

The crystal structure of a light-harvesting protein that interacts with photosystem II is reported.
Keywords: photosynthesis, light-harvesting proteins
Abstract
Cyanobacteria and red algae use light-harvesting pigments bound by proteins to capture solar radiation and to channel excitation energy into their reaction centres. In most cyanobacteria, a multi-megadalton soluble structure known as the phycobilisome is a major light-harvesting system. Allophycocyanin is the main component of the phycobilisome core, forming a link between the rest of the phycobilisome and the reaction-centre core. The crystal structure of allophycocyanin from Thermosynechococcus elongatus (TeAPC) has been determined and refined at 3.5 Å resolution to a crystallographic R value of 26.0% (R free = 28.5%). The structure was solved by molecular replacement using the allophycocyanin structure from Spirulina platensis as the search model. The asymmetric unit contains an (αβ) monomer which is expanded by symmetry to a crystallographic trimer.
1. Introduction
The thermophilic cyanobacterium Thermosynechococcus elongatus has emerged as the model organism of choice for structural studies of photosynthetic complexes. It has yielded atomic models of photosystem I (PSI; Jordan et al., 2001 ▶) and photosystem II (Ferreira et al., 2004 ▶; Loll et al., 2005 ▶). A crystal structure of the cytochrome bf complex has been determined from another thermophilic cyanobacterium, Mastigocladus laminosus (Kurisu et al., 2003 ▶). As with most cyanobacteria and red algae, T. elongatus contains an extrinsic light-harvesting structure known as the phycobilisome (PBS; reviewed by Adir, 2005 ▶). Although phycobilisomes are diverse, they follow a basic morphology. Phycobiliproteins (PBPs) form (α/β)6 discs. A core of short stacks of discs is associated with the reaction centres, from which radiate rods of stacked discs. The structure is held together by linker polypeptides, which link cylinders into rods, the core to the rods and rods to rods (Krogmann et al., 2007 ▶).
The PBS core is composed mainly of allophycocyanin (APC) and in T. elongatus the rods contain phycocyanin (PC). Energy absorbed by the pigments in PC is transferred by nonradiative transfer into APC and from there into chlorophyll a, with an efficiency approaching 100%. In the absence of the photosynthetic reaction centre (RC), the PBPs are strongly fluorescent. In both PC and APC, phycocyanobilin, an open-chain tetrapyrrole pigment, is covalently bound to the protein matrix. The pigments in phycobiliproteins absorb light in the visible parts of the spectrum, where the absorbance of chlorophyll a is low. This enables the organisms to use more of the available solar spectrum for photosynthesis.
The crystal structure of phycocyanin from T. elongatus has been reported by Nield et al. (2003 ▶). We now report the structure of allophycocyanin (APC) from T. elongatus, thus providing structural models of both major PBPs from this organism. There are three other published APC structures: those from Spirulina platensis (Brejc et al., 1995 ▶), Porphyra yezoensis (Liu et al., 1999 ▶) and M. laminosus (Reuter et al., 1999 ▶).
2. Materials and methods
2.1. Purification and crystallization
T. elongatus was grown in 20 l carboys at 328 K to an OD of 1.5 at 680 nm in medium D (Castenholz, 1969 ▶) while bubbled with 5% CO2. Cells were harvested by concentration using a Sartorius Sartocon membrane concentrator system followed by centrifugation at 11 700g. The cells were washed in buffer 1 [40 mM MES pH 6.5, 15 mM MgCl2, 15 mM CaCl2, 1.2 M betaine, 10%(v/v) glycerol] before being broken by two passages through a French pressure cell (cooled on ice prior to usage) at 40 MPa in buffer 1 containing 0.2%(w/v) BSA, 50 mg l−1 DNAse I, 1 mM aminocaproic acid and 1 mM benzamidine. Unbroken cells were removed by spinning at 1000g for 5 min. The lysate was spun at 180 000g for 20 min to pellet the thylakoid membranes. The pellet was resuspended in buffer 1 and respun at 180 000g for 20 min twice. The supernatants from these steps, the ‘thylakoid wash’, were used as a source of APC.
The thylakoid washes were pooled and adjusted to pH 9 with 1 M Tris buffer and then diluted fourfold to reduce the ionic strength. The diluted wash was loaded onto a column of Toyopearl DEAE-650S (Tosoh Bioscience) anion-exchange resin of bed volume 50 ml, which was then washed with buffer A (20 mM Tris pH 9). TeAPC was eluted with a linear gradient of NaCl in buffer A to 160 mM NaCl. The TeAPC eluted after the more abundant phycocyanin and was detected by its absorption maximum at 652 nm, compared with 620 nm for phycocyanin. The APC fractions were pooled and concentrated to 10 mg ml−1 protein using a Vivaspin centrifugal concentrator with a nominal 30 kDa molecular-weight cutoff.
TeAPC was screened for crystallization using hanging-drop vapour diffusion against a sparse-matrix screen (Jancarik & Kim, 1991 ▶) and hits were then optimized. Several conditions in the screen gave hexagonal plate crystals of up to several hundred micrometres across but only a few micrometres thick. The crystal used for structure determination grew in 1 M ammonium sulfate mixed in an equal volume with the protein solution and reached its final size after a few days. Typical crystals are shown in Fig. 1 ▶. The crystal was cryoprotected in its mother liquor with 40%(v/v) glycerol added and was then flash-cooled in liquid nitrogen.
Figure 1.

Crystals of T. elongatus allophycocyanin belonging to space group R32. The crystal dimensions are ∼400 × 400 × 10 µm.
2.2. Data collection, structure solution and refinement
Data were collected on beamline X06S at the Swiss Light Source and were reduced using MOSFLM (Leslie, 1992 ▶), SCALA (Evans, 1993 ▶) and programs from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The diffraction images were indexed using LABELIT (Sauter et al., 2004 ▶). The data were integrated in space group R32 to a resolution of 3.5 Å. Data-collection statistics are shown in Table 1 ▶.
Table 1. Data-collection and refinement statistics for TeAPC.
Values in parentheses are for the outer resolution shell. 〈I〉/〈σ(I)〉 fell to 2 at 4.2 Å resolution.
| PDB code | 2v8a |
| Data collection | |
| Beamline | SLS X06S |
| Detector | MAR CCD |
| Wavelength (Å) | 0.90500 |
| Space group | R32 |
| Unit-cell parameters (Å, °) | a = b = 102.18, c = 193.98, α = β = 90, γ = 120 |
| Resolution (Å) | 43.15–3.50 (3.69–3.50) |
| Total No. of observations | 22898 (3371) |
| Unique observations | 5071 (719) |
| Multiplicity | 4.5 (4.7) |
| Completeness (%) | 99.5 (99.9) |
| Wilson B (Å2) | 53 |
| 〈I/σ(I)〉 | 7.2 (3.4) |
| Rmerge† | 0.22 (0.37) |
| 〈I〉/〈σ(I)〉 | 2.7 (1.7) |
| Refinement | |
| Rcryst/Rfree‡ (%) | 26.0/28.5 |
| R.m.s. deviation from ideal values | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.4 |
| Dihedrals (°) | 3.4 |
| Mean B value (Å2) | |
| All atoms | 44.5 |
| Main chain | 44.4 |
| Side chain | 44.7 |
R
merge = 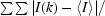
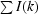 , where I(k) and 〈I〉 represent the kth observation of I and the mean value, respectively.
, where I(k) and 〈I〉 represent the kth observation of I and the mean value, respectively.
R
cryst = 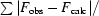
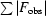 ; R
free is R
cryst for the 5% cross-validation test set.
; R
free is R
cryst for the 5% cross-validation test set.
The 2.3 Å structure of APC from S. platensis was used as the search model (Brejc et al., 1995 ▶). An (αβ) monomer was found using Phaser (McCoy et al., 2007 ▶). The sequence was mutated to the T. elongatus sequence using Coot (Emsley & Cowtan, 2004 ▶) and then subjected to rounds of rebuilding and refinement in Coot and REFMAC (Murshudov et al., 1997 ▶), respectively. 5% of the reflections were removed from the refinement and used for cross-validation (Brünger, 1992 ▶). The final R value was 26.0% (R free = 28.5%); other refinement statistics are shown in Table 1 ▶.
The structure was validated with MOLPROBITY (Lovell et al., 2003 ▶). Structures were superposed using secondary-structure matching (Krissinel & Henrick, 2004 ▶). Hexamers of TeAPC were constructed using the P. yezoensis APC crystallographic hexamers as a model and the core model was constructed using the crystal lattice as a model for the packing of hexamers. Buried surface areas were calculated using the program AREAIMOL (Lee & Richards, 1971 ▶). Electron density and atomic models were visualized using PyMOL (DeLano, 2002 ▶).
3. Results and discussion
3.1. Overall structure
The asymmetric unit contains an (α/β) monomer which forms crystallographic trimers. The trimers form sheets which stack to form sheets of offset trimers. As expected (Liu et al., 1999 ▶), TeAPC adopts the classic phycobiliprotein fold, which is a globin fold similar to that of haemoglobin.
All but residue Thr74 of the β subunit fall into the allowed region of the Ramachandran plot, as defined by MOLPROBITY (Lovell et al., 2003 ▶). This residue is in a loop region and is also a Ramachandran outlier in all phycobiliprotein structures currently (October, 2007) deposited in the PDB. In the phycocyanin of the primitive cyanobacterium Gloeobacter violaceus, the equivalent residue is a proline (Nakamura et al., 2003 ▶), suggesting a different solution to the strain problem, as proline has a different backbone conformation to the other amino acids. In all the deposited structures including that presented here, the backbone N atom of Thr74 makes a hydrogen bond to an O atom on a pigment on an adjacent α subunit (shown in Fig. 2 ▶). Therefore, it seems likely that in this case specific requirements of the protein fold and ligand interaction override the usual Ramachandran constraints. Asn71 in the β chain was modelled as γ-N-methylasparagine. This post-translational modification (Klotz et al., 1986 ▶; Klotz & Glazer, 1987 ▶) has only been found in phycobiliproteins and is associated with the β allophycocyanin subunit in all species so far examined.
Figure 2.

A 2F o − F c, σA-weighted electron-density map of a model perturbed by shifts in each atom of up to 0.2 Å with the chromophore omitted. This map should show an unbiased view of the chromophore. The map is contoured at 1.2σ around Cys84 of the α subunit and the covalently attached phycocyanobilin chromophore (in blue) and the Thr74 of chain B (in green), with the hydrogen bond indicated between the chromophore and the backbone N atom of Thr74.
The resolution of the data is rather low and so is the overall 〈I〉/〈σ(I)〉, although the multiplicity of the data improves the 〈I/σ(I)〉 somewhat (Table 1 ▶). However, the search model used was from a highly similar protein solved at much better resolution (2.3 Å). Therefore, after refinement using tight restraints and using rotamer side-chain positions, the quality indicators are good. There is only one bad rotamer and a low clash score (14.7) in the model (Lovell et al., 2003 ▶). An omit map of the pigment attached to the α subunit is shown in Fig. 2 ▶.
3.2. Crystal packing and oligomeric state
In the phycobilisome, it is thought that the phycobiliproteins are present as (α/β)6 hexamers. This is supported by a recent reconstruction of the entire phycobilisome by electron microscopy (Yi et al., 2005 ▶). These hexamers are held together with linker peptides. In solution, the hexamers dissociate to monomers or trimers. However, most phycobiliprotein structures contain hexamers in the crystal, which may be generated by combinations of crystallographic and noncrystallographic symmetry operators.
In this crystal form, the trimers are in sheets of alternating direction, such that the ‘top’ surfaces composed of β chains are in contact, as are the ‘bottom’ surfaces composed of α chains. A view of the crystal lattice is shown in Fig. 3 ▶. Hexamers are not present, unlike in the published structure of APC from the red algae P. yezoensis. It is likely that the association into hexamers in that crystal form is a consequence of crystallization, the hexameric disc being thermodynamically favoured under supersaturating conditions. The other two published APC structures, which are from cyanobacteria, contain a trimer and a trimer–linker complex, respectively (Brejc et al., 1995 ▶; Reuter et al., 1999 ▶).
Figure 3.

View of the crystal packing of TeAPC, showing two sheets of trimers. In the back sheet α subunits are magenta and β subunits are cyan; in the front sheet α subunits are red and β subunits are blue. (a) View down the trimer axis; (b) view perpendicular to the trimer axis. In the physiological hexamer, the α subunits are on the inside and the β subunits are on the outside.
In the (α/β) monomer present in the asymmetric unit of TeAPC deposited in the PDB, the α–β interface buries 1465 Å2. The interface between α and β′, where β′ is the crystallographically related β subunit in the same trimer, buries 720 Å2. The trimer–hexamer interface, which is actually formed by (α) units, buries 2645 Å2, which is 882 Å2 per monomer. Therefore, a trimer buries 6555 Å2 of surface, in comparison with 2645 Å2 for the trimer–hexamer interface. If the binding energy is related positively to the buried area, then this could explain why the trimer is more stable than the hexamer in the absence of the linkers and is the form present in the crystal. More speculatively, the P. yezoensis APC crystal lattice contains sheets of hexamers, of which a trimer of hexamers can be taken as a model of part of the phycobilisome core; such a model is shown in Fig. 5. This is different to the approach taken by Yi et al. (2005 ▶) in their phycobilisome core model, in which hexamers of APC were manually placed to form models of the APC core. If our model based on the crystal packing of the P. yezoensis APC is correct, three APC hexamers bury 1772 Å2 relative to a single hexamer. This relatively small interaction area for such a large complex will be supplemented in the phycobilisome in vivo with strong specific interactions from peptide linkers.
3.3. Relationship to the other APC structures
The individual subunits are highly conserved; the red algae sequences are very similar to those of cyanobacteria. The α and β subunits have appeared by duplication of an ancestral gene, with only a single amino-acid sequence insertion between the α and β subunits. An alignment of the APC sequences from the published structures is shown in Fig. 4 ▶ and the r.m.s.d. over Cα atoms is shown in Table 2 ▶. The TeAPC structure is essentially identical within error to the other structures.
Figure 4.

An alignment of the α and β APC sequences from the published structures, produced with ClustalX (Thompson et al., 1997 ▶) and TeXshade (Beitz, 2000 ▶). Amino-acid residues are coloured by type; putative γ-methylasparagine residues are shaded red, the conserved Ramachandran outlier Thr74 is shaded cyan and the cysteine residues to which the chromophores are attached are shaded yellow. α-Helical regions of the two subunits of the T. elongatus structure are shown as grey boxes. There is 32% identity between all subunits shown; the α subunits share 78% identity and the β subunits 88%.
Table 2. Pairwise r.m.s.d. values in Å for Cα atoms for the α and β subunits between the three published APC structures and the T. elongatus APC structure.
The extreme conservation of the sequence and hence the structure is likely to be a consequence of the many constraints on the structure. The APC peptide chains must associate to form monomers, trimers and hexamers, which must in turn interact with linker peptides, other hexamers and the RC core. APC must also covalently bind the chromophores and interact specifically with the enzymes that ligate the chromophore to the protein. Therefore, there are many constraints on both the specific surfaces of the protein and the overall shape. It is notable that the β subunit responsible for the hexamer–hexamer rod contacts is even more conserved than the α subunit.
3.4. Interaction with photosystem II
Barber et al. (2003 ▶) argued that the APC core interacted with PSII at a region identified in an electron-microscopy projection map as having a lower electron density than the surrounding regions. In the light of subsequent atomic models of PSII, this low electron-density region is identified as a cavity linking the stromal surface to the Qb site, a feature commented on by Loll et al. (2005 ▶) and Murray & Barber (2007 ▶). It is possible that this cavity provides a site for phycobilisome binding. PSII is a dimeric complex and the two cavities are about 110 Å apart, which is approximately the same distance as the two tips of the basal APC hexamers in the APC core model shown in Fig. 5 ▶. This is therefore the optimum match for an interaction between this cavity and linker peptides extending from the APC core. In the future it is hoped that this PSII–phycobilisome core complex can be isolated and subjected to single-particle reconstruction using electron microscopy. In this event, the atomic models could be fitted into the resulting electron-density maps, thus leading to a more detailed structural understanding of how the APC core interacts with PSII.
Figure 5.

A manually constructed model of the PSII reaction centre (Ferreira et al., 2004 ▶) and APC core complex.
Supplementary Material
PDB reference: allophycocyanin, 2v8a, r2v8asf
Acknowledgments
We acknowledge the data-collection facilities and staff of the Swiss Light Source and the financial support of the Biotechnology and Biological Sciences Research Council.
References
- Adir, N. (2005). Photosynth. Res.85, 15–32. [DOI] [PubMed]
- Barber, J., Morris, E. P. & da Fonseca, P. C. (2003). Photochem. Photobiol. Sci.2, 536–541. [DOI] [PubMed]
- Beitz, E. (2000). Bioinformatics, 16, 135–139. [DOI] [PubMed]
- Brejc, K., Ficner, R., Huber, R. & Steinbacher, S. (1995). J. Mol. Biol.249, 424–440. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Castenholz, R. W. (1969). Bacteriol. Rev.33, 476–504. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122.
- Ferreira, K. N., Iverson, T. M., Maghlaoui, K., Barber, J. & Iwata, S. (2004). Science, 303, 1831–1838. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W. & Krauss, N. (2001). Nature (London), 411, 909–917. [DOI] [PubMed]
- Klotz, A. V. & Glazer, A. N. (1987). J. Biol. Chem.262, 17350–17355. [PubMed]
- Klotz, A. V., Leary, J. A. & Glazer, A. N. (1986). J. Biol. Chem.261, 15891–15894. [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Krogmann, D., Pérez-Gómez, B., Gutiérrez-Cirlos, E., Chagolla-López, A., González de la Vara, L. & Gómez-Lojero, C. (2007). Photosynth. Res.93, 27–43. [DOI] [PubMed]
- Kurisu, G., Zhang, H., Smith, J. L. & Cramer, W. A. (2003). Science, 302, 1009–1014. [DOI] [PubMed]
- Lee, B. & Richards, F. M. (1971). J. Mol. Biol.55, 379–400. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Liu, J. Y., Jiang, T., Zhang, J. P. & Liang, D. C. (1999). J. Biol. Chem.274, 16945–16952. [DOI] [PubMed]
- Loll, B., Kern, J., Saenger, W., Zouni, A. & Biesiadka, J. (2005). Nature (London), 438, 1040–1044. [DOI] [PubMed]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Murray, J. W. & Barber, J. (2007). J. Struct. Biol.159, 228–237. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nakamura, Y. et al. (2003). DNA Res.10, 137–145. [DOI] [PubMed]
- Nield, J., Rizkallah, P. J., Barber, J. & Chayen, N. E. (2003). J. Struct. Biol.141, 149–155. [DOI] [PubMed]
- Reuter, W., Wiegand, G., Huber, R. & Than, M. E. (1999). Proc. Natl Acad. Sci. USA, 96, 1363–1368. [DOI] [PMC free article] [PubMed]
- Sauter, N. K., Grosse-Kunstleve, R. W. & Adams, P. D. (2004). J. Appl. Cryst.37, 399–409. [DOI] [PMC free article] [PubMed]
- Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. (1997). Nucleic Acids Res.25, 4876–4882. [DOI] [PMC free article] [PubMed]
- Yi, Z. W., Huang, H., Kuang, T. Y. & Sui, S. F. (2005). FEBS Lett.579, 3569–3573. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: allophycocyanin, 2v8a, r2v8asf