

Abstract
The rubredoxin protein from the hyperthermophilic archaebacterium Pyrococcus furiosus was examined by a hydrogen exchange method. Even though the protein does not exhibit reversible thermal unfolding, one can determine its stability parameters—free energy, enthalpy, entropy, and melting temperature—and also the distribution of stability throughout the protein, by using hydrogen exchange to measure the reversible cycling of the protein between native and unfolded states that occurs even under native conditions.
Hyperthermophilic organisms thrive at temperatures near and even above 100°C (1, 2). The exceedingly stable protein molecules that make this possible provide important targets for study (3, 4). Unfortunately, the usual methods for studying thermodynamic stability are largely ineffective for these proteins (4, 5). The measurement of stability parameters requires reversible unfolding, but the thermal transition of thermophilic proteins from their native to their unfolded state is almost always irreversible (6–8) (one exception is known; ref. 9), and denaturant driven unfolding is often not possible. A method based on hydrogen exchange is able to measure the reversible cycling of protein molecules between native and unfolded states that occurs continually even under native conditions. The measurements can directly determine global stability parameters (10–12) and can provide site-resolved information on the components of stability throughout a protein molecule (13). Results obtained for a rubredoxin protein (RdPf) from the hyperthermophile Pyrococcus furiosus (14), an organism that grows optimally at 100°C (15), show that RdPf has unusually high stability that decreases above 100°C, pointing to an equilibrium melting transition just below 200°C.
The structure of RdPf (Fig. 1) has been determined by solution NMR (16) and x-ray crystallography (17). This 53-aa protein has three antiparallel β strands joined by two loops, each containing two cysteine residues liganded to a metal ion to form a common loop structure known as a “knuckle” (Cys-X-X-Cys-X-X) (18). The normally occurring paramagnetic iron was replaced in vitro by a diamagnetic zinc, which is favorable for NMR studies (14, 16). The Fe and Zn forms have virtually identical structures (16, 17, 19). Fig. 2 shows that the far ultraviolet circular dichroism of Zn RdPf is insensitive to temperature over a wide range as is the proton NMR spectrum (8).
Figure 1.

molscript representation (35) of the rubredoxin from P. furiosus (16, 17). The H-bonding pattern of the most slowly exchanging hydrogens is Cys-5NH to Tyr-10CO and Cys-8NH to Cys-5S in the first knuckle of the protein, and symmetrically, Cys-38NH to Ala-43CO and Cys-41NH to Cys-38S in the second knuckle.
Figure 2.

Far ultraviolet circular dichroism of Zn RdPf. The ellipticity measured at 224 nm decreases by only 4% between 2°C and 94°C, after correction for the 4% temperature-dependent increase in solution volume. The different symbols indicate readings of RdPf at 10 μM concentration in solution at pH 6.5, 11.6, and in 3 M GdmCl at pH 6.5. RdPf was purified as previously described (14).
Fig. 3 shows hydrogen exchange (HX) data obtained for some residue NHs in D2O at the conditions indicated. The resonances of the slowly exchanging amide NHs of RdPf are well resolved in the two-dimensional correlated spectroscopy spectrum, and many can even be resolved in the one-dimensional proton NMR spectrum after brief exchange in D2O to remove faster NHs, especially so at high temperature. Thus their exchange with solvent deuterons can be followed by simple spectroscopy.
Figure 3.

Typical kinetic hydrogen-deuterium exchange data measured by NMR. HX rate constants (kex) were determined by uniexponential fitting of the time-dependent decrease in resonance amplitude and translated into free energy values for the determining structural opening reaction by use of Eq. 1. Exchange of the different NHs could be moved into a convenient time window by adjusting pD between 6.5 and 9.4, made possible by the invariance of unfolding ΔG with solution pD. Nonunit amplitudes are due to partial overlap (Cys-8) and dead time losses (Tyr-10).
Eq. 1 was used to translate measured HX rates (kex) into the free energy for the H-bond opening reaction that allows each NH to exchange (ΔGHX):
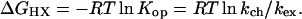 |
1 |
Here kch is the chemical exchange rate expected for each NH in the absence of structural protection (20–22). The ratio, kch/kex, gives the structural protection factor for each NH. Its inverse is the equilibrium constant (Kop) for the transient structural opening reaction that separates the protecting hydrogen bond and exposes the NH to exchange. These relationships hold in the so-called EX2 (bimolecular exchange) limit where structural reclosing is fast compared with kch (23). The EX2 condition was confirmed for readings up to 123°C by three independent criteria: the ideal pH dependence observed, the agreement among NHs with different intrinsic kch values (as described by Bai et al., ref. 10), and the m value obtained from the denaturant dependence of the global ΔGHX (see below). At higher temperatures, exchange enters the EX1 regime (unimolecular exchange; ref. 23) where structural reclosing is slower than kch so that kex measures the opening rate.
The various peptide NH hydrogens in RdPf exchange over a range of rates differing by more than 10 orders of magnitude. The experimental HX time window can be adjusted to include faster or slower NHs by appropriately setting the ambient pH and temperature. The chemical rate constant, kch, changes by a factor of 10 per pH unit due to catalysis by OH− ion. Accordingly, a range of 1010 in HX rates can be subsumed by a factor of 105 in the pH adjustment (e.g., pH 5–10) together with a factor of 105 in the accessible time scale (minutes to months). Further factors of 10 in rate can be obtained by serial ≈20°C increases in temperature (21), and denaturant concentration can supply another useful variable (10, 24). The ability to accurately determine ΔGHX for the various NHs in RdPf was facilitated by the fact that the protein structure and stability do not change over a wide pH range. (NMR chemical shifts are independent of pH between pH 5.5 to 9.5 at least; data not shown).
Free energy values obtained in this way are shown in Fig. 4 as a function of temperature and in Fig. 5 as a function of the concentration of guanidinium chloride (GdmCl) denaturant at 60°C. The temperature dependence of ΔG provides the entropy (dΔ G/dT = −ΔS) and therefore also the enthalpy (ΔH = ΔG + TΔS) of the HX rate-determining unfolding reaction. The denaturant dependence relates to the surface exposed in the determining unfolding reaction (25), encoded in the slope dΔG/d[GdmCl] = m.
Figure 4.

Dependence on temperature of the free energy of the opening reactions that determine hydrogen exchange of various slowly exchanging NHs. For H-D exchange, samples were incubated in a water-ethylene glycol bath at temperatures up to 123°C simply by holding them in a tightly sealed NMR tube. At higher temperature exchange entered the EX1 regime.
Figure 5.

Dependence of the free energy of opening reactions on denaturant concentration, measured at 60°C. Most NHs are dominated by small local fluctuations (m ≈ 0) at low GdmCl. At increased GdmCl the curves merge into HX isotherms that indicate large unfolding reactions (large m and extrapolated ΔG), including a high energy global unfolding and at least one large subglobal unfolding (see Bai et al., refs. 10 and 11). The highest energy unfolding curve points to an experimentally unreachable midpoint GdmCl concentration for global unfolding of 10 M at 60°C. The GdmCl-dependent HX curves were fit with an equation (24) that considers the dependence on denaturant (den) of local (Kloc), subglobal (Ksg), and global (Kgl) equilibrium opening, as follows: ΔGHX = − RT ln {Kloc(den) + Ksg(den) + Kgl(den)} = − RT ln {Kloc + Ksg(0) exp (msg [GdmCl]/RT + Kgl(0) exp(mgl [GdmCl]/RT)}. This derives from Eq. 1 together with the usual linear dependence of unfolding free energy on denaturant concentration, namely ΔG(den) = ΔG(0) −m[den], so that K(den) = K(0) exp(m[den]/RT).
The computed ΔGHX values exhibit little dependence on temperature or denaturant concentration when exchange is measured at low temperature or low denaturant. The protein opening reactions that mediate exchange under these conditions have small ΔS and m values (above equations) and therefore represent small fluctuations that expose little new surface. Accordingly, the different hydrogens respond only to local determinants and can exchange with very different rates. As temperature or denaturant concentration is increased, larger unfolding reactions (large ΔS and m) are selectively promoted and come to dominate the exchange of many NHs. This appears as a merging of the ΔGHX curves for different hydrogens into a common HX isotherm (Figs. 4 and 5). The protein segments involved in each transient large scale unfolding can be identified by the residue NHs that join the isotherm (10).
The highest lying isotherm involves the four cysteine NHs, which are the slowest exchanging hydrogens in RdPf. The different cysteine NH hydrogens exchange at differing rates at low temperature and low GdmCl, but they merge into a common HX isotherm at higher temperature (Fig. 4) and at higher GdmCl concentration (Fig. 5). This behavior reveals a high free energy unfolding reaction that transiently and reversibly exposes even the most protected cysteine NHs. An HX isotherm that points to a large opening reaction with somewhat lower free energy also is indicated in Fig. 5. These results appear to implicate the metal ligation and surrounding knuckle structure as the seat of thermostability. It should be noted, however, that this structure in the homologous rubredoxin from mesophilic Clostridium pasteurianum does not display the same enhanced stability (data not shown) and that similar zinc knuckle-containing proteins (18) have not been noted to be unusually thermostable.
Here we are concerned with the global unfolding reaction. Many cases are now known in which the slowest NHs in a protein exchange by way of transient global unfolding (24, 26–32). The exchange of these hydrogens, measured under fully native conditions, displays the same large denaturant dependence (m value) and close to the same ΔG value for global stability as is obtained by measurement of the classical global unfolding transition. Similarly, the present results for RdPf exhibit a major high energy unfolding reaction with m value consistent with the global unfolding of a protein this size (m = 1.7 kcal mol−1M−1 from Fig. 5). This unfolding reaction exposes even the most protected NHs to exchange, namely the four cysteine NHs that crosslink the knuckle structures. It seems likely that this behavior reflects the transient global unfolding reaction. If the HX data do not yet represent global unfolding, then the true global stability and the extrapolated melting temperature will be even higher.
ΔGHX values found for the major unfolding reaction in RdPf are shown as a function of temperature in Fig. 6. The data can be fit in different ways. The arched curve in Fig. 6 fits the data to the integrated form of the van’t Hoff equation. Alternatively, the data can be fit with the linear curves drawn. In either case, these results specify the equilibrium unfolding parameters at the physiological temperature (100°C) as follows: ΔG = 15 kcal/mol, ΔH = 70 kcal/mol, TΔS = 55 kcal/mol. Also m(GdmCl) is 1.7 kcal/mol per molar. The fitted curves point to a global melting temperature, the temperature at which ΔG for unfolding passes through zero, of 176 to 195°C.
Figure 6.

Thermal stability curves. The data, from the global unfolding isotherm in Fig. 4, represent the concerted exchange of the four Cys-NHs at temperatures above 100°C and the exchange of Cys-5NH at lower temperature. The RdPf data may be fit by straight lines as shown or by the usual integrated van’t Hoff equation (arched curve), given by ΔG = ΔH −TΔS + ΔCp{(T − Tm) − T ln T/Tm)}, where ΔCp is the temperature-independent specific heat and Tm is the midpoint melting temperature. Similar curves for some high Tm proteins with nearly identical size (53 to 56 residues) from mesophilic organisms are shown for comparison. GB1 (36) is the B1 version of the IgG binding domain of streptococcal protein G, and BPTI (37) is the basic pancreatic trypsin inhibitor.
In principle, the extremely high melting temperature for hyperthermophilic proteins may be due to unusually high intrinsic stability or to factors that can shift the right limb of the ΔG vs. temperature curve to higher temperatures without imposing extreme stability (33). The right to left placement of the van’t Hoff curve depends on the balance of enthalpic and entropic components for the stabilizing interactions; the relative flatness or width of the curve depends also on the specific heat parameter and its variation at high temperature (34). Fig. 6 includes global stability curves for some exceptionally stable proteins with nearly identical size from conventional mesophilic organisms. In comparison, the curve for RdPf is upshifted, indicating enhanced ΔG, presumably due to the presence of added stabilizing interactions in the protein. In addition, the RdPf curve appears to be right-shifted so that the temperature of maximum stability approximates the physiological temperature while the apparent melting temperature approaches 200°C.
In summary, the hydrogen exchange behavior of rubredoxin from P. furiosus can identify cooperative global and perhaps subglobal unfolding reactions and can measure their quantitative thermodynamic parameters—ΔG, ΔH, ΔS, and Tm—and also a steric parameter, the m value. These results illustrate an approach that can determine structural stability by measurements on the native protein, without exposure to extreme denaturing conditions, and thus can quantify and help to locate the factors that contribute to the unusual stability and the high melting temperature characteristic of hyperthermophilic proteins.
Acknowledgments
This work was supported by National Institutes of Health Grants DK11295 to S.W.E. and GM50736 to M.W.W.A.
ABBREVIATIONS
- RdPf
the rubredoxin protein from Pyrococcus furiosus
- HX
hydrogen exchange
- GdmCl
guanidinium chloride
References
- 1.Blochl E, Burggraf S, Fiala F, Lauerer G, Huber G, Huber R, Rachel R, Segerer A, Stetter K O, Volkl P. World J Microbiol Biotechnol. 1995;11:9–16. doi: 10.1007/BF00339133. [DOI] [PubMed] [Google Scholar]
- 2.Stetter K O. FEMS Microbiol Rev. 1996;18:149–158. doi: 10.1111/j.1574-6976.2000.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 3.Adams M W W. Annu Rev Microbiol. 1993;47:627–658. doi: 10.1146/annurev.mi.47.100193.003211. [DOI] [PubMed] [Google Scholar]
- 4.Adams, M. W. W. (1996) Adv. Protein Chem. 48. [DOI] [PubMed]
- 5.Jaenicke R. FASEB J. 1996;10:84–92. doi: 10.1096/fasebj.10.1.8566552. [DOI] [PubMed] [Google Scholar]
- 6.Klump H, DiRuggiero J, Kerssel M, Park J-B, Adams M W W, Robb F T. J Biol Chem. 1992;267:22681–22685. [PubMed] [Google Scholar]
- 7.Klump K H, Adams M W W, Robb F T. Pure Appl Chem. 1994;66:485–489. [Google Scholar]
- 8.Cavagnero S, Zhou Z H, Adams M W W, Chan S I. Biochemistry. 1995;34:9865–9873. doi: 10.1021/bi00031a007. [DOI] [PubMed] [Google Scholar]
- 9.McCrary B S, Edmondson S P, Shriver J W. J Mol Biol. 1996;264:784–805. doi: 10.1006/jmbi.1996.0677. [DOI] [PubMed] [Google Scholar]
- 10.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai Y, Englander S W. Proteins Struct Funct Genet. 1996;24:145–151. doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain A K, Handel T M, Marqusee S. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 13.Englander S W, Kallenbach N R. Q Rev Biophys. 1984;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 14.Blake P R, Bryant F O, Aono S, Magnuson J K, E, E, Howard J B, Adams M W W. Biochemistry. 1991;30:10885–10891. doi: 10.1021/bi00109a012. [DOI] [PubMed] [Google Scholar]
- 15.Fiala G, Stetter K O. Arch Microbiol. 1986;145:56–61. [Google Scholar]
- 16.Blake P R, Park J B, Zhou Z H, Hare D R, Adams M W W, Summers M F. Protein Sci. 1992;1:1508–1521. doi: 10.1002/pro.5560011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Day M W, Hsu B T, Joshua-Tor L, Park J B, Zhou Z H, Adams M W W, Rees D C. Protein Sci. 1992;1:1494–1507. doi: 10.1002/pro.5560011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwabe J W R, Klug A. Nat Struct Biol. 1994;1:345–349. doi: 10.1038/nsb0694-345. [DOI] [PubMed] [Google Scholar]
- 19.Blake P R, Day M W, Hsu B T, Joshua-Tor L, Park J B, Hare D R, Adams M W W, Rees D C, Summers M F. Protein Sci. 1992;1:1522–1525. doi: 10.1002/pro.5560011113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molday R S, Englander S W, Kallen R G. Biochemistry. 1972;11:150–158. doi: 10.1021/bi00752a003. [DOI] [PubMed] [Google Scholar]
- 21.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connelly G P, Bai Y, Jeng M-F, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- 23.Hvidt A, Nielsen S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 24.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1994;20:4–14. doi: 10.1002/prot.340200103. [DOI] [PubMed] [Google Scholar]
- 25.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loh S N, Prehoda K E, Wang J, Markley J L. Biochemistry. 1993;32:11022–11028. doi: 10.1021/bi00092a011. [DOI] [PubMed] [Google Scholar]
- 27.Kim K S, Fuchs J A, Woodward C K. Biochemistry. 1993;32:9600–9608. doi: 10.1021/bi00088a012. [DOI] [PubMed] [Google Scholar]
- 28.Orban J, Alexander P, Bryan P. Biochemistry. 1994;33:5702–5710. doi: 10.1021/bi00185a006. [DOI] [PubMed] [Google Scholar]
- 29.Perrett S, Clarke J, Hounslow A M, Fersht A R. Biochemistry. 1995;34:9288–9298. doi: 10.1021/bi00029a003. [DOI] [PubMed] [Google Scholar]
- 30.Bai Y, Englander J J, Mayne L, Milne J S, Englander S W. Methods Enzymol. 1995;259:344–356. doi: 10.1016/0076-6879(95)59051-x. [DOI] [PubMed] [Google Scholar]
- 31.Swint-Kruse L, Robertson A D. Biochemistry. 1996;35:171–180. doi: 10.1021/bi9517603. [DOI] [PubMed] [Google Scholar]
- 32.Englander S W, Sosnick T R, Englander J J, Mayne L. Curr Opin Struct Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becktel W J, Schellman J A. Biopolymers. 1987;26:1859–1877. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- 34.Privalov P L. J Mol Biol. 1996;258:707–725. doi: 10.1006/jmbi.1996.0280. [DOI] [PubMed] [Google Scholar]
- 35.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 36.Alexander P, Orban J, Bryan P. Biochemistry. 1992;31:7243–7248. doi: 10.1021/bi00147a006. [DOI] [PubMed] [Google Scholar]
- 37.Makhatadze G I, Kim K S, Woodward C K, Privalov P L. Protein Sci. 1993;2:2028–2036. doi: 10.1002/pro.5560021204. [DOI] [PMC free article] [PubMed] [Google Scholar]