

Abstract
In vitro selection experiments have produced nucleic acid ligands (aptamers) that bind tightly and specifically to a great variety of target biomolecules. The utility of aptamers is often limited by their vulnerability to nucleases present in biological materials. One way to circumvent this problem is to select an aptamer that binds the enantiomer of the target, then synthesize the enantiomer of the aptamer as a nuclease-insensitive ligand of the normal target. We have so identified a mirror-image single-stranded DNA that binds the peptide hormone vasopressin and have demonstrated its stability to nucleases and its bioactivity as a vasopressin antagonist in cell culture.
The enantiomer of an autogenously folding macromolecule will assume the mirror-image structure of the parent molecule, as has been documented for proteins and nucleic acids (1, 2). The corollary that the enantiomers of two complex-forming molecules will interact identically also has been demonstrated, for example by inverting all chiral centers in both an enzyme and its substrate (3). The principle has application to a problem in biotechnology, namely, that the peptide or nucleic acid ligands generated by phage display or in vitro selection are susceptible to enzymatic degradation; for example, a DNA oligonucleotide injected i.v. is degraded with a half-life of ≈5 min (4–7). The principle of chiral inversion can be coupled to the powerful reiterable selection methods to generate ligands that are insusceptible to degradative enzymes. First, a peptide or nucleic acid ligand is generated that binds the synthetic enantiomer of the target molecule. Then, the enantiomer of the selected ligand is synthesized from d amino acids or l nucleotides. The resulting reflected ligand will then bind and possibly inhibit the target molecule (Fig. 1). This “selection–reflection” strategy has been used recently in the identification of a d peptide ligand of a Src homology 3 domain (8) and l-RNA ligands of arginine and adenosine (9, 10).
Figure 1.

Selection–reflection. A bimolecular complex is revealed by in vitro selection. Inversion of all chiral centers in both partners yields a symmetrical complex of biotechnological interest, in which a biological target is bound by a ligand insusceptible to enzymatic degradation.
In our study, the vertebrate hormone arginine vasopressin (VP), a 9-residue cyclic l peptide, was chosen as a target for its ease of bioassay, its ease of synthesis in all-d form, and its cyclic structure that restricts conformational degrees of freedom relative to a linear peptide and may thus allow selection of a tighter binding ligand. A stable ligand of VP could be useful as a diagnostic reagent or as a therapeutic antagonist in diseases associated with excessive levels of VP in blood or cerebrospinal fluid. DNA was chosen as ligand material because much higher pool diversity can be achieved than for peptide selection, and the chemical stability of DNA is superior to that of RNA.
We have identified a mirror image DNA that binds VP and have shown that this ligand is bioactive, specifically antagonizing the VP response of cultured kidney cells. Selection–reflection thus joins the approach of using modified RNA polymerase substrates (11–14) as a viable means of coupling the power of in vitro selection to the production of stable bioactive ligands.
MATERIALS AND METHODS
Peptides.
d-VP was synthesized by conventional fluorenylmethoxycarbonyl chemistry from d amino acids. Aliquots were biotinylated or radiolabeled at the NH2 terminus while on the resin, using the N-hydroxysuccinimide–long chain–biotin reagent (Pierce) or [3H]-acetic anhydride (DuPont/NEN). After cleavage from the resin, peptides were air-oxidized (incubation with shaking for 3 days at 0.5 mg/ml in 0.1 M Tris⋅HCl, pH 8) and purified by reverse-phase HPLC. The identity of each purified peptide was confirmed by mass spectrometry. Affinity resin was prepared by incubating streptavidin–agarose (Pierce or Sigma) with biotinylated d-VP (≈100 nmol d-VP per milliliter of packed resin). l-VP and oxytocin were from Sigma; [3H]-l-VP was from DuPont/NEN.
DNAs.
All DNAs were purified by gel electrophoresis and denatured and renatured before use by incubation in H2O for 5 min at 70° and then 30 min at room temperature. The four l-deoxynucleoside phosphoramidites and l-dA resin were custom-synthesized by Chem-Genes (Waltham, MA). Because it would not otherwise be a substrate for T4 polynucleotide kinase (15), one preparation of l-aptamer contained a 5′ tail of two d-thymidine residues; this modification had no detectable effect on binding activity. The control l-DNA differed from the l-DNA aptamer in two segments, which had reversed 5′ to 3′ polarity [sequence 5′-T-(54–26)-(19–25)-(18–2)-A; numbers refer to segments of the aptamer of Fig. 2F]. l-aptamer was salvaged from equilibrium dialysis samples by 10-min incubation in 10 mM DTT, five serial phenol extractions, and two ethanol precipitations.
Figure 2.

A DNA motif that binds d-VP. Paired segments (orange and pink) enclose a large asymmetric internal loop (yellow), each position of which exhibits marked base preference. (A and B) The two sequences found in the final pool of the first selection experiment, aligned by large regions of sequence identity (capital letters). The central 60 positions of the initial pool were randomized; underlined sequence, serving in PCR primer binding, did not vary. A limited truncation study showed that variants lacking the boxed 3′ regions were fully active in d-VP chromatography; derivatives of sequence A lacking either 18 nt from the 5′ end or 26 nt from the 3′ end were inactive. (C and D) Aptamer variants isolated in a second selection experiment. Positions originally doped at 30% are shown aligned to the parental 68-nt segment from sequence B. Bases differing from the parental sequence are boxed. Dots mark positions where the parental base was retained in at least 35 of the 41 variants [P, the probability (binomial distribution tail) that a neutral base would be retained at this level, is 0.019]. Nearly half (43%) of the variation at these 31 conserved positions occurred in the three sequences of D. D sequences have at least nine changes from the parental base at these positions (no sequence of C has more than three such changes) and were inactive in d-VP chromatography. Coloring of boxed changes within the proposed pairing segments distinguishes whether the change would result in Watson–Crick (green), wobble (blue), or no (red) pairing; this color coding also denotes effects on pairing status of proposed partner positions that retain the parental base. It is useful to note that the ratio of changes forming new Watson–Crick pairs (green) to those disrupting pairing (red) would be 3:40 at a 30% doped Watson–Crick pair before selection. (E) Secondary structure model of sequence B. Small circles indicate putative GA:GA dinucleotide pairing. Dashes indicate where Watson–Crick or wobble pairing is maintained in ≥30 of the 38 C sequences (P = 0.011 for a parental Watson–Crick pair); dashes are colored green at very highly conserved pairs (pairing maintained in ≥33 sequences; P = 0.00031 for parental Watson–Crick pair). Thickening of dashes indicates pairs where nonparental Watson–Crick pairs occurred at significantly high levels [P ≤ 0.012 (see Materials and Methods)]. (F) Aptamer synthesized in both enantiomeric forms for subsequent study.
Aptamer Selection.
A 96-nt single-stranded DNA pool was synthesized with the sequence 5′-TCTAACGTCAATGATAGA-N60-TTAACTTATTCGACCAAA [where N is the position added from a mixture of phosphoramidites (20.2 mM A, 20.3 mM C, 20.0 mM G, and 16.2 mM T) empirically formulated to give near equal representation of each base]. Because previously described DNA aptamers are G-rich (16–18), a second pool of the same sequence was synthesized with the relative concentration of G phosphoramidite in the N mixture raised by 20%. The copyable fraction of each pool (0.32 and 0.37, respectively) was measured by extension of trace- radiolabeled, negative-strand primer in the conditions of a single PCR cycle. Portions (5 × 1015 copyable molecules) of each pool were mixed, and a small fraction was radiolabeled and added back. This starting pool was denatured and renatured, brought to 10 ml of buffer A (20 mM sodium 2-[bis(2-hydroxyethyl)amino]ethane sulfonate, pH 7.3/140 mM NaCl/5 mM KCl/5 mM MgCl2/1 mM CaCl2/0.02% Triton X-100), and loaded onto 2 ml of d-VP agarose. The column was washed with 11 ml of buffer A and eluted with three loadings of 2 ml of 0.5 mM d-VP in buffer A; there were 15-min waiting periods after each loading, followed by 4 ml of buffer A. The recovered material was amplified by PCR with a negative-strand primer-terminator as described (19), and the 96-nt positive strands were separated from the 118-nt negative strands in a denaturing gel. Twelve additional cycles were performed and scaled down to the use of a 0.1-ml d-VP column using a 0.1-ml streptavidin–agarose precolumn. The pool was cloned using a T vector cloning kit (Novagen).
The starting pool for the second selection had 68 nt from sequence B (Fig. 2) doped at 30% and flanked by AGGCTAGGAAGATCAATT and TTAACTTATTCGACCAAA. The four doping solutions were prepared from 86 mM A, 83 mM C, 96 mM G, and 67 mM T phosphoramidite stocks (estimation of concentrations yielding equal base representation had been slightly revised since synthesis of the first pool) by mixing seven equivalents of one stock with one equivalent of each of the other three stocks. The pool (1015 copyable molecules) was subjected to a first cycle as above on a 0.5-ml d-VP column and then to three more cycles without precolumns using buffer B (like buffer A but with 1.5 mM MgCl2), after which the pool was cloned.
Base Pairing Analysis.
For the 38 aligned sequences of Fig. 2C, base frequencies (Ai, Ci, Gi, and Ti) at each doped sequence position i were determined. Base pairing potential was assessed for all 2278 pair-wise combinations of these positions by computer analysis. For each test couple ij, (i) Wij, the number of sequences with nonparental Watson–Crick couples, was determined, (ii) Fij, the expected frequency of nonparental Watson-Crick couples, was calculated from the observed base frequencies Fij = AiTj + CiGj + GiCj + TiAj, omitting the appropriate term if ij was Watson–Crick in the parental sequence, (iii) a binomial distribution was calculated using p = Fij and n = 38, and (iv) the tail of the distribution just containing Wij was taken as the probability P that Wij would be observed if the bases at the two positions assorted randomly. P values were used to rank couples.
Binding Assay.
In an equilibrium dialysis unit (Hoefer), pairs of 0.1-ml chambers were separated by a dialysis membrane with a 6- to 8-kDa cutoff (traversed by the peptides but not by the DNAs) and loaded with 0.1 ml of buffer B containing in one chamber DNA and in the other chamber [3H]-peptide (2 μM N-[3H]-acetyl-d-VP or 0.05 μM [3H]-l-VP) with or without unlabeled competitor peptide. The unit was incubated for 24 h at room temperature with rotation. Radioactivity in the DNA chamber (RD) and in the peptide chamber (RP) was measured by scintillation counting, RP was corrected for a small nondiffusing fraction (≈1% for N-[3H]-acetyl-d-VP and ≈4% for [3H]-l-VP) measured in each experiment by a non-DNA control, and the fraction F of labeled peptide bound was calculated using F = (RD − RP)/(RD + RP). Assuming that (i) the DNA–peptide dissociation constant Kd is identical for labeled and unlabeled peptides, (ii) complex stoichiometry is 1:1, and (iii) the unlabeled peptide is fully active, then
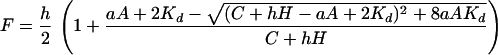 |
where A, C, are H concentrations of aptamer, competitor peptide, and [3H]-peptide, respectively, and a and h are active fractions of aptamer and [3H]-peptide, respectively. For each complex, data from varying-A and varying-C curves were combined to fit a, h, and Kd in the above equation using matlab (Mathworks, Natick, MA), yielding a = 0.68 for d-aptamer and 0.49 for l-aptamer, h = 0.68 for N-[3H]-acetyl-d-VP and 0.54 for [3H]-l-VP, and Kd = 0.85 μM for d-aptamer/d-VP and 1.17 μM for l-aptamer/l-VP. (More complex analyses that did not assume equal affinity for N-acetyl-d-VP and d-VP did not significantly improve fitting nor change the resultant value of Kd for d-VP.) The fraction of active [3H]-peptide bound to aptamer (Fa = F/h) is presented in Fig. 3.
Figure 3.

Equilibrium dialysis studies of mirror image aptamers (see Fig. 2F). Fa, the fraction of active radiolabeled peptide bound to DNA, was determined, and curves were fitted as described in Materials and Methods. Radiolabeled peptide was either N-[3H]-acetyl-d-VP (filled symbols) or [3H]-l-VP (open symbols). (A) d-aptamer, no competitor peptide. (B) d-aptamer (10 μM), d-VP competitor. (C) l-aptamer, no competitor peptide. (D) l-aptamer (8 μM), l-VP or oxytocin competitor.
Nuclease Susceptibility Assay.
d- or l-aptamer DNAs or a control d-DNA (5′-GAGGGCTCTAAGCAGGTTATTAATCATCTAAAGCGTAGTTTTCGTCGTTTGAGACTATTTT) was incubated at 37° at 1 μM in 0.1 units/μl DNase I (Promega), 40 mM Tris⋅HCl (pH 8), 10 mM NaCl, 6 mM MgCl2, 10 mM CaCl2 or 8 units/μl S1 nuclease (Boehringer Mannheim), 50 mM NaOAc (pH 5.0), 20 mM NaCl, and 1 mM ZnSO4 or at 5 μM in 95% male human (Sigma) or fetal calf (JRH Biosciences, Lenexa, KS) serum, and samples (10 μl for purified nucleases, 2 μl for sera) were stopped by mixing with 10 μl of 8 M urea, 25 mM EDTA and freezing on dry ice. These samples and a dilution series of the aptamer were run in a 10% polyacrylamide, 8-M urea gel and stained with ethidium bromide, and degradation rates were obtained after quantitation on the Bio-Rad Gel Doc 1000. Retention of purified nuclease activity after a 10-day incubation was confirmed by adding 10 μl of the 10-day l-DNA nuclease treatments to 10 pmol of a 69-nt d-DNA version of the aptamer and incubating 0.5 min at 37°; this d-DNA was 80% digested by the DNase I sample and fully digested by the S1 nuclease sample.
Bioassay.
LLC-PK1 cells (ATCC CL101) were grown as a monolayer on plastic Petri dishes in DMEM supplemented with 10% fetal calf serum at 37°C in a humidified 5% CO2 atmosphere. At confluent growth, cells were washed once with PBS and treated with 0.5 mg/ml trypsin/7 mM EDTA for 10–15 min at 37°, and 5000 cells were replated with 0.1 ml of medium on the flat 0.4-cm2 bottom of each test well. After growth for 2 days to 90–95% confluence, cells were washed with DMEM lacking serum at room temperature, and peptides and DNAs were added in 20 μl of DMEM lacking serum. After a 25-min incubation at room temperature, the medium was taken and stored at −20° until radioimmunoassay for cAMP according to the kit manufacturer’s (DuPont) instructions, interpolating from a cAMP standard curve conducted with each assay. Previous studies have demonstrated the validity of this assay for the secretion of the cAMP in response to vasopressin and a number of its analogs (20, 21).
RESULTS AND DISCUSSION
The first step in selection–reflection is to select a ligand that binds the enantiomer of the target molecule. Accordingly, mirror-image VP was synthesized from d-amino acids, both with an amino-terminal biotinyl group to prepare a matrix for affinity chromatography and in the free form to serve as an affinity eluent. We used in vitro selection (22) to isolate ligands of d-VP from an initial pool of 1016 different single-stranded DNAs, in which each molecule of the initial pool had a 60-nt central block of random sequence. Each selective cycle consisted of d-VP chromatography, amplification by PCR, and purification of positive DNA strands based on the use of a negative-strand PCR primer with an uncopyable tail (19). The pool exhibited no detectable d-VP elution peak in early cycles but by 13 cycles had reached a state where one-half could be bound to, and eluted from, d-VP agarose. The pool was cloned and found to be dominated by a single sequence (Fig. 2A); a restriction digestion screen revealed one of 55 clones with a different sequence (Fig. 2B). The two sequences share a remarkably large (23-nt) block of identical sequence in the originally randomized region, which suggested, as was later confirmed, that they belong to the same structural class. Both aptamers probably were present in the original pool and were independently selected (although it cannot be ruled out that one arose from the other by a double-recombination event that precisely preserved the original chain length). The independent selection of only two aptamers, of a single class, would be consistent with this aptamer class being better than any others of equal or lesser complexity.
With the goals of finding more active variants, determining aptamer secondary structure, and identifying unimportant regions that might be deleted, a second selection experiment was initiated with a partially randomized pool based on the aptamer. To simplify pool construction, a brief truncation study was performed to identify terminal segments unnecessary for activity (Fig. 2 A and B). This revealed that 27 nt could be deleted from the 3′ terminus of aptamer B without loss of activity. The truncated aptamer B sequence was doped at 30%, i.e., at each doped position, the parental base was present at a frequency of 0.7, and each of the other three bases was present at 0.1. After four cycles of selection, the pool became highly active in d-VP binding.
Sequence analysis of this pool identified highly conserved residues likely to be important for aptamer function (Fig. 2 C and D); 31 of the doped positions (Fig 2, dotted line) showed significantly higher conservation of the parental base than expected from the doping rate. Three of the 41 sequences (Fig. 2D) had a disproportionate number of changes at these highly conserved positions, suggesting that they either do not bind tightly to d-VP or they use novel modes of d-VP binding. When these three molecules were passed over a d-VP column, nearly all of the material flowed through; there was some trailing behind the void volume peak (a possible basis for their retention in the final pool) but no elution by free d-VP of the residual DNA. Subsequent analysis of covariation was therefore restricted to the 38 sequences of Fig. 2C. Although none of these was individually assayed for d-VP binding, the aggregate activity of the pool from which they were cloned indicates that a large majority of these sequences is active.
A global search for candidate base pairing positions was designed, scoring for base variations or covariations that could form new Watson–Crick pairs, while avoiding skewing by the very high base conservation (see Materials and Methods). The four top scoring position pairs of all 2278 tested (thick green dashes in Fig. 2E) identified two base pairing regions, P1 and P2. Conservation of base pairing is not absolute for any pairs in P1 or P2, but for most it is significantly higher than would be expected if pairing were not important for binding. Mismatching could be explained by the possible inclusion of inactive clones or a certain degree of tolerance of mismatches and bulges. In some sequences, P1 or P2 appear to extend beyond the marked regions, sometimes by bulging a nucleotide out of the stem.
Opposed GA dinucleotides were flanked by well established Watson–Crick pairs in P1 (Fig. 2E). Such opposed GA dinucleotides can form stable tandem G:A pairs of surprising stability within DNA helices, especially in the sequence context found in the aptamer (5′ T and 3′ A flanking each GA) (23, 24). The observation that the same P1 tandem G:A pairs (rather than Watson–Crick pairs) were present in both aptamers of the original selection experiment suggested that the distinctive structural features expected for the GA region (25–27) might contribute to binding. However, some sequences from the doped selection indicated that Watson–Crick pairs could be tolerated (Fig. 2C). We confirmed that conversion of the G:A pairs to Watson–Crick pairs (replacing both As with Cs) did not affect d-VP chromatography.
The paired regions flank an asymmetric internal loop (L1) of 20 extremely conserved nucleotides. A few single- and double-base changes occurred in this region but none at high enough levels to suggest that they would significantly improve d-VP binding. In contrast, the loop L2 showed little sequence conservation, indicating that its content is largely irrelevant for d-VP binding. We found that replacement of this loop with a short sequence known to form a stable hairpin in single-stranded DNA (28) did not change d-VP chromatography.
In summary, the comparison of sequences selected from the doped pool defined the secondary structure of the aptamer and identified an internal region that could be deleted. It did not reveal base substitutions improving aptamer activity but instead showed that the identities of all or nearly all bases in the 20-nt L1 loop are important for aptamer function. Constraint on the identities of 16 contiguous residues, as appears to be the case within L1′, is very unusual for RNA or single-stranded DNA structures and suggests an elaborate network of important DNA–DNA and DNA–peptide interactions involving these L1′ bases.
A 55-nt aptamer (Fig. 2F) was designed according to the above information and used in subsequent experiments. Equilibrium dialysis experiments (29) measured a dissociation constant of 0.9 μM for the aptamer/d-VP complex (Fig. 3 A and B; see Materials and Methods). Affinity for l-VP is at least 1000-fold lower. Other equilibrium dialysis experiments (not shown) revealed that binding is (i) abolished in 10 mM DTT, suggesting that the peptide disulfide is critical, (ii) somewhat aided by, but not absolutely dependent on, divalent cations, (iii) unaffected by replacing potassium with sodium ions or vice versa, and (iv) dependent on at least one thymine methyl group as determined by loss of activity upon substitution of all thymine bases with uridine.
After identifying a ligand of the target enantiomer, the second step of selection–reflection is to synthesize the enantiomer of the ligand; this reflected ligand will bind the normal target. In our case, the ligand was normal DNA (d-DNA), so it was necessary to synthesize l-DNA. Each nucleotide in DNA contains three chiral centers, all in the sugar moiety; the four bases are achiral. The four l-deoxynucleoside phosphoramidites can be prepared from the natural sugar l-arabinose (2, 30); these were used to synthesize the l-DNA enantiomer of the d-VP aptamer. Equilibrium dialysis experiments demonstrated that the l-aptamer binds l-VP but not d-VP (Fig. 3 C and D). The dissociation constant measurement of the l-aptamer/l-VP complex (1.2 μM) matched that of the d-aptamer/d-VP complex (0.9 μM), within experimental error. The l-aptamer showed marked (>100-fold) preference for VP over oxytocin (Fig. 3D), which is the closest known human analog of VP, differing at two residues.
The practical advantage of l-DNAs is their insusceptibility to most nucleases. We tested the l- and d-aptamers using purified S1 nuclease (a single-stranded endonuclease) and DNase I (a double-stranded endonuclease) and found that the l-aptamer was unaffected after incubation for 10 days with either nuclease under conditions in which the d-aptamer wasd degraded in under 10 s (see Materials and Methods). Others have performed similar tests with S1 nuclease (15, 31), P1 nuclease (a single-stranded endonuclease) (15, 30), spleen phosphodiesterase (a 5′ exonuclease) (15, 31, 32), and venom phosphodiesterase (a 3′ exonuclease) (15, 33). Of these, only venom phosphodiesterase was found to degrade l-DNA; however, hydrolysis was 10,000 times slower than for d-DNA (15, 33).
The l-aptamer showed no hint of degradation in human serum (Table 1), even at our latest time point (7 days). However, calf serum exhibited very slow exonucleolytic activity toward the l-aptamer (per-nucleotide half-time of ≈2 days). In this context, it is useful to note that derivatization of the 3′ end has been shown to completely protect l-DNA from venom phosphodiesterase (30). Such derivatization of l-aptamers could conceivably make them invulnerable to all naturally occurring degradative enzymes. The d-aptamer was degraded slowly in human and calf serum, with half-lives of 33 and 2.5 h, respectively. The long terminal P1 pairing of the aptamer appears to have prevented the more commonly noted stepwise degradation pattern associated with 3′ exonuclease activity (34, 35); exonuclease-like degradation was detected for a control d-DNA predicted to be relatively unstructured at its 3′ terminus (Table 1).
It should be noted that in vitro studies with serum preparations probably have little bearing on the question of i.v. stability. It is unclear to what extent the nuclease content of a serum preparation represents that of the blood plasma in the organism; the main source of nuclease may be the cytoplasm of blood cells lysed during serum preparation (4). More importantly, the slow-acting serum nucleases would appear to be a relatively insignificant subset of the nucleases that DNA must face when injected i.v. Degradation of d-DNA to monomers occurs i.v. within 5 min or less (4–7), even when the DNA is chemically protected against the 3′ exonuclease found in serum (6).
Bioactivity of the l-aptamer was demonstrated with the cell line LLC-PK1 from porcine renal epithelium, which bears V2 vasopressin receptors and exhibits a roughly linear response of cAMP release upon stimulation with up to 5 nM VP (Fig. 4A). Cells were treated at 5 nM VP with increasing amounts of l-aptamer, which at 10 μM acted to reduce the VP response 2.6-fold (Fig. 4B); the corresponding value for a control l-DNA with the same base composition and secondary structure as the aptamer was only 1.1-fold. The l-aptamer is not a general inhibitor of cAMP release in response to hormones; it did not diminish cAMP release induced by oxytocin in LLC-PK1 (data not shown).
Figure 4.

Bioactivity of l-aptamer. Release of cAMP from LLC-PK1 cells in response to (A) VP alone or (B) 5 nM VP in the presence of l-DNA. Means and SD are shown for triplicate samples.
We have shown that selection–reflection is a viable means of producing bioactive and nuclease-insusceptible ligands of biological target molecules, but some limitations should be noted. The l-nucleoside phosphoramidites required for the synthesis of reflected aptamers are not readily available for the time being. Potential targets are limited to those that can be chemically synthesized in mirror-image form although achiral molecules and domains within macromolecules could also be viable targets. The tradeoff between molecular weight and yield in the synthesis of the target favors smaller targets. However, aptamers selected to bind peptides and smaller targets generally have lower affinities than those selected to bind larger targets (22). VP was chosen as a target for selection–reflection in part for its humoral action; an l-aptamer can easily be administered i.v. and may even survive oral delivery. However, it is unclear what immune responses or other adverse effects (36) l-DNA might elicit, and the pharmacokinetics of l-DNA also is unknown. It can be anticipated from studies of phosphorothioate oligonucleotides (37) that l-DNA will survive substantially longer in the blood than d-DNA but that urinary excretion and other clearance processes will control its lifetime. Should clearance or adverse effects limit the therapeutic use of l-aptamers, they still may be useful in diagnostic applications such as the in vitro detection of target molecules in biological samples.
Table 1.
Susceptibility of DNAs to serum nucleases
| Serum source | Decay mode* | Half-time of decay
|
||
|---|---|---|---|---|
| Control d-DNA | d-aptamer | l-aptamer | ||
| Human male | exonuclease | 2h/nt | not dominant | no decay |
| endonuclease | 33 h | 33 h | no decay | |
| Fetal calf | exonuclease | 0.2 h/nt | not dominant | 50 h/nt |
| endonuclease | 1.9 h | 2.5 h | no decay | |
Decay mode: exonuclease, apparent stepwise degradation from end; endonuclease, broad size distribution of degradation products.
Acknowledgments
We thank M. Burgess for synthesis of d-VP and P. Unrau for help with curve fitting and comments on the manuscript. This work was supported by a grant from Innovir Laboratories.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: VP, arginine vasopressin.
References
- 1.Zawadzke L E, Berg J M. Proteins. 1993;16:301–305. doi: 10.1002/prot.340160308. [DOI] [PubMed] [Google Scholar]
- 2.Urata H, Ogura E, Shinohara K, Ueda Y, Akagi M. Nucleic Acids Res. 1992;20:3325–3332. doi: 10.1093/nar/20.13.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milton R C D, Milton S C F, Kent S B H. Science. 1992;256:1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 4.Goodchild J, Kim B, Zamecnik P C. Antisense Res Dev. 1991;1:153–160. doi: 10.1089/ard.1991.1.153. [DOI] [PubMed] [Google Scholar]
- 5.Sands H, Gorey-Feret L J, Cocuzza A J, Hobbs F W, Chidester D, Trainor G L. Mol Pharmacol. 1994;45:932–943. [PubMed] [Google Scholar]
- 6.Sands H, Gorey-Feret L J, Ho S P, Bao Y, Cocuzza A J, Chidester D, Hobbs F W. Mol Pharmacol. 1995;47:636–646. [PubMed] [Google Scholar]
- 7.Agrawal S, Temsamani J, Galbraith W, Tang J. Clin Pharmacokinet. 1995;28:7–16. doi: 10.2165/00003088-199528010-00002. [DOI] [PubMed] [Google Scholar]
- 8.Schumacher T N M, Mayr L M, Minor D L, Milhollen M A, Burgess M W, Kim P S. Science. 1996;271:1854–1856. doi: 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- 9.Kluβmann S, Nolte A, Bald R, Erdmann V A, Fürste J P. Nature Biotechnology. 1996;14:1112–1115. doi: 10.1038/nbt0996-1112. [DOI] [PubMed] [Google Scholar]
- 10.Nolte A, Kluβmann S, Bald R, Erdmann V A, Fürste J P. Nat Biotechnol. 1996;14:1116–1119. doi: 10.1038/nbt0996-1116. [DOI] [PubMed] [Google Scholar]
- 11.Pan W, Craven R C, Qiu Q, Wilson C B, Wills J W, Golovine S, Wang J F. Proc Natl Acad Sci USA. 1995;92:11509–11513. doi: 10.1073/pnas.92.25.11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pagratis N C, Bell C, Chang Y-F, Jennings S, Fitzwater T, Jellinek D, Dang C. Nat Biotechnol. 1997;15:68–73. doi: 10.1038/nbt0197-68. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Qiu Q, Gill S C, Jayasena S D. Nucleic Acids Res. 1994;22:5229–5234. doi: 10.1093/nar/22.24.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jellinek D, Green L S, Bell C, Lynott C, Gill N, Vargeese C, Kirschenheuter G, McGee D P, Abesinghe P, Pieken W A, Rifkin D B, Moscatelli D, Janjic N. Biochemistry. 1995;34:11363–11372. doi: 10.1021/bi00036a009. [DOI] [PubMed] [Google Scholar]
- 15.Damha M J, Giannaris P A, Marfey P, Reid L S. Tetrahedron Lett. 1991;32:2573–2576. [Google Scholar]
- 16.Bock L C, Griffin L C, Latham J A, Vermaas E H, Toole J J. Nature (London) 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 17.Ellington A D, Szostak J W. Nature (London) 1992;355:850–852. doi: 10.1038/355850a0. [DOI] [PubMed] [Google Scholar]
- 18.Huizenga D E, Szostak J W. Biochemistry. 1995;34:656–665. doi: 10.1021/bi00002a033. [DOI] [PubMed] [Google Scholar]
- 19.Williams K P, Bartel D P. Nucleic Acids Res. 1995;23:4220–4221. doi: 10.1093/nar/23.20.4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy C, Ausiello D A. J Biol Chem. 1981;256:3415–3422. [PubMed] [Google Scholar]
- 21.Roy C, Hall D, Karish M, Ausiello D A. J Biol Chem. 1981;256:3423–3427. [PubMed] [Google Scholar]
- 22.Gold L, Polisky B, Uhlenbeck O C, Yarus M. Annu Rev Biochem. 1995;64:763–797. doi: 10.1146/annurev.bi.64.070195.003555. [DOI] [PubMed] [Google Scholar]
- 23.Ebel S, Lane A, Brown T. Biochemistry. 1992;31:12083–12086. doi: 10.1021/bi00163a017. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Agrawal S. Biochemistry. 1995;34:10056–10062. doi: 10.1021/bi00031a030. [DOI] [PubMed] [Google Scholar]
- 25.Greene K L, Jones R L, Li Y, Robinson H, Wang A H, Zon G, Wilson W D. Biochemistry. 1994;33:1053–1062. doi: 10.1021/bi00171a003. [DOI] [PubMed] [Google Scholar]
- 26.Lane A N, Martin S R, Ebel S, Brown T. Biochemistry. 1992;31:12087–12095. doi: 10.1021/bi00163a018. [DOI] [PubMed] [Google Scholar]
- 27.Chou S H, Cheng J W, Reid B R. J Mol Biol. 1992;228:138–155. doi: 10.1016/0022-2836(92)90497-8. [DOI] [PubMed] [Google Scholar]
- 28.Hirao I, Kawai G, Yoshizawa S, Nishimura Y, Ishido Y, Watanabe K, Miura K-i. Nucleic Acids Res. 1994;22:576–582. doi: 10.1093/nar/22.4.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breslow E, Walter R. Mol Pharmacol. 1972;8:75–81. [PubMed] [Google Scholar]
- 30.Asseline U, Hau J-F, Czernecki S, Diguarher T L, Perlat M-C, Valery J-M, Thuong N T. Nucleic Acids Res. 1991;19:4067–4074. doi: 10.1093/nar/19.15.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morvan F, Génu C, Rayner B, Gosselin G, Imbach J-L. Biochem Biophys Res Commun. 1990;172:537–543. doi: 10.1016/0006-291x(90)90706-s. [DOI] [PubMed] [Google Scholar]
- 32.Fujimori S, Shudo K, Hashimoto Y. J Am Chem Soc. 1990;112:7436–7238. [Google Scholar]
- 33.Anderson D J, Reischer R J, Taylor A J, Wechter W J. Nucleosides Nucleotides. 1984;3:499–512. [Google Scholar]
- 34.Shaw J-P, Kent K, Bird J, Fishback J, Froehler B. Nucleic Acids Res. 1991;19:747–750. doi: 10.1093/nar/19.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang J Y, Temsamani J, Agrawal S. Nucleic Acids Res. 1993;21:2729–2735. doi: 10.1093/nar/21.11.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agrawal S, Rustagi P K, Shaw D R. Toxicol Lett. 1995;82:431–434. doi: 10.1016/0378-4274(95)03573-7. [DOI] [PubMed] [Google Scholar]
- 37.Zhang R, Diasio R B, Lu Z, Liu T, Jiang Z, Galbraith W M, Agrawal S. Biochem Pharmacol. 1995;49:929–939. doi: 10.1016/0006-2952(95)00010-w. [DOI] [PubMed] [Google Scholar]