

Abstract
Zinc has anti-inflammatory properties and is crucial for the integrity of vascular endothelial cells, and the development and homeostasis of the cardiovascular system. The aryl hydrocarbon receptor (AhR) which is expressed in the vascular endothelium also plays an important role in responses to xenobiotic exposure and cardiovascular development. We hypothesize that cellular zinc can modulate induction of AhR responsive genes in endothelial cells. To determine if zinc deficiency can alter responses to AhR ligands, aortic endothelial cells were exposed to the AhR ligands 3,3’,4,4’-tetrachlorobiphenyl (PCB77) or beta-naphthoflavone (β-NF) alone or in combination with the membrane permeable zinc chelator TPEN, followed by measurements of the AhR responsive cytochrome P450 enzymes CYP1A1 and 1B1. Compared to vehicle treated cells, both PCB77-induced CYP1A1 activity (EROD) and mRNA expression were significantly reduced during zinc deficiency. In addition, PCB77 and β-NF-mediated upregulation of CYP1A1 and CYP1B1 protein expression was significantly reduced in zinc-deficient endothelial cells. The inhibition of CYP1A1 and CYP1B1 protein expression caused by zinc deficiency was reversible by cellular zinc supplementation. Overall, our results strongly suggest that nutrition can modulate an environmental toxicant-induced biological outcome and that adequate levels of individual nutrients such as zinc are necessary for induction of AhR responsive genes in vascular endothelial cells.
Keywords: Zinc deficiency, AhR, CYP1A1, CYP1B1, EROD
1. Introduction
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that is normally found in the cytoplasm and complexed with Hsp90, XAP2, Ara9 and p23. Upon activation, the AhR complex goes through a conformational change that exposes a nuclear localization signal domain and triggers translocation from the cytosol to the cell nucleus where it forms a complex with ARNT/HIF-1β. This complex recognizes specific enhancer domain sequences in the promoter regions of responsive genes that are known as xenobiotic response elements (XREs). The AhR/ARNT heterodimers stimulate transcription of Phase I and Phase II xenobiotic metabolizing enzymes. Cytochrome P450 genes, specifically those belonging to the CYP1 family (e.g. CYP1A1/2 and CYP1B1) are highly inducible by AhR activation, and the molecular mechanisms involved in their regulation by AhR have been well characterized (Fujii-Kuriyama and Mimura 2005; Puga et al. 2005). Although most of the research performed on AhR has focused on its role in the molecular, biochemical, and toxic responses to xenobiotic ligands, recent studies have also shown that the AhR plays a critical role in the development of various organ systems and cardiovascular homeostasis (Savouret et al. 2003). For example, mice that lack the AhR gene have been shown to suffer from cardiac fibrosis, hypertrophy, increased left ventricular mass, increased expression of the cardiac hypertrophy markers β-myosin heavy chain, and β-myosin light chain 2V and increased plasma levels of the vasoactive agents angiotensin II and endothelin-1 (Fernandez-Salguero et al. 1997; Lund et al. 2003; Lund et al. 2006). Such findings combined with the high degree of conservation of AhR among species suggest that, in addition to orchestrating responses to exposure to xenobiotic ligands, the AhR plays an important role in systemic homeostasis and development (Fujii-Kuriyama and Mimura 2005). Little is known about nutritional modulation of AhR-mediated cell signaling. The current study focuses on the micronutrient zinc, because of its importance in regulating protein structure and cell signaling (Reiterer et al. 2006)
Zinc has multiple roles in maintaining the physiological conditions of the cardiovasculature (Reiterer et al. 2006), and zinc may be critical in normal vascular development. For example, zinc deficiency leads to decreased function of transcription factors associated with cardiovascular development and homeostasis (e.g. PPARs □ and □ and GATA-4) (Duffy et al. 2004; Reiterer et al. 2004). Furthermore, a threshold activity of the zinc finger transcription factors GATA4 and GATA6 is required for gene expression in the developing cardiovascular system (Xin et al. 2006). There is also evidence that zinc may be critical for normal AhR signaling. For example, both AhR and ARNT can interact with the zinc finger domain of Sp1 via their basic HLH/PAS domains (Kobayashi et al. 1996), and AhR can participate in induction of the zinc finger transcription factor Slug, which, in turn, regulates cellular physiology including cell adhesion and migration (Ikuta and Kawajiri 2006).
