

The crystal structure of the minimized α/β-hydrolase fold protein encoded by the gene TTHA1544 from T. thermophilus HB8 has been determined at 2.0 Å resolution.
Keywords: T. thermophilus HB8, TTHA1544, α/β-hydrolase fold, singleton
Abstract
The gene encoding TTHA1544 is a singleton found in the Thermus thermophilus HB8 genome and encodes a 131-amino-acid protein. The crystal structure of TTHA1544 has been determined at 2.0 Å resolution by the single-wavelength anomalous dispersion method in order to elucidate its function. There are two molecules in the asymmetric unit. Each molecule consists of four α-helices and six β-strands, with the β-strands composing a central β-sheet. A structural homology search revealed that the overall structure of TTHA1544 resembles the α/β-hydrolase fold, although TTHA1544 lacks the catalytic residues of a hydrolase. These results suggest that TTHA1544 represents the minimized α/β-hydrolase fold and that an additional component would be required for its activity.
1. Introduction
Genome projects provide information about the number of genes and the sequences of coding frames; of these, nearly half are proteins of unknown function. Structural genomic projects and proteomic studies are expected to generate information about protein function. Genome projects for Thermus thermophilus are being performed in Germany for strain HB27 (Project ID 10617; Henne et al., 2004 ▶) and in Japan for strain HB8 (Project ID 13202; Yokoyama et al., 2000 ▶). T. thermophilus HB27 has one chromosome (NC 005835, 1976 proteins, 1.89 Mbp) and one plasmid (NC 005838, 228 proteins, 0.23 Mbp), while T. thermophilus HB8 has one chromosome (NC 006461, 1973 proteins, 1.8 Mbp) and two plasmids (NC 006463, 14 proteins, 9.3 kbp; NC 006462, 251 proteins, 0.25 Mbp). Although the genome sizes and number of genes differ, the corresponding sequences are almost identical. TTHA1544 is one of 864 proteins of unknown function from T. thermophilus HB8 and is encoded in the chromosome. The corresponding gene of T. thermophilus HB27 is TTC1178 (Henne et al., 2004 ▶); its gene arrangement is also similar to that of TTHA1544. No significant homologue was found by BLAST searches of the NCBI database. We have selected TTHA1544 as a target for a structural genomics study and have determined its crystal structure.
2. Materials and methods
2.1. Expression, purification and crystallization
The TTHA1544 gene was amplified by PCR from T. thermophilus HB8 genomic DNA and cloned into the pET11a expression vector (Novagen). Selenomethionine (SeMet) substituted TTHA1544 was produced in Escherichia coli cultivated in LeMaster medium (LeMaster & Richards, 1985 ▶) containing SeMet at 310 K. After disruption of the cells by sonication, the lysate was subjected to heat treatment for 30 min at 343 K and was centrifuged to remove the denatured proteins. Purification of TTHA1544 was performed by serial chromatography steps with Resource ISO, Resource Q (GE Healthcare) and Hydroxyapatite CHT5 (Bio-Rad). After size-exclusion chromatography on a HiLoad 16/60 Superdex-75 (GE Healthcare) column equilibrated with 20 mM Tris–HCl buffer pH 7.5 containing 150 mM NaCl and 1 mM DTT, TTHA1544 was purified to homogeneity and concentrated to 8.2 mg ml−1 using a Centricon filter unit (Millipore).
Crystallization was carried out by the sitting-drop vapour-diffusion method at 293 K. Crystals were obtained in a drop consisting of 0.5 µl protein solution mixed with 0.5 µl reservoir solution, which contained 0.9 M 1,6-hexanediol, 100 mM bis-Tris pH 5.0, 10% glycerol and 10 mM CoCl2.
2.2. Diffraction data collection and structure determination
An anomalous data set was collected on beamline 22ID at the Advanced Photon Source (APS), Argonne National Laboratory. The X-ray diffraction data were collected to 2.7 Å resolution and were indexed, scaled and merged using the HKL-2000 software (Otwinowski & Minor, 1997 ▶). Phasing of the data was carried out by the single-wavelength dispersion (SAD) method. The positions of the Se atoms in the asymmetric unit and the initial phases were calculated using the program SOLVE (Terwilliger & Berendzen, 1999 ▶) and the phases were improved using the program RESOLVE (Terwilliger, 2000 ▶), yielding an overall figure of merit (FOM) of 0.72. Model building was performed using the program O (Jones et al., 1991 ▶). Structure refinement was performed using the CNS program (Brünger et al., 1998 ▶) with a 2.0 Å resolution X-ray diffraction data set collected on beamline BL26B1 at the SPring-8 synchrotron facility (Harima, Japan). The stereochemical quality of the final model was assessed using PROCHECK (Laskowski et al., 1993 ▶) and WHATIF (Vriend, 1990 ▶). The crystallographic data, refinement and the Ramachandran plot statistics are summarized in Tables 1 ▶ and 2 ▶.
Table 1. Crystallographic data.
Values in parentheses are for the highest resolution shell.
| X-ray resource | APS (SAD) | SPring-8 |
|---|---|---|
| Wavelength () | 0.9724 | 1.0000 |
| Resolution range () | 502.70 (2.802.70) | 502.00 (2.112.00) |
| Total reflections | 48073 | 58010 |
| Total unique reflections | 6698 | 15276 |
| Completeness (%) | 98.2 (97.5) | 93.5 (80.3) |
| R merge † | 0.07 (0.216) | 0.064 (0.151) |
| Redundancy | 7.2 (6.5) | 3.8 (3.4) |
| I/(I) | 27.4 (8.7) | 20.4 (6.6) |
R
merge = 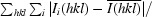
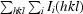 , where Ii(hkl) is the observed integrated intensity,
, where Ii(hkl) is the observed integrated intensity, 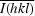 is the average integrated intensity obtained from multiple measurements and the summation is the overall observed reflections.
is the average integrated intensity obtained from multiple measurements and the summation is the overall observed reflections.
Table 2. Refinement statistics.
| Resolution range () | 31.682.00 |
| No. of reflections | 13671 |
| No. of atoms | 1858 |
| No. of waters | 146 |
| R factor/free R factor† (%) | 20.1/24.5 |
| Average B factor (2) | 35.5 |
| R.m.s. deviations | |
| Bond lengths () | 0.011 |
| Bond angles () | 1.60 |
| Dihedral angles () | 24.70 |
| Improper angles () | 1.02 |
| Ramachandran plot statistics | |
| Most favoured regions (%) | 91.6 |
| Additional allowed regions (%) | 7.4 |
| Generously allowed regions (%) | 1.1 |
| Disallowed regions (%) | 0 |
R factor = 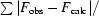
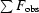 . The free R factor was calculated using 10% of reflections omitted from refinement.
. The free R factor was calculated using 10% of reflections omitted from refinement.
3. Results and discussion
The crystal of TTHA1544 belongs to space group P21, with unit-cell parameters a = 31.71, b = 65.82, c = 59.32 Å, β = 92.61°. There are two molecules in the asymmetric unit, giving a Matthews coefficient of 2.2 Å3 Da−1 (Matthews, 1968 ▶) with an approximate solvent content of 44%. The main-chain r.m.s. deviation (r.m.s.d.) value between the two molecules is 0.8 Å. Each molecule contains residues Arg2–Gly123. The surface areas buried in the dimer interface are 420.3 Å2 for molecule A and 407.6 Å2 for molecule B, as calculated using the Protein–Protein Interaction Server (Jones & Thornton, 1996 ▶). The electron densities of Met1 and the C-terminal eight residues were not observed because of disorder. TTHA1544 consists of six β-strands and four α-helices, including one short span of 310-helix (Fig. 1 ▶ a). All of the β-strands, including the five mutually parallel strands (β1, β3, β4, β5 and β6) and one antiparallel strand (β2), compose a central β-sheet. Helices η1, α1 and α2 are located between β3 and β4, β4 and β5, and β5 and β6, respectively.
Figure 1.

(a) Stereoview of the overall structure of TTHA1544, shown using a ribbon presentation. Helices, strands and loops are shown in red, green and grey, respectively. (b) Stereoview of the superposition of TTHA1544 and human CCG1/TAF(II)250-interacting factor B (CIB). TTHA1544 and CIB are shown in red and sky blue, respectively. The 2F o − F c map of residue Tyr43 of TTHA1544, which is conserved in CIB, is contoured at 1σ. Tyr43, Ser111, Asp162 and His188 are shown as ball-and-stick models. The C, N and O atoms are coloured black, green and red, respectively. (a) and (b) were prepared with the programs MOLSCRIPT (Kraulis, 1991 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶). The electron-density map was prepared with the program CONSCRIPT (Lawrence & Bourke, 2000 ▶). (c) Amino-acid structural sequence alignment of TTHA1544 with CIB using the DALI server. Identical residues are shown as white letters on a red background, while similar residues are shown as red letters. The secondary-structural elements determined on the basis of the crystal structure of TTHA1544 are indicated with arrows for β-strands and coils for α-helices at the top of the alignment. The residues of the CIB active site are marked by red triangles under the alignment.
Structural homology searches using the DALI server (Holm & Park, 2000 ▶) revealed five structural homologues of TTHA1544: human CCG1/TAF(II)250-interacting factor B (CIB; PDB code 1imj; SCOP entry 75285; Padmanabhan et al., 2004 ▶), murine liver epoxide hydrolase (PDB code 1cr6; Argiriadi et al., 1999 ▶), Agrobacterium radiobacter AD1 epoxide hydrolase (Ephy; PDB code 1ehy; Nardini et al., 1999 ▶), Burkholderia sp. FA1 fluoroacetate dehalogenase (PDB code 1y37) and Rhodococcus sp. MB1 cocaine esterase (PDB code 1ju3; Larsen et al., 2002 ▶). The Z scores, r.m.s.d.s and amino-acid identities between TTHA1544 and each protein are summarized in Table 3 ▶. Many of these proteins belong to the α/β-hydrolase superfamily (SCOP entry 53474) and share significant amino-acid identity within the partial region exhibiting structural similarity. The highest amino-acid identity, 38%, is found with CIB. Superposition of the three-dimensional structures of TTHA1544 and CIB and an amino-acid structural sequence alignment with the secondary structures revealed a large insertion between β6 and the C-terminal helix (Figs. 1 ▶ b and 1 ▶ c). The structure of CIB includes an α/β-hydrolase fold, with a catalytic triad composed of Ser111, Asp162 and His188. Hydrolase activity was observed by in vitro studies, despite the lack of a binding-site excursion which facilitates substrate selectivity. Although the overall folds of the two proteins are similar, the residues of the CIB active site are not conserved in TTHA1544. The important residues, Asp162 and His188, are located in the inserted region (Figs. 1 ▶ b and 1 ▶ c).
Table 3. Statistics of structural homologues of TTHA1544.
| Protein name | PDB code | Z score | R.m.s.d. () | Residues aligned | Sequence identity (%) |
|---|---|---|---|---|---|
| Murine liver epoxide hydrolase | 1cr6 | 8.0 | 2.0 | 119 | 18 |
| A. radiobacter AD1 epoxide hydrolase | 1ehy | 11.1 | 2.0 | 119 | 17 |
| Burkholderia sp. FA1 fluoroacetate dehalogenase | 1y37 | 10.9 | 2.0 | 119 | 18 |
| Rhodococcus sp. MB1 cocaine esterase | 1ju3 | 10.4 | 2.7 | 116 | 14 |
| Human CCG1/TAF(II)250-interacting factor B | 1imj | 10.6 | 2.5 | 119 | 23 |
Superpositions with the four other proteins are shown in Fig. 2 ▶: Ephy (PDB code 1ehy; Fig. 2 ▶ a), murine liver epoxide hydrolase (PDB code 1cr6; Fig. 2 ▶ b), Burkholderia sp. FA1 fluoroacetate dehalogenase (PDB code 1y37; Fig. 2 ▶ c) and Rhodococcus sp. MB1 cocaine esterase (PDB code 1ju3; Fig. 2 ▶ d). Each superposition shows a large inserted region between β6 and α3 of TTHA1544 (Fig. 2 ▶). The catalytic sites of these proteins and CIB are located in the inserted regions. Ephy consists of two domains: the core domain and the cap domain (Fig. 2 ▶ a). The core domain comprises residues 1–137 and 219–294, which catalyze the cofactor-independent hydrolysis of epoxides. Since TTHA1544 corresponds to the cap domain, comprising residues 138–218, the active-site residues in the core domain are not conserved in TTHA1544.
Figure 2.

Superimposition of the structure of TTHA1544 (red) with those of four other proteins: (a) A. radiobacter AD1 epoxide hydrolase (PDB code 1ehy, blue), (b) murine liver epoxide hydrolase (PDB code 1cr6, grey), (c) Burkholderia sp. FA1 fluoroacetate dehalogenase (PDB code 1y37, cyan) and (d) Rhodococcus sp. MB1 cocaine esterase (PDB code 1ju3, light green), shown in a ribbon presentation. These figures were prepared with the programs MOLSCRIPT (Kraulis, 1991 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶).
In conclusion, the overall structure of TTHA1544 can be described as the minimized α/β-hydrolase fold; an additional component containing catalytic residues would be required for its hydrolase activity.
Supplementary Material
PDB reference: minimized α/β-hydrolase fold protein, 2dst, r2dstsf
Acknowledgments
The authors are grateful to the beamline staff of the Southeast Regional Collaborative Access Team (SER-CAT) at the 22-ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory. Use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. We also thank Drs Kawamoto and Yamamoto for the use of BL26B1 at SPring-8. This study was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI) and the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Argiriadi, M. A., Morisseau, C., Hammock, B. D. & Christianson, D. W. (1999). Proc. Natl Acad. Sci. USA, 96, 10637–10642. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Henne, A. et al. (2004). Nature Biotechnol. 22, 547–553. [DOI] [PubMed]
- Holm, L. & Park, J. (2000). Bioinformatics, 16, 566–567. [DOI] [PubMed]
- Jones, S. & Thornton, J. M. (1996). Proc. Natl Acad. Sci. USA, 93, 13–20.
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst. 24, 946–950.
- Larsen, N. A, Turner, J. M., Stevens, J., Rosser, S. J., Basran, A., Lerner, R. A., Bruce, N. C. & Wilson, I. A. (2002). Nature Struct. Biol. 9, 17–21. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lawrence, M. C. & Bourke, P. (2000). J. Appl. Cryst. 33, 990–991.
- LeMaster, D. M. & Richards, F. M. (1985). Biochemistry, 24, 7263–7268. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol. 277, 505–524. [DOI] [PubMed]
- Nardini, M., Ridder, I. S., Rozeboom, H. J., Kalk, K. H., Rink, R., Janssen, D. B. & Dijkstra, B. W. (1999). J. Biol. Chem. 274, 14579–14586. [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 277, 505–524. [DOI] [PubMed]
- Padmanabhan, B., Kuzuhara, T., Adachi, N. & Horikoshi, M. (2004). J. Biol. Chem. 279, 9615–9624. [DOI] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Vriend, G. (1990). J. Mol. Graph. 8, 52–56. [DOI] [PubMed]
- Yokoyama, S., Hirota, H., Kigawa, T., Yabuki, T., Shirouzu, M., Terada, T., Ito, Y., Matsuo, Y., Kuroda, Y., Nishimura, Y., Kyogoku, Y., Miki, K., Masui, R. & Kuramitsu, S. (2000). Nature Struct. Biol. 7, 943–945. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: minimized α/β-hydrolase fold protein, 2dst, r2dstsf