

I-Dmo-I is a well characterized homing endonuclease from the archaeon Desulfurococcus mobilis. The enzyme was cloned and overexpressed in Escherichia coli. Crystallization experiments of I-Dmo-I in complex with its DNA target in the presence of Ca2+ and Mg2+ yielded crystals that were suitable for X-ray diffraction analysis.
Keywords: homing endonucleases, I-Dmo-I
Abstract
Homing endonucleases are highly specific DNA-cleaving enzymes that recognize long stretches of base pairs. The availability of these enzymes has opened novel perspectives for genome engineering in a wide range of fields, including gene therapy, by taking advantage of the homologous gene-targeting enhancement induced by a double-strand break. I-Dmo-I is a well characterized homing endonuclease from the archaeon Desulfurococcus mobilis. The enzyme was cloned and overexpressed in Escherichia coli. Crystallization experiments of I-Dmo-I in complex with its DNA target in the presence of Ca2+ and Mg2+ yielded crystals that were suitable for X-ray diffraction analysis. The crystals belonged to the monoclinic space group P21, with unit-cell parameters a = 106.75, b = 70.18, c = 106.85 Å, α = γ = 90, β = 119.93°. The self-rotation function and the Matthews coefficient suggested the presence of three protein–DNA complexes per asymmetric unit. The crystals diffracted to a resolution limit of 2.6 Å using synchrotron radiation at the Swiss Light Source (SLS) and the European Synchrotron Radiation Facility (ESRF).
1. Introduction
Homing endonucleases (HEs), also known as meganucleases, produce double-strand breaks in DNA which induce the transposition of mobile intervening sequences, either introns or inteins, containing the endonuclease open reading frame into cognate alleles that lack this sequence, a process known as homing (Belfort & Roberts, 1997 ▶). They are highly sequence-specific enzymes, with recognition sites 12–45 base pairs in length. HEs have a very low frequency of cleavage even in complex genomes. Because of this rare-cutting property, they are very powerful tools for the manipulation of the genomes of mammalian cells and plants (Choulika et al., 1994 ▶, 1995 ▶; Rouet et al., 1994 ▶). The use of meganuclease-induced recombination has long been limited by the repertoire of natural meganucleases.
Sequence homology has been used to classify HEs into four families, the largest of which has the conserved LAGLIDADG sequence motif (Chevalier & Stoddard, 2001 ▶). Homing endonucleases with only one such motif, such as I-CreI (Wang et al., 1997 ▶), function as homodimers. In contrast, larger HEs containing two motifs, such as I-SceI (Jacquier & Dujon, 1985 ▶) or I-Dmo-I (Dalgaard et al., 1993 ▶), are single-chain proteins. The three-dimensional structures of several LAGLIDADG endonucleases (Chevalier et al., 2001 ▶; Duan et al., 1997 ▶; Flick et al., 1997 ▶; Ichiyanagi et al., 2000 ▶; Jurica et al., 1998 ▶; Moure et al., 2003 ▶; Nakayama et al., 2006 ▶; Poland et al., 2000 ▶; Silva et al., 1999 ▶; Spiegel et al., 2006 ▶; Werner et al., 2002 ▶) indicate that these proteins adopt a similar active conformation as homodimers or as monomers with two separate domains. The last acidic residue of the LAGLIDADG motif participates in DNA cleavage by a metal-dependent mechanism of phosphodiester hydrolysis (Chevalier et al., 2001 ▶).
The structures of I-Dmo-I and of its N-terminal domain fused to I-Cre-I bound to a chimeric DNA target have been solved (Silva et al., 1999 ▶; Chevalier et al., 2002 ▶). Owing to its monomeric nature, I-Dmo-I is ideal for the production of tailored specificities in an LAGLIDAG scaffold that could cleave specific DNA sequences. Therefore, owing to the sparse structural information available on the native enzyme in complex with its target DNA, a detailed characterization of the cleavage and DNA-recognition mechanisms of I-Dmo-I is required in order to engineer new specificities using this meganuclease as a scaffold. Here, we report the crystallization of this enzyme in complex with its DNA target in the presence of Ca2+ and Mg2+ as a first step towards solving its structure, which may open up new possibilities for engineering custom specificities using I-Dmo-I as a scaffold.
2. Materials and methods
2.1. Protein expression and purification
E. coli Rosetta(DE3)pLysS cells were transformed with plasmid pET24d(+) containing the I-Dmo-I ORF with a 6×His tag at the C-terminus. His-tagged I-Dmo-I was overexpressed in LB medium at 298 K for 5 h after addition of 0.3 mM IPTG when the OD600 was around 0.6–0.8. Selenomethionine-labelled I-Dmo-I was expressed using the same strain. The cells were collected from a 50 ml overnight culture grown in LB medium containing 30 µg ml−1 kanamycin until OD600 ≃ 1.0; at this point the cells were spun down, washed once with M9 minimal medium and finally resuspended in M9 minimal medium supplemented with thiamine (0.01 mg ml−1), glucose [0.4%(w/v)], CaCl2 (0.0147 mg ml−1), MgSO4 (0.246 mg ml−1) and kanamycin (30 µg ml−1). The culture was shaken at 310 K for 30 min and selenomethionine (50 mg ml−1; Molecular Dimensions) was then added together with lysine hydrochloride, threonine, phenylalanine, leucine, isoleucine and valine as described in Van Duyne et al. (1993 ▶). After an additional 15 min of shaking, protein expression was induced for 5 h at 298 K by the addition of 0.3 mM IPTG. The bacterial pellet was resuspended and the cells were disrupted by sonication in 50 mM sodium phosphate pH 8.0, 300 mM NaCl and 5% glycerol including protease inhibitors (Complete EDTA-free tablets, Roche). The lysate was clarified by centrifugation (20 000g for 1 h).
The supernatant was applied onto a Co2+-loaded HiTrap Chelating HP column (GE Healthcare) and the protein was eluted using an imidazole gradient (0–0.5 M). The fractions containing I-Dmo-I were collected and the pH was adjusted to 6.0. The sample was loaded onto a 5 ml HiTrap Heparin HP column (GE Healthcare) previously equilibrated with 20 mM sodium phosphate pH 6.0. The sample was eluted with a continuous gradient from 0 to 1 M NaCl in 20 mM sodium phosphate pH 6.0 buffer. The purified protein was subsequently concentrated using an Amicon Ultra system equipped with a 10 kDa cutoff filter and loaded onto a PD-10 Desalting column (GE Healthcare) pre-equilibrated with 5 mM Tris–HCl pH 8.0 and 150 mM NaCl. The protein was concentrated to 16 mg ml−1, flash-frozen in liquid nitrogen and stored at 193 K. The protein concentration was determined from the absorbance at 280 nm. The purity of the samples was checked by SDS–PAGE and their homogeneity was evaluated using dynamic light scattering. Finally, the incorporation of selenomethionine was tested by mass spectrometry (data not shown).
2.2. I-Dmo-I–DNA complex formation
The I-Dmo-I target DNA was purchased from Proligo and consisted of two strands of sequence 5′-GCCTTGCCGGGTAAGTTCCGGCGCG-3′ and 5′-CGCGCCGGAACTTACCCGGCAAGGC-3′. The construct forms a 25 bp blunt-end duplex. Because of the stability of I-Dmo-I at high temperature (T m ≃ 363 K, data not shown), the I-Dmo-I–DNA complex was formed after pre-warming the meganuclease and the oligonucleotide samples to 338 K and then mixing them in a 1.5:1 molar ratio (DNA:protein). The mixture was incubated for 50 min and then spun down for 5 min. The supernatant was stored at room temperature to avoid precipitation. To assess the presence of DNA in the complex with I-Dmo-I, the purified complex was analyzed by running a 15% SDS–PAGE and staining first with Coomassie and subsequently with SYBR Safe (Invitrogen; Fig. 1 ▶ a). The same protocol was followed in the presence of 2 mM Ca2+ or Mg2+.
Figure 1.

Formation and crystallization of the I-Dmo-I–DNA target complex. (a) A 15% SDS–PAGE gel of the purified I-Dmo-I–DNA complex was stained with Coomassie (left) and SYBR Safe (right) to detect protein and DNA, respectively. Lane 1, protein molecular-weight markers (kDa); lane 2, soluble I-Dmo-I; lane 3, I-DmoI cocrystallized with DNA in the presence of Ca2+; lane 4, I-DmoI cocrystallized with DNA in the presence of Mg2+; lane 5, dsDNA (25 bp); lane 6, 50 bp ladder (1031–50 bp). (b) I-Dmo-I–DNA complex crystals grown in the presence of Mg2+. (c) I-Dmo-I–DNA complex crystals stained with SYBR Gold, revealing the presence of DNA.
2.3. Crystallization
Crystallization screening was performed immediately after complex formation using a Cartesian MicroSys robot (Genomic Solutions) and the sitting-drop method (96-well MRC plates) with nanodrops of 0.1 µl protein solution plus 0.1 µl reservoir solution and a reservoir volume of 60 µl. The initial screens tested were Crystal Screens I and II, Crystal Screen Cryo and Crystal Screen Lite (Hampton Research), Wizard I and II, Wizard Cryo I and II, Precipitant Synergy Primary, Precipitant Synergy Expanded 67% and Precipitant Synergy Expanded 33% (Emerald BioSystems). The final concentration of I-Dmo-I in the DNA–protein complex solution was 6 mg ml−1. Crystals were obtained in the nanodrops under several conditions (Crystal Screen I conditions 15 and 36, Crystal Screen II conditions 22, 35, 37 and 43, Crystal Screen Cryo conditions 15, 20 and 37, Crystal Screen Lite conditions 18, 28 and 41, Wizard I condition 21, Wizard Cryo I conditions 40 and 47, Wizard Cryo II condition 10, Precipitant Synergy Primary conditions 42 and 52 and Precipitant Synergy Expanded 67% condition 51). These small crystals were collected under Al’s oil and tested for diffraction using synchrotron radiation. The best diffracting crystals were obtained using condition 37 of Hampton Research Crystal Screen Cryo (5.6% PEG 4000, 0.07 M sodium acetate pH 4.6, 30% glycerol). Crystals grown under these conditions were subjected to three cycles of optimization using 24-well Linbro plates with droplets containing 1 µl protein solution plus 1 µl reservoir solution and a reservoir volume of 500 µl. The best crystals were those that grew using 5.6% PEG 4000, 0.07 M sodium acetate with a pH ranging from 4.5 to 5.5 and 30% glycerol (Fig. 1 ▶ b). Plate-shaped clusters (approximately 0.2–0.4 × 0.1 × 0.05 mm) grew in 5–15 d and were easily disrupted into single crystals using an acupuncture needle. Further changes in the crystallization conditions did not lead to single crystals (changes in the PEG and buffer, the use of Hampton Additive Screen and one cycle of seeding were attempted). The presence of DNA in the crystals was confirmed by fluorescent detection using SYBR Gold (Invitrogen; Kettenberger & Cramer, 2006 ▶; Fig. 1 ▶ c). Finally, crystallization trials were performed in the presence of 2 mM CaCl2 or MgCl2.
2.4. Data collection
Crystals were removed directly from the drop and flash-frozen in liquid nitrogen. The crystals were tested in-house using a Bruker FR-591 generator and diffracted to 3.5 Å resolution. Further data sets for the I-Dmo-I–DNA complex were collected using synchrotron radiation at the ID-29 (ESRF) and PX (SLS) beamlines. The diffraction data in Table 1 ▶ were recorded using an ADSC-Q315 detector at ID-29. The best data set was collected using Δϕ = 1° and a wavelength of 0.979 Å. Processing and scaling were accomplished using HKL-2000 (Otwinowski & Minor, 1997 ▶). The statistics of the crystallographic data are summarized in Table 1 ▶.
Table 1. Data-collection statistics of the native I-Dmo-I–DNA crystals grown in 2 mM Mg2+ .
| Resolution limits (Å) | 〈I/σ(I)〉 | Completeness (%) | Multiplicity | Observed reflections | Unique reflections | Rsym† |
|---|---|---|---|---|---|---|
| 50.0–8.22 | 27.9 | 94.0 | 2.8 | 3697 | 1329 | 0.021 |
| 8.22–5.81 | 19.2 | 98.7 | 2.7 | 6782 | 2475 | 0.032 |
| 5.81–4.75 | 15.7 | 99.5 | 2.8 | 9102 | 3196 | 0.038 |
| 4.75–4.11 | 19.4 | 99.8 | 2.9 | 10912 | 3479 | 0.031 |
| 4.11–3.68 | 19.3 | 99.8 | 2.9 | 12542 | 4279 | 0.035 |
| 3.68–3.36 | 17.7 | 99.9 | 3.0 | 13892 | 4721 | 0.038 |
| 3.36–3.11 | 12.9 | 99.8 | 2.9 | 15187 | 5144 | 0.055 |
| 3.11–2.91 | 6.9 | 99.7 | 2.7 | 15985 | 5455 | 0.106 |
| 2.91–2.74 | 4.6 | 97.2 | 2.4 | 15516 | 5676 | 0.160 |
| 2.74–2.60 | 3.8 | 77.2 | 2.4 | 11598 | 4758 | 0.196 |
| Overall | 14.4 | 95.9 | 2.8 | 115213 | 40782 | 0.043 |
R
sym = 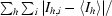
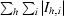 .
.
3. Results and discussion
The recombinant I-Dmo-I construct used in this work is 200 residues in length with a theoretical molecular weight of 23 353 Da. However, the purified protein has a measured molecular weight of 23 221 Da, indicating that the initial methionine was processed during expression. This construct differs from the native I-Dmo-I protein in that it contains an extra Ala residue at the C-terminus, lacks the last six residues at the C-terminus and incorporates the sequence AAALEHHHHHH at the C-terminal end for affinity purification. This enzyme is active (Epinat et al., 2003 ▶) and none of the missing residues were observed in the X-ray structure of the free enzyme (Silva et al., 1999 ▶). The enzyme was expressed in E. coli with typical yields of 7 mg pure protein per litre of culture. The recombinant protein was subjected to His-tag and heparin affinity purification and a final gel-filtration purification step. The purified I-Dmo-I was concentrated and used for crystallization assays. Initial hits were clusters of crystals (Fig. 1 ▶ b) and several rounds of refinement did not yield single crystals. Therefore, the clusters were disrupted with the help of an acupuncture needle to obtain single crystals that were suitable in size and quality for diffraction experiments (Fig. 1 ▶ c).
Initial diffraction tests were performed using an in-house source, but diffraction data could only be recorded to 3.5 Å resolution (data not shown). Therefore, the crystals were tested using a synchrotron-radiation source in order to obtain higher resolution data. Several high-resolution native data sets were collected at 100 K at the PX beamline (SLS, Villigen) and multiple anomalous dispersion (MAD) data sets at the Se K edge were collected at the ID-29 beamline (ESRF, Grenoble). On these undulator-equipped beamlines the crystals diffracted to a maximum resolution of 1.9 Å (at the SLS). The statistics for the data set collected to 2.6 Å on ID-29 are given in Table 1 ▶.
The crystals belong to the monoclinic space group P21, with unit-cell parameters a = 106.75, b = 70.18, c = 106.85 Å, α = γ = 90, β = 119.93°. The Matthews coefficient (V M = 3.00 Å3 Da−1) and the self-rotation function (data not shown) suggested the presence of three protein–DNA complexes per asymmetric unit and a solvent content of 60%. The collected diffraction data (Fig. 2 ▶) were 95.4% complete, with a multiplicity of 2.8 and an overall I/σ(I) of 14.4 (see Table 1 ▶ for details). Crystals of the selenomethionine-derived protein were used to collect data in order to solve the structure using multiple anomalous dispersion at the Se K edge.
Figure 2.

Diffraction pattern from the native crystals using synchrotron radiation at the PX beamline (SLS). Circles are labelled with the resolution limits in angstroms.
These are the first crystals of I-Dmo-I in complex with DNA. We believe that these studies will help to elucidate the molecular mechanisms of DNA recognition and the cleavage mechanisms of this meganuclease. These findings will facilitate the use of I-Dmo-I as a new scaffold that should help in the production of intelligent molecular scalpels that recognize and substitute certain DNA sequences.
Acknowledgments
We would like to thank CELLECTIS SA for the cDNA coding for the I-Dmo-I gene and Inés Muñoz and the ESRF and SLS biocrystallography beamline personnel for help with crystallization and data collection. Financial support was obtained through the Megatools EU project (LSHG-CT-2006-037226).
References
- Belfort, M. & Roberts, R. J. (1997). Nucleic Acids Res.25, 3379–3388. [DOI] [PMC free article] [PubMed]
- Chevalier, B. S., Kortemme, T., Chadsey, M. S., Baker, D., Monnat, R. J. & Stoddard, B. L. (2002). Mol. Cell, 10, 895–905. [DOI] [PubMed]
- Chevalier, B. S., Monnat, R. J. Jr & Stoddard, B. L. (2001). Nature Struct. Biol.8, 312–316. [DOI] [PubMed]
- Chevalier, B. S. & Stoddard, B. L. (2001). Nucleic Acids Res.29, 3757–3774. [DOI] [PMC free article] [PubMed]
- Choulika, A., Perrin, A., Dujon, B. & Nicolas, J. F. (1994). C. R. Acad. Sci. III, 317, 1013–1019. [PubMed]
- Choulika, A., Perrin, A., Dujon, B. & Nicolas, J. F. (1995). Mol. Cell. Biol.15, 1968–1973. [DOI] [PMC free article] [PubMed]
- Dalgaard, J. Z., Garrett, R. A. & Belfort, M. (1993). Proc. Natl Acad. Sci. USA, 90, 5414–5417. [DOI] [PMC free article] [PubMed]
- Duan, X., Gimble, F. S. & Quiocho, F. A. (1997). Cell, 89, 555–564. [DOI] [PubMed]
- Epinat, J. C., Arnould, S., Chames, P., Rochaix, P., Desfontaines, D., Puzin, C., Patin, A., Zanghellini, A., Paques, F. & Lacroix, E. (2003). Nucleic Acids Res.31, 2952–2962. [DOI] [PMC free article] [PubMed]
- Flick, K. E., McHugh, D., Heath, J. D., Stephens, K. M., Monnat, R. J. Jr & Stoddard, B. L. (1997). Protein Sci.6, 2677–2680. [DOI] [PMC free article] [PubMed]
- Ichiyanagi, K., Ishino, Y., Ariyoshi, M., Komori, K. & Morikawa, K. (2000). J. Mol. Biol.300, 889–901. [DOI] [PubMed]
- Jacquier, A. & Dujon, B. (1985). Cell, 41, 383–394. [DOI] [PubMed]
- Jurica, M. S., Monnat, R. J. Jr. & Stoddard, B. L. (1998). Mol. Cell, 2, 469–476. [DOI] [PubMed]
- Kettenberger, H. & Cramer, P. (2006). Acta Cryst. D62, 146–150. [DOI] [PubMed]
- Moure, C. M., Gimble, F. S. & Quiocho, F. A. (2003). J. Mol. Biol.334, 685–695. [DOI] [PubMed]
- Nakayama, H., Shimamura, T., Imagawa, T., Shirai, N., Itoh, T., Sako, Y., Miyano, M., Sakuraba, H., Ohshima, T., Nomura, N. & Tsuge, H. (2006). J. Mol. Biol.365, 362–378. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Poland, B. W., Xu, M. Q. & Quiocho, F. A. (2000). J. Biol. Chem.275, 16408–16413. [DOI] [PubMed]
- Rouet, P., Smih, F. & Jasin, M. (1994). Proc. Natl Acad. Sci. USA, 91, 6064–6068. [DOI] [PMC free article] [PubMed]
- Silva, G. H., Dalgaard, J. Z., Belfort, M. & Van Roey, P. (1999). J. Mol. Biol.286, 1123–1136. [DOI] [PubMed]
- Spiegel, P. C., Chevalier, B., Sussman, D., Turmel, M., Lemieux, C. & Stoddard, B. L. (2006). Structure, 14, 869–880. [DOI] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol.229, 105–124. [DOI] [PubMed]
- Wang, J., Kim, H. H., Yuan, X. & Herrin, D. L. (1997). Nucleic Acids Res.25, 3767–3776. [DOI] [PMC free article] [PubMed]
- Werner, E., Wende, W., Pingoud, A. & Heinemann, U. (2002). Nucleic Acids Res.30, 3962–3971. [DOI] [PMC free article] [PubMed]