The objectives of the experiments described below were to determine if zinc plays a critical role in AhR function in the vascular endothelium. Our data strongly suggests that zinc is required for induction of the AhR responsive genes CYP1A1 and CYP1B1 upon endothelial cell exposure to xenobiotic and non-toxic AhR ligands. Alterations in AhR function and transcription present a novel mechanism for understanding induction of vascular diseases associated with zinc deficiency and exposure to environmental pollutants such as AhR ligands.
2. Materials and Methods
2.1. Cell culture and experimental media
Endothelial cells were isolated from porcine pulmonary arteries and cultured as previously described (Hennig et al. 1984). Cells were exposed to experimental media containing the membrane-permeable zinc chelator N, N, N′, N′-Tetrakis - (2-pyridylmethyl) ethylenediamine (TPEN) (Sigma-Aldrich, St. Louis, MO) with or without zinc supplementation (20 µmol/L) and/or the AhR ligands PCB77 or β-naphthoflavone (β-NF) (Sigma-Aldrich, St. Louis, MO) for 24 h. PCB77 was kindly provided by Dr. Larry W. Robertson (University of Iowa). The control media were composed of culture media containing 0.05% of ethanol and up to 0.04% of DMSO.
2.2. Measurement of CYP1A activity
Cellular cytochrome P450 1A (CYP1A) activity was measured in intact endothelial cells grown in 48 well plates (Costar, Corning Incorporated, NY) by ethoxyresorufin-o-deethylase (EROD) activity assay as previously described (Ramadass et al. 2003; Stegeman et al. 1995). 7-Ethoxyresorufin was used as a CYP1A substrate. CYP1A activity indicated by the fluorescence of resorufin generated was measured using a Cytofluor 4000 plate reader (PE Biosystems, Foster City, CA) with excitation and emission at 530 nm and 590 nm, respectively.
2.3. Measurement of CYP1A1 gene expression
Total RNA was extracted with Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s directions. cDNA was generated using the Reverse Transcription System (Promega, Madison, WI). Gene expression of CYP1A1 was determined by RT-PCR. β-Actin was used as an endogenous control for normalizing the expression of CYP1A1. Specific primer sequences were synthesized by IDT Technologies, Inc, San Jose, CA. The primers used were: CYP1A1, forward, 5'-TGGAGAGGCAAGAGTAGTTGG-3', and reverse, 5'-GGCACAACGGAGTAGCTCATA-3; β-Actin, forward, 5’-GGGACCTGACCGACTACCTC-3’, and reverse, 5’-GGGCGATGATCTTGATCTTC-3’.
2.4. Measurement of CYP1A1 and CYP1B1 protein expression
Cellular protein was extracted as previously described (Slim et al. 1999). Protein extracts were electrophoresed on 8–10% SDS-polyacrylamide gels followed by transfer to nitrocellulose membranes. The membranes were incubated in blocking solution for 1 hour followed by incubation with a 1:1000 dilution of CYP1A1 goat polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA) or CYP1B1 rabbit polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA) or a 1:4000 dilution of β-Actin rabbit polyclonal IgG (Sigma, St. Louis, MO) in blocking buffer overnight at 4°C. β-Actin was used as an endogenous control to normalize the expression of proteins of interest. The membranes were then incubated with a mouse anti-goat or goat anti-rabbit secondary antibody conjugated to horseradish peroxidase. Signals of the blots were measured using the enhanced chemiluminescence (ECL) detection system (GE Healthcare, Piscataway, NJ).
2.5. Statistical analysis
Statistical analysis was performed with SPSS 12.0 (SPSS, Inc., Chicago, IL). Data were analyzed using one way ANOVA with post hoc comparisons of the means by LSD procedure. Differences were considered significant at p < 0.05. Data are presented as means ± standard error of the mean (SEM).
3. Results
3.1. Zinc deficiency reduces PCB77-induced CYP1A activity and CYP1A1 mRNA expression in vascular endothelial cells
To determine if zinc deficiency can alter induction of the AhR responsive enzyme CYP1A1, we first measured PCB77 induction of CYP1A1 activity by the EROD assay. As expected, PCB77, a potent AhR agonist, significantly increased cellular CYP1A1 activity. Zinc deficiency caused by TPEN treatment did not change basal CYP1A1 activity but significantly reduced PCB77-induced CYP1A1 activity in vascular endothelial cells (Figure 1A). To determine if zinc deficiency alters CYP1A1 induction, we measured CYP1A1 mRNA expression in endothelial cells treated with PCB77 alone or in combination with TPEN. The PCB77-mediated upregulation of CYP1A1 mRNA expression was significantly reduced during zinc deficiency (Figure 1B).
Figure 1. Zinc deficiency reduces PCB77-induced CYP1A activity and CYP1A1 mRNA expression in vascular endothelial cells.
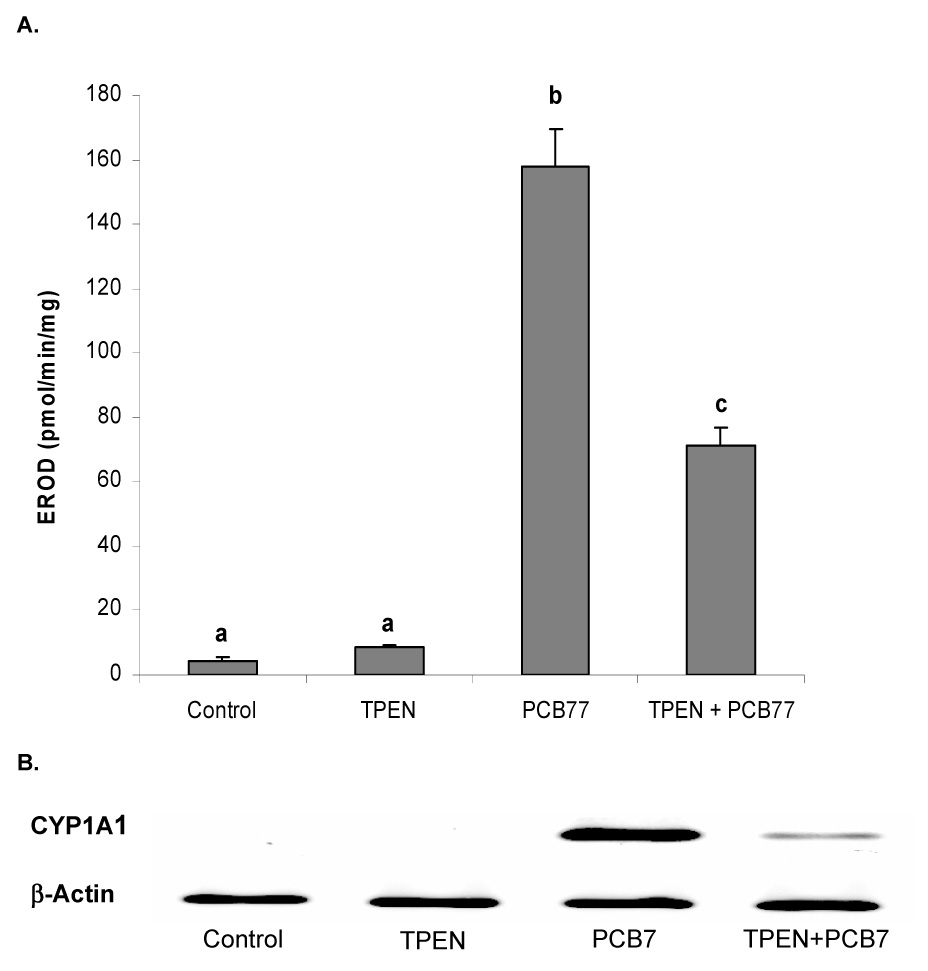
A. CYP1A activity measured by EROD assay. Cells were exposed to vehicle control (0.05 % of ethanol and 0.04 % of DMSO), TPEN (1.0 µM), PCB77 (0.04 µM), or TPEN (1.0 µM) plus PCB77 for 24 hours. Bars with different letters (a, b, c) are statistically different from each other (p < 0.05). n = 8. B. CYP1A1 mRNA expression measured by RT-PCR. Cells were exposed to vehicle control, TPEN, PCB77 (3.4 µM), or TPEN plus PCB77 for 24 hours. The figure is representative of the typical outcome of three repeated RT-PCR experiments.
3.2. Zinc deficiency compromises PCB77-induced CYP1A1 and CYP1B1 protein expression in vascular endothelial cells, which can be reversed with zinc supplementation
Western blot analysis demonstrated that the compromising effect of zinc deficiency on AhR responsive protein expression could be reversed by zinc supplementation. Specifically, PCB77 significantly increased the cellular protein levels of CYP1A1 and CYP1B1, both major PCB-inducible CYP1 enzymes. TPEN treatment alone did not affect expression of the two proteins, but co-treatment with TPEN and PCB77 led to a significant reduction in the expression of both enzymes. However, zinc supplementation of the TPEN-treated cell culture media reversed the reduction of CYP1A1 and CYP1B1 observed during zinc deficiency (Figure 2).
Figure 2. Zinc deficiency compromises PCB77-induced CYP1A1 and CYP1B1 protein expression in vascular endothelial cells.
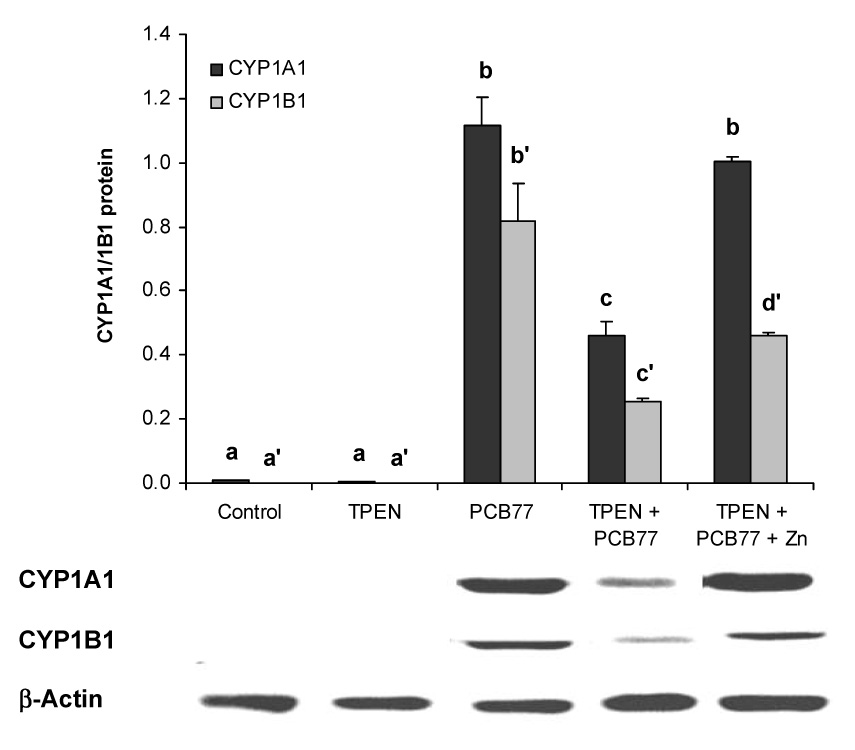
Similar to Figure 1, cells were exposed to vehicle control, TPEN, PCB77 (3.4 µM), TPEN plus PCB77, or TPEN with zinc supplementation (20 µM) plus PCB77 for 24 hours. The values are ratios of the densitometric units of CYP1A1 or CYP1B1 over those of β-Actin. Bars with different letters (a, b, c for CYP1A1 and a’, b’ c’ for CYP1B1) are statistically different from each other (p < 0.05). n = 3. The figure is a representative of the typical outcome of three repeated western blot experiments.
To determine if the observations made above were specific not only to PCB77 exposure but also relevant to other AhR ligands, similar experiments were performed using the non-toxic AhR agonistβ-NF. Treatment with β-NF significantly induced CYP1A1 protein expression, which was significantly reduced in TPEN-treated endothelial cells. When zinc was added back to the cell cultures, CYP1A1 protein induction by β-NF was completely reversible (Figure 3).
Figure 3. Zinc deficiency compromises β-naphthoflavone-induced CYP1A1 protein expression in vascular endothelial cells.
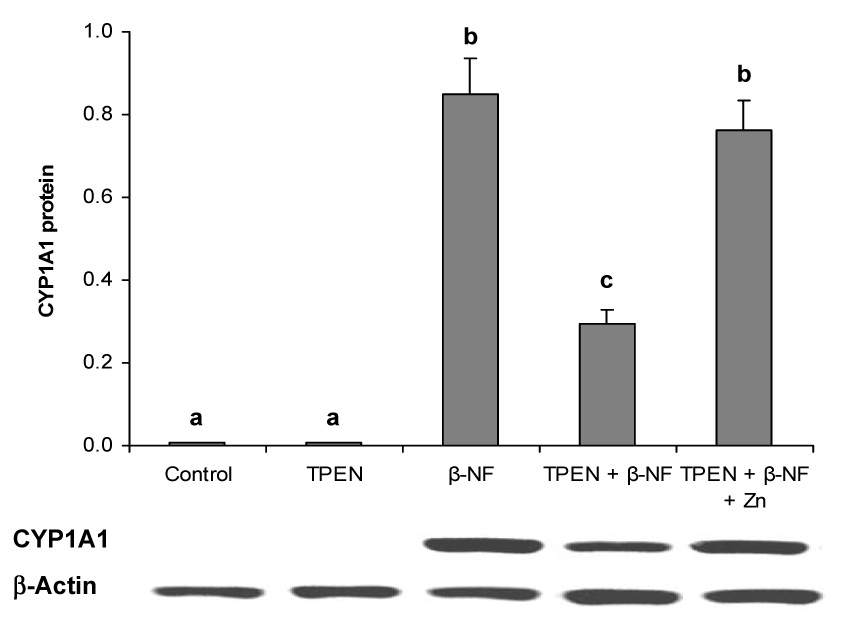
Cells were exposed to vehicle control, TPEN, β- NF (1.0 µM), TPEN plus β- NF, or TPEN with zinc supplementation plus β- NF for 24 hours. The values are ratios of the densitometric units of CYP1A1 over those of β-Actin. Bars with different letters (a, b, c) are statistically different from each other (p < 0.05). n = 3. The figure is a representative of the typical outcome of three repeated western blot experiments.
4. Discussion
The results from the experiments described above suggest that induction of AhR responsive genes in the endothelium is dependent on zinc availability, i.e., our data provide evidence that zinc is required for proper induction of the AhR-CYP1 pathway. As predicted, both toxic (PCB77) and non-toxic (β-NF) AhR ligands can markedly induce both mRNA and protein of CYP1A1, as well as activity of CYP1A1. Induction of the CYP1A1 gene was markedly down-regulated during zinc deficiency. This suggests that zinc deficiency can impair enzyme function, or that zinc is critical for proper transcriptional or translational induction of gene expression. Our data also suggest that the dysfunction of the AhR pathway is zinc specific because we were able to reverse the reduction in protein of CYP1A1 (and CYP1B1) by zinc supplementation of TPEN-treated cells.
Zinc has a critical role in protein structure, enzyme activity and gene regulation. Most of the genes that are zinc regulated are involved in signal transduction, responses to stress and redox changes, growth and energy utilization (Cousins et al. 2003). Thus, zinc has a role not only in tertiary protein structure but also in the capacity of proteins to interact with DNA, RNA and other proteins. For protein-DNA and protein-RNA interactions, zinc is commonly found as a zinc finger motif in transcription factors (Klug and Schwabe 1995). Classical zinc fingers have also been shown to interact with RNA and DNA/RNA complexes (Shi and Berg 1995). The susceptibility of zinc fingers to zinc deprivation is not well understood. For example, certain zinc-finger transcription factors, such as Sp1, EGR-1, vitamin D3 receptor and retinoid X receptor have been found to be inhibited due to loss of zinc as a response to zinc replacement by nitric oxide (Kroncke and Carlberg 2000). Following translation, a gene is also subject to zinc dependency during protein folding. For example, zinc is essential for certain chaperones, such as heat shock protein 40 (Hsp40) (Linke et al. 2003), and Hsp40, Hsp60, and Hsp70 mRNA expression were down-regulated during zinc deficiency (Cousins et al. 2003).
One possible mechanism for our observed compromising effect of zinc deficiency on PCB77-induced CYP1A1 and CYP1B1 expression and CYP1A1 activity could have been due to changes in AhR expression. However, western blot analysis showed no effect of either zinc deficiency or PCB77 or both on AhR expression (data not shown). This suggests that zinc deficiency may affect cell signaling downstream of AhR. Studies from other groups also suggest that AhR ligand-induced DNA binding is not altered by depletion of metal ions, including zinc (Denison and Deal 1990; Mahon and Gasiewicz 1992). However, these studies did not measure the effects of metal depletion on AhR dependent gene regulation, which may be a major target during zinc deficiency.
We provide evidence that induction of the AhR responsive P450 genes CYP1A1 and CYP1B1 is sensitive to cellular zinc depletion. Mechanisms for inhibition of AhR-dependent gene up-regulation during zinc deficiency could be possible by modulating ARNT nuclear translocation (Chun et al. 2001) or by effecting AhR-DRE binding (Saatcioglu et al. 1990). Another mechanism could be inhibition of zinc-dependent AhR co-factors that are necessary for transcriptional initiation and gene induction. One of these necessary interactions occurs with the transcription co-factor Sp1, which binds to GC-rich regions in the promoter of responsive genes and contains three Cys2His2 Zn fingers on its C-terminal region (Li et al. 2004; Suske 1999). It has been demonstrated that Sp1 expression and function is significantly reduced by cellular depletion of zinc and other metals (Denison and Deal 1990; Thiesen and Bach 1991) and that CYP1A1 induction requires AhR/ARNT interactions with Sp1 (Kobayashi et al. 1996). Our preliminary data suggest a correlation between Sp1 expression and reduced AhR signaling during zinc deficiency. This may be critical in understanding the involvement of the AhR in the regulation of cardiovascular functions.
Zinc finger DNA-binding proteins such as members of the Sp1 family also contain redox-sensitive thiol groups (Wu et al. 1996). For example, attenuation of cardiac dysfunction by PPAR-□ agonists is associated with down-regulation of redox-regulated transcription factors, including Sp1, NF-□B, and AP-1 (Ichihara et al. 2006). The regulation of redox-regulated transcription factors by zinc and the involvement of PPAR signaling further support our data that zinc is also required for the anti-inflammatory properties of both PPAR-□ and -□ agonists (Reiterer et al. 2004).
In summary, there is clear evidence that AhR function plays a critical role in the development and homeostasis of the cardiovascular system. Our results demonstrate that zinc deficiency can inhibit AhR-dependent gene induction. Impairment of the AhR pathway presents an additional molecular mechanism by which zinc deficiency negatively alters transcription factor function and homeostasis of the vascular system.
Acknowledgements
This study was supported by grants from NIH (P42ES07380), the University of Kentucky AES and the University of Kentucky Lyman T. Johnson Postdoctoral Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chun YS, Choi E, Yeo EJ, Lee JH, Kim MS, Park JW. A new HIF-1 alpha variant induced by zinc ion suppresses HIF-1-mediated hypoxic responses. J Cell Sci. 2001;114(Pt 22):4051–4061. doi: 10.1242/jcs.114.22.4051. [DOI] [PubMed] [Google Scholar]
- Cousins RJ, Blanchard RK, Moore JB, Cui L, Green CL, Liuzzi JP, Cao J, Bobo JA. Regulation of zinc metabolism and genomic outcomes. J Nutr. 2003;133(5 Suppl 1):1521S–1526S. doi: 10.1093/jn/133.5.1521S. [DOI] [PubMed] [Google Scholar]
- Denison MS, Deal RM. The binding of transformed aromatic hydrocarbon (Ah) receptor to its DNA recognition site is not affected by metal depletion. Mol Cell Endocrinol. 1990;69(1):51–57. doi: 10.1016/0303-7207(90)90088-p. [DOI] [PubMed] [Google Scholar]
- Duffy JY, Overmann GJ, Keen CL, Clegg MS, Daston GP. Cardiac abnormalities induced by zinc deficiency are associated with alterations in the expression of genes regulated by the zinc-finger transcription factor GATA-4. Birth Defects Res B Dev Reprod Toxicol. 2004;71(2):102–109. doi: 10.1002/bdrb.20004. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol. 1997;34(6):605–614. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- Fujii-Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. 2005;338(1):311–317. doi: 10.1016/j.bbrc.2005.08.162. [DOI] [PubMed] [Google Scholar]
- Hennig B, Shasby DM, Fulton AB, Spector AA. Exposure to free fatty acid increases the transfer of albumin across cultured endothelial monolayers. Arteriosclerosis. 1984;4(5):489–497. doi: 10.1161/01.atv.4.5.489. [DOI] [PubMed] [Google Scholar]
- Ichihara S, Obata K, Yamada Y, Nagata K, Noda A, Ichihara G, Yamada A, Kato T, Izawa H, Murohara T, et al. Attenuation of cardiac dysfunction by a PPAR-alpha agonist is associated with down-regulation of redox-regulated transcription factors. J Mol Cell Cardiol. 2006;41(2):318–329. doi: 10.1016/j.yjmcc.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Kawajiri K. Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp Cell Res. 2006;312(18):3585–3594. doi: 10.1016/j.yexcr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Klug A, Schwabe JW. Protein motifs 5. Zinc fingers. Faseb J. 1995;9(8):597–604. [PubMed] [Google Scholar]
- Kobayashi A, Sogawa K, Fujii-Kuriyama Y. Cooperative interaction between AhR.Arnt and Sp1 for the drug-inducible expression of CYP1A1 gene. J Biol Chem. 1996;271(21):12310–12316. doi: 10.1074/jbc.271.21.12310. [DOI] [PubMed] [Google Scholar]
- Kroncke KD, Carlberg C. Inactivation of zinc finger transcription factors provides a mechanism for a gene regulatory role of nitric oxide. Faseb J. 2000;14(1):166–173. doi: 10.1096/fasebj.14.1.166. [DOI] [PubMed] [Google Scholar]
- Li L, He S, Sun JM, Davie JR. Gene regulation by Sp1 and Sp3. Biochem Cell Biol. 2004;82(4):460–471. doi: 10.1139/o04-045. [DOI] [PubMed] [Google Scholar]
- Linke K, Wolfram T, Bussemer J, Jakob U. The roles of the two zinc binding sites in DnaJ. J Biol Chem. 2003;278(45):44457–44466. doi: 10.1074/jbc.M307491200. [DOI] [PubMed] [Google Scholar]
- Lund AK, Goens MB, Kanagy NL, Walker MK. Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol Appl Pharmacol. 2003;193(2):177–187. doi: 10.1016/j.taap.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Lund AK, Goens MB, