigma) to separate rat IgG from the supernatant of F4/80 hybridomas (American Type Culture Collection, Bethesda, USA). The concentration of affinity-isolated IgGs was determined by enzyme-linked immunosorbent assay, and samples were diluted to 0.1-0.2 μg ml−1 for use in immunohistochemistry. Immunohistochemistry was performed as described previously (Wehling et al. 2001).
Myeloid cell concentrations in experimental and control soleus muscles were measured using standard stereological techniques. The entire cross-section of each muscle was examined by light microscopy using a calibrated eyepiece micrometer. The total numbers of neutrophils or macrophages were then counted for the entire cross-section and the area of each section was measured. The number of cells per area of section was calculated, and that value was converted to a volume density by dividing by the section thickness (10 μm). Each value is expressed as the mean ±s.e.m. Values are compared using Student's t test, with P < 0.05 taken to indicate statistical significance.
Measurement of muscle fibre area
Fibre cross-sectional area was measured for every fibre in complete cross-sections of each soleus muscle using a digitized imaging system (Bioquant, Nashville, TN, USA). Fibre cross-sectional area was used as an index of muscle atrophy or growth instead of using muscle mass, because the mass of mouse soleus muscles is approximately 10 mg, and they experience a 20–30 % mass change in the unloading protocol used here. Small differences in dissection could therefore cause differences in mass that overwhelm any experimental treatment effects. Each value is expressed as the mean ±s.e.m. Values are compared using Student's t test, with P < 0.05 taken to indicate statistical significance.
RESULTS
Null mutation of gp91phox reduces neutrophil lysis of mouse skeletal muscle cells in vitro
Peritoneal neutrophils collected from C57 mice and gp91phox null mutant mice were analysed with Western blots to confirm the lack of gp91phox expression in the mutants. Neutrophils from C57 mice showed a prominent immunoreactive band at the mass of gp91phox (58 kDa), but no detectable immunoreactive band was observed in Western analysis of neutrophils from gp91phox null mice (Fig. 1). In addition, stimulation of neutrophils with PMA or FMLP to activate the respiratory burst had no effect on superoxide production in co-cultures of muscle cells with gp91phox null neutrophils, although large increases in superoxide production in wild-type neutrophils occurred (Fig. 1). Cytotoxicity assays using neutrophils from wild-type or gp91phox null mice showed an increase in the lysis of myotubes with increasing concentrations of neutrophils. However, muscle cell lysis by gp91phox null neutrophils was 60–85 % less (at 25 000 neutrophils mm−2) than in co-cultures with wild-type neutrophils (Fig. 2). This indicates that most of the cytotoxicity was attributable to neutrophil-derived superoxide.
Figure 1. Neutrophils that are null mutants for the 58 kDa subunit of NADPH oxidase lack inducible superoxide production.
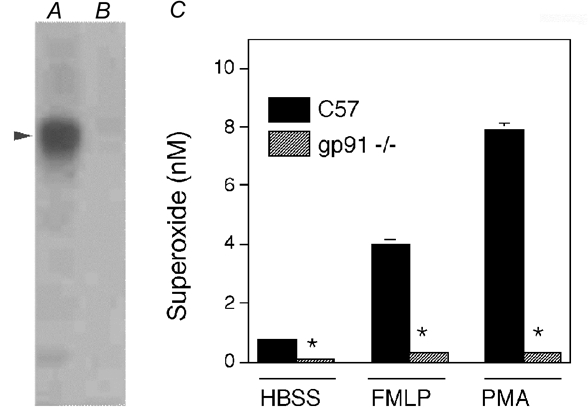
Western analysis was used to confirm that peritoneal cells collected from C57 mice expressed the 58 kDa subunit of NADPH oxidase (A; arrowhead indicates 58 kDa) and that gp91phox null mutant mice did not express the 58 kDa subunit (B). In addition, assays of superoxide production were performed on C57 neutrophils and gp91phox null neutrophils (C) to confirm that the mutation prevented the production of superoxide that was induced by FMLP or by PMA. Superoxide release assays were performed using 10 000 neutrophils mm−2. *Significantly different from the value for C57 neutrophils under the same culture conditions (P < 0.05). Bars = s.e.m.
Figure 2. The null mutation of gp91phox greatly reduces muscle cell lysis by neutrophils in vitro.
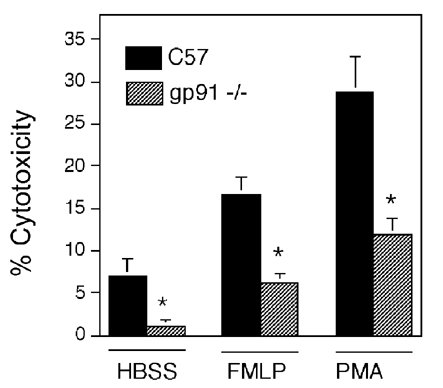
Neutrophils at 25 000 neutrophils mm−2 from C57 control mice or gp91phox null mutant mice that are activated by receptor-mediated (FMLP) or receptor-independent (PMA) mechanisms were used as effectors in cytotoxicity assays with C2C12 myotubes as targets. *Significantly different from the value for C57 neutrophils under same culture conditions (P < 0.05). Bars = s.e.m.
Null mutation of gp91phox prevents lysis of skeletal muscle fibres during modified muscle loading in vivo
Muscle membrane injury during modified muscle use was assessed by assaying for the presence of procion orange, a fluorescent extracellular marker dye, in the intracellular space of muscle fibres. The proportion of total fibres in muscle cross-sections that were brightly fluorescent did not differ between wild-type or gp91phox mice in either the ambulatory control groups or the 10 day unloaded group (Fig. 3 and Fig. 4). However, gp91phox mutant mice had 90 % fewer fibres that showed bright intracellular fluorescence when subjected to either 6 or 24 h reloading after the 10 day unloading period (P < 0.05). There was no increase in the proportion of injured fibres between 6 and 24 h of reloading in either the wild-type or gp91phox mutant mice.
Figure 3. Identification of muscle fibre membrane lesions by the presence of intracellular procion orange.
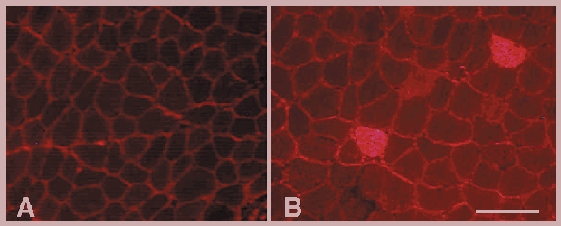
A, cross-section of a control soleus muscle that was incubated with the extracellular marker dye procion orange. In healthy control muscle the procion orange is located in the extracellular space that outlines each muscle fibre. B, cross-section of a soleus muscle from a C57 mouse that experienced 24 h of muscle reloading after 10 days of unloading. Two fibres are brightly fluorescent because of the entry of procion orange through membrane lesions. Note also that the general level of intracellular fluorescence in muscle fibres in reloaded soleus (B) is higher than in control fibres (A). Bar = 100 μm.
Figure 4. A null mutation of gp91phox decreases the concentration of injured fibres in reloaded muscle.
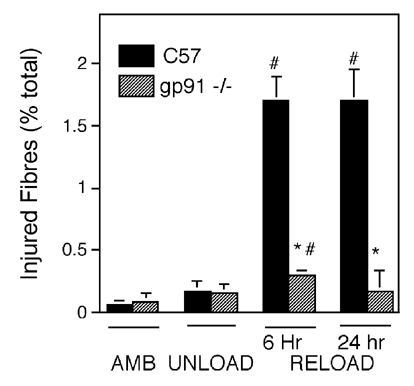
The concentration of brightly fluorescent fibres (see Fig. 3B) in cross-sections of soleus muscles that were incubated in procion orange was determined. AMB, control ambulatory group (i.e. did not undergo hindlimb unloading); UNLOAD, animals were subjected to hindlimb unloading and then immediately sacrificed; RELOAD, animals were subjected to hindlimb unloading followed by 6 or 24 h of reloading before being sacrificed. #Concentrations that differed significantly from unloaded muscles of same genotype (P < 0.05); *concentrations that differed significantly from C57 muscles in the same treatment group (P < 0.05). Bars = s.e.m.
Membrane lesions were also assessed by measuring the fluorescence intensity of every individual fibre in each complete cross-sections of each soleus muscle. Fluorescence intensity was measured in approximately 760 fibres in each muscle analysed in the investigation. Muscle membrane injury was then determined by testing for significant shifts in fluorescence for the entire population of muscle fibres. The frequency distribution of fluorescence intensities of all fibres showed a small shift to higher average values in C57 mice at 6 h of reloading compared to the values for gp91phox null mice, although the mean fluorescent intensity did not differ significantly between the two groups. At 24 h of reloading, mean fluorescence intensity was significantly greater in C57 fibres than in fibres from gp91phox mutant mice (P < 0.05), which indicates that gp91phox null mice were protected from membrane damage (Fig. 5). No significant difference in mean fluorescence intensity between C57 and gp91phox null mice was observed in the ambulatory control or the groups experiencing 0 h reloading following unloading.
Figure 5. Frequency distributions of muscle fibres over the range of intensities of intracellular fluorescence.
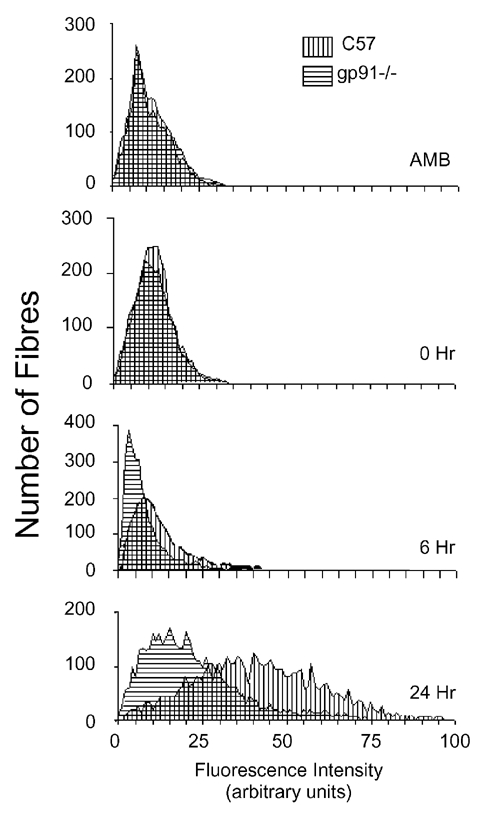
Intracellular fluorescence was measured for individual fibres in entire cross-sections of soleus muscles from each treatment group (six animals per treatment group). Treatments consisted of ambulatory controls, animals experiencing 10 days of hindlimb unloading only (0 h), or 10 days unloading followed by 6 h (6 h) or 24 h reloading (24 h). A rightward shift of peaks on the abscissa indicates an increase in the frequency of fibres with muscle membrane lesions.
Null mutation of gp91phox does not affect the muscle inflammation caused by modified muscle use in vivo
We tested whether the reduction of muscle membrane damage in reloaded mouse soleus muscles of gp91phox null mice resulted from reductions in inflammatory cell infiltration rather than reductions in superoxide-mediated cytotoxicity. We assessed this possibility by measuring the concentrations of neutrophils and macrophages in ambulatory, unloaded and reloaded muscles. Our data show that the concentrations of neutrophils and macrophages in wild-type and gp91phox mutant mice were identical in control and unloaded-only groups. Neither macrophage nor neutrophil concentrations differed significantly between wild-type and gp91phox mutant mice at either 6 or 24 h reloaded groups (Fig. 6).
Figure 6. Myeloid cell invasion is not affected by the gp91phox null mutation.
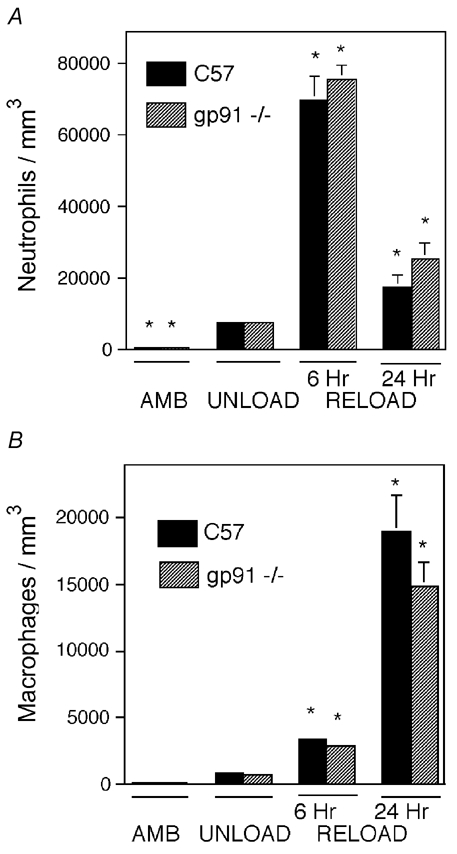
Muscles from ambulatory control mice experiencing 10 days unloading only, or unloading followed by 6 h or 24 h reloading were assayed for neutrophil concentration (A) and macrophage concentration (B). Each treatment group included six animals. *Concentrations that differed significantly from unloaded muscles of the same genotype (P < 0.05). No significant differences were found between gp91phox and C57 mice in any experimental group. Bars = s.e.m.
Muscle growth during muscle reloading is not affected by null mutation of gp91phox and does not require muscle membrane damage
Measurements of muscle fibre cross-sectional area showed that null mutation of gp91phox did not affect the size of fibres in ambulatory controls. In addition, neither the decrease in fibre cross-sectional area during unloading (35 % in C57; 38 % in gp91phox mutants) nor the increase in fibre cross-sectional area during reloading (30 % in C57; 32 % in gp91phox mutants) were affected by the absence of gp91phox, or by the reduction of membrane injury during the reloading period in gp91phox null mice (Fig. 7). Fewer than 1.5 % of the fibres present in any muscle assayed showed either a rounded profile or hyaline cytoplasm, which indicate fibre swelling. All other fibres showed dense, uniform arrays of myofibrils when examined with the aid of Nomarski optics.
Figure 7. Rates of muscle atrophy and growth are not affected by the gp91phox null mutation.
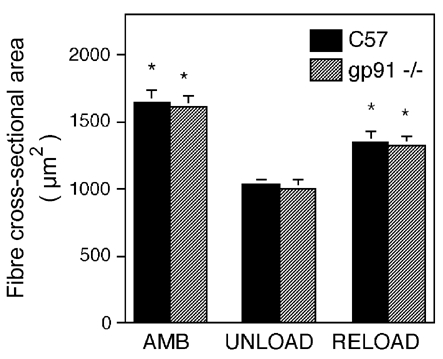
Values are means ±s.e.m. of soleus muscle fibre cross-sectional areas for all fibres in the entire cross-section for each treatment group (six animals per treatment group). *Significant difference from unloaded muscles of same genotype (P < 0.05). No significant differences were found between gp91phox and C57 mice in any experimental group.
DISCUSSION
The results of the present investigation show that superoxide mediates the majority of muscle membrane lysis that occurs in the unloading/reloading model of muscle injury. Null mutations of gp91phox eliminated the increased occurrence of membrane lesions during the 24 h reloading period that normally follows 10 days of unloading. During this period of reloading, neutrophils rapidly invade muscle at high concentrations and provide a rich source of superoxide production. Although macrophages also invade the muscle within the first 24 h of reloading, they generate relatively low levels of superoxide and they do not lyse muscle cells through superoxide-dependent mechanisms (Nguyen & Tidball, 2003a). In addition, the results of the present investigation show that most lysis of muscle cells by neutrophils is superoxide-dependent in cytotoxicity assays, and that the superoxide that mediates lysis is generated by neutrophils. Although superoxide production in other cells such as macrophages and muscle is impaired in gp91phox null mice, when taken together, the findings of the present investigation support the conclusion that neutrophil-derived superoxide or a superoxide derivative causes most membrane lysis in muscles experiencing reloading following a period of unloading. However, it is still feasible that the superoxide that is derived from sources other than neutrophils contributes to muscle membrane lysis during muscle reloading in this in vivo model.
Although the present results show that a null mutation of gp91phox prevents the occurrence of significant increases in muscle membrane damage during muscle reloading in vivo, we are not able to conclude whether superoxide directly mediates membrane lysis in vivo. Superoxide can be rapidly converted into other, more highly reactive molecules that may cause membrane lysis, such as peroxynitrite or hydrogen peroxide. In addition, decreased concentrations of superoxide may cause a shift in the redox status of the muscle, which can influence membrane stability through more remote processes. Superoxide also has the ability to influence signalling mechanisms in cells (Hancock et al. 2001), so it is feasible that changes in cell signalling could ultimately have indirect effects on membrane stability. Although any of these potential mechanisms individually or in aggregate can possibly affect the occurrence of membrane lysis, the central role of superoxide in mediating muscle membrane lesions in the present model is clear.
The present investigation also shows that the release of chemoattractants through muscle membrane lesions may not be necessary to promote muscle inflammation. Previous investigations have shown that homogenates of injured muscle contain chemoattractants for neutrophils and macrophages in vitro (Robertson et al. 1993). Other findings have indicated that muscle membrane lesions can cause the release of basic fibroblast growth factor (Clarke et al. 1993), which can function as a chemoattractant. However, the reduction in muscle membrane lysis in reloaded muscles of gp91phox null mice, without a decrease in neutrophil or macrophage invasion, indicates that release of cytosolic proteins into the extracellular space may not be required for myeloid cell chemotaxis in this model of muscle injury. Instead, activation of the complement system through mechanisms that are independent of muscle membrane lesions may lead to the recruitment of inflammatory cells into muscle experiencing unloading/ reloading. Previous findings have shown that complement activation occurs in muscle unloading/ reloading, and that inhibition of complement activation reduces the neutrophil invasion of reloaded muscle (Frenette et al. 2000). Although the mechanism through which this complement activation occurs in reloaded muscle is unknown, activation may result from disruption of free radical production in muscle. For example, exposure of complement component C5 to hydrogen peroxide modifies C5 structure so that it is similar to the active form C5a (Shingu & Nobunaga, 1984; Vogt et al. 1986), which can attract and activate neutrophils to injury sites (Shingu & Nobunaga, 1984). Although modified muscle loading can produce changes in free radical concentrations in muscle (Powers et al. 1994; Bejma & Ji, 1999; Lawler et al. 2003), whether those changes are sufficient for complement activation is unknown.
As more is learned about the mechanisms of muscle injury during muscle reloading, similarities to injury mechanisms in muscle experiencing ischaemia followed by reperfusion have become more evident. Both perturbations result in a rapid influx of neutrophils that is concomitant with muscle damage (Formigli et al. 1992; Tidball et al. 1999). Muscle injury in both models is mediated by superoxide (Korthuis et al. 1985; Belkin et al. 1989; Yokota et al. 1989; present study) and complement activation (Rubin et al. 1990; Weiser et al. 1996; Kyriakides et al. 1999; Frenette et al. 2000). In addition, most damage associated with inflammatory cells occurs in the first few hours of reperfusion (Merchant et al. 2003) or reloading (Tidball et al. 1999). However, the role of superoxide in the two injury processes is not identical. In ischaemia/ reperfusion, administration of superoxide dismutase during reperfusion causes significant decreases in neutrophil interactions with endothelial cells and causes reductions in vascular damage and leakage (Menger et al. 1992). Together, these effects of superoxide dismutase treatment reduce neutrophil extravasation into reperfused muscle. In unloading/reloading, no reduction in the concentration of extravasated neutrophils was observed in gp91phox null mice, which indicates that the protective effect of the null mutation results from reduced cytotoxicity rather than reduced extravasation.
The results of the present investigation also show that muscle membrane lesions may not be necessary for muscle growth during muscle reloading. Generally, teleological arguments have supported the expectation that muscle damage is a necessary component of the muscle growth and hypertrophy that follows modified muscle loading, although there is little direct experimental evidence to show that injury is a requirement for growth. In vitro assays have indicated that muscle contains solutes that can promote muscle cell growth and proliferation when released by muscle homogenization and applied to muscle cells in vitro (Chen & Quinn, 1992; Chen et al. 1994). However, it is not known whether release of those factors through membrane lesions in vivo is required for muscle growth in vivo. The present findings show that the greatly reduced membrane damage in reloaded muscle of gp91phox null mice has no effect on the rate of fibre growth during 24 h of muscle reloading. Thus, in at least this model of muscle injury and repair, growth appears to be independent of the presence of membrane lesions. However, reductions of membrane lesions during reloading may have influenced satellite cell proliferation. This possibility will be tested in continuing studies.
In conclusion, this investigation provides evidence that much of the muscle membrane injury that occurs in muscle reloading results from superoxide-mediated lysis, and supports the view that neutrophils actively promote muscle damage during the early stages of modified muscle use. Furthermore, membrane lysis does not promote myeloid cell chemotaxis or muscle fibre growth during 24 h of reloading. If neutrophils are the major source of the superoxide that mediates muscle membrane lysis, these findings suggest a deleterious role for neutrophils in the response of muscle to modified loading. However, these findings are not sufficient to address whether neutrophils have a net negative effect on the response of muscle to modified loading. Previous investigators have speculated that the presence of neutrophils in injured muscle is important for the removal of damaged cellular debris, so that growth and repair can occur (Fielding et al. 1993; Tiidus, 1998). Those potential beneficial effects may occur through superoxide-independent processes and may not have been affected by the null mutation of gp91phox.
Acknowledgments
This work was supported by grants from the NIH (AR47721, AR47855) and NASA (NAG5-4837).
REFERENCES
- Bejma J, Ji LL. Aging and acute exercise enhance free radical generation in rat skeletal muscle. J Appl Physiol. 1999;87:465–470. doi: 10.1152/jappl.1999.87.1.465. [DOI] [PubMed] [Google Scholar]
- Belkin M, Lamorte WL, Wright JG, Hobson RW. The role of leukocytes in the pathophysiology of skeletal muscle ischemic injury. J Vasc Surg. 1989;10:14–19. doi: 10.1067/mva.1989.vs0100014. [DOI] [PubMed] [Google Scholar]
- Burnette WN. ‘Western blotting’: electrophoretic transfer of proteins from sodium dodecyl sulfate polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Cantini M, Carraro U. Macrophage-released factor stimulates selectively myogenic cells in primary muscle culture. J Neuropathol Exp Neurol. 1995;54:121–128. doi: 10.1097/00005072-199501000-00014. [DOI] [PubMed] [Google Scholar]
- Chen G, Birnbaum RS, Yablonka-Reuveni Z, Quinn LS. Separation of mouse crushed muscle extract into distinct mitogenic activities by heparin affinity chromatography. J Cell Physiol. 1994;160:563–572. doi: 10.1002/jcp.1041600320. [DOI] [PubMed] [Google Scholar]
- Chen G, Quinn LS. Partial characterization of skeletal myoblast mitogens in mouse crushed muscle extract. J Cell Physiol. 1992;153:563–574. doi: 10.1002/jcp.1041530318. [DOI] [PubMed] [Google Scholar]
- Clarke MS, Khakee R, McNeil PL. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J Cell Sci. 1993;106:121–133. doi: 10.1242/jcs.106.1.121. [DOI] [PubMed] [Google Scholar]
- Fielding RA, Manfredi TJ, Ding W, Fiatarone MA, Evans WJ, Cannon JG. Acute phase response in exercise. III. Neutrophil and IL-1β accumulation in skeletal muscle. Am J Physiol. 1993;265:R166–172. doi: 10.1152/ajpregu.1993.265.1.R166. [DOI] [PubMed] [Google Scholar]
- Florence JM, Fox PT, Planer GJ, Brooke MH. Activity, creatine kinase, and myoglobin in Duchenne muscular dystrophy: a clue to etiology. Neurology. 1985;35:758–761. doi: 10.1212/wnl.35.5.758. [DOI] [PubMed] [Google Scholar]
- Formigli L, Lombardo L, Adembri C, Brunelleschi S, Ferrari E, Novelli G. Neutrophils as mediators of human skeletal muscle ischemia-reperfusion syndrome. Hum Pathol. 1992;23:627–634. doi: 10.1016/0046-8177(92)90317-v. [DOI] [PubMed] [Google Scholar]
- Frenette J, Cai B, Tidball JG. Complement activation promotes muscle inflammation during modified muscle use. Am J Pathol. 2000;156:2103–2110. doi: 10.1016/S0002-9440(10)65081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greelish JP, Su LT, Lankford EB, Burkman JM, Chen H, Konig SK, Mercier IM, Desjardins PR, Mitchell MA, Zheng XG, Leferovich J, Gao GP, Balice-Gordon RJ, Wilson JM, Stedman HH. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat Med. 1999;5:439–443. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- Hack AA, Lam MJ, Cordier L, Shoturma DI, Ly CT, Hadhazy MA, Hadhazy HR, Sweeney HL, McNally EM. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci. 2000;113:2535–2544. doi: 10.1242/jcs.113.14.2535. [DOI] [PubMed] [Google Scholar]
- Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans. 2001;29:345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- Kasper CE. Sarcolemmal disruption in reloaded atrophic skeletal muscle. J Appl Physiol. 1995;79:607–614. doi: 10.1152/jappl.1995.79.2.607. [DOI] [PubMed] [Google Scholar]
- Korthuis RJ, Granger DN, Townsley MI, Taylor AE. The role of oxygen-derived free radicals in ischemia-induced increases in canine skeletal muscle vascular permeability. Circ Res. 1985;57:599–609. doi: 10.1161/01.res.57.4.599. [DOI] [PubMed] [Google Scholar]
- Krippendorf BB, Riley DA. Distinguishing unloading- versus reloading-induced changes in rat soleus muscle. Muscle Nerve. 1993;16:99–108. doi: 10.1002/mus.880160116. [DOI] [PubMed] [Google Scholar]
- Krippendorf BB, Riley DA. Temporal changes in sarcomere lesions of rat adductor longus muscle during hindlimb reloading. Anat Rec. 1994;238:304–210. doi: 10.1002/ar.1092380304. [DOI] [PubMed] [Google Scholar]
- Kyriakides C, Austen W, Wang Y, Favuzza J, Kobzik L, Moore FD, Hechtman HB. Skeletal muscle reperfusion injury is mediated by neutrophils and the complement membrane attack complex. Am J Physiol. 1999;277:C1263–1268. doi: 10.1152/ajpcell.1999.277.6.C1263. [DOI] [PubMed] [Google Scholar]
- Laemmli K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lawler JM, Song W, Demaree SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic Biol Med. 2003;35:9–16. doi: 10.1016/s0891-5849(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Lescaudron L, Peltekian E, Fontaine-Perus J, Paulin D, Zampieri M, Garcia L, Parrish E. Blood borne macrophages are essential for the triggering of muscle regeneration following muscle transplant. Neuromuscul Disord. 1999;9:72–80. doi: 10.1016/s0960-8966(98)00111-4. [DOI] [PubMed] [Google Scholar]
- Malm C, Nyberg P, Engstrom M, Sjodin B, Lenkei R, Ekblom B, Lundberg I. Immunological changes in human skeletal muscle and blood after eccentric exercise and multiple biopsies. J Physiol. 2000;529:243–262. doi: 10.1111/j.1469-7793.2000.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menger MD, Pelikan S, Steiner D, Messmer K. Microvascular ischemia-reperfusion injury in striated muscle: significance of ‘reflow paradox’. Am J Physiol. 1992;263:H1901–1906. doi: 10.1152/ajpheart.1992.263.6.H1901. [DOI] [PubMed] [Google Scholar]
- Merchant SH, Gurule DM, Larson RS. Amelioration of ischemia-reperfusion injury with cyclic peptide blockade of IVAM-1. Am J Physiol. 2003;284:H1260–1268. doi: 10.1152/ajpheart.00840.2002. [DOI] [PubMed] [Google Scholar]
- Merly F, Lescaudron L, Rouaud T, Crossin F, Gardahaut MF. Macrophages enhance muscle satellite cell proliferation and delay their differentiation. Muscle Nerve. 1999;22:724–732. doi: 10.1002/(sici)1097-4598(199906)22:6<724::aid-mus9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Minamide S, Bamburg JR. A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal Biochem. 1990;190:66–70. doi: 10.1016/0003-2697(90)90134-u. [DOI] [PubMed] [Google Scholar]
- Morey-Holton ER, Globus RK. Hindlimb unloading rodent model: technical aspects. J Appl Physiol. 2002;92:1367–1377. doi: 10.1152/japplphysiol.00969.2001. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Jones DA, Edwards RHT. Large delayed plasma creatine kinase changes after stepping exercise. Muscle Nerve. 1983;6:380–385. doi: 10.1002/mus.880060507. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG. Interactions between neutrophils and macrophages promote macrophage killing of muscle cells in vitro. J Physiol. 2003a;547:125–132. doi: 10.1113/jphysiol.2002.031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG. Expression of a muscle-specific, nitric oxide synthase transgene prevents muscle membrane injury and reduces muscle inflammation during modified muscle use in mice. J Physiol. 2003b;550:347–356. doi: 10.1113/jphysiol.2003.040907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MAC, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- Powers SK, Criswell D, Lawler J, Ji LL, Martin D, Herb RA, Dudley G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am J Physiol. 1994;266:R375–380. doi: 10.1152/ajpregu.1994.266.2.R375. [DOI] [PubMed] [Google Scholar]
- Robertson TA, Maley MAL, Grounds MD, Papadimitriou J. The role of macrophages in skeletal muscle regeneration with particular reference to chemotaxis. Exp Cell Res. 1993;207:321–331. doi: 10.1006/excr.1993.1199. [DOI] [PubMed] [Google Scholar]
- Rubin BB, Smith A, Liauw S, Idenman D, Romaschin AD, Walker PM. Complement activation and white cell sequestration in postischemic skeletal muscle. Am J Physiol. 1990;259:H525–531. doi: 10.1152/ajpheart.1990.259.2.H525. [DOI] [PubMed] [Google Scholar]
- Shingu M, Nobunaga M. Chemotactic activity generated in human serum from the fifth component of complement by hydrogen peroxide. Am J Pathol. 1984;117:201–206. [PMC free article] [PubMed] [Google Scholar]
- Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995;270:10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- St Pierre B, Tidball JG. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J Appl Physiol. 1994;77:290–297. doi: 10.1152/jappl.1994.77.1.290. [DOI] [PubMed] [Google Scholar]
- Talpain E, Armstrong RA, Coleman RA, Vardey CJ. Characterization of the PGE receptor subtype mediating inhibition of superoxide production in human neutrophils. Br J Pharm. 1995;114:1459–1465. doi: 10.1111/j.1476-5381.1995.tb13370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Berchenko E, Frenette J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J Leukoc Biol. 1999;65:492–498. [PubMed] [Google Scholar]
- Tiidus PM. Radical species in inflammation and overtraining. Can J Physiol Pharmacol. 1998;76:533–538. doi: 10.1139/cjpp-76-5-533. [DOI] [PubMed] [Google Scholar]
- Vogt W, Von Zabern I, Hesse D, Nolte R, Haller Y. Generation of an activated form of human C5 (C5b-like C5) by oxygen radicals. Immunol Lett. 1986;14:209–215. doi: 10.1016/0165-2478(87)90103-9. [DOI] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser MR, Williams JP, Moore FD, Kobzik L, Ma M, Hechtman HB, Carroll MC. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183:2343–2348. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota J, Minei JP, Fantini GA, Shires GT. Role of leukocytes in reperfusion injury of skeletal muscle after partial ischemia. Am J Physiol. 1989;257:H1068–1075. doi: 10.1152/ajpheart.1989.257.4.H1068. [DOI] [PubMed] [Google Scholar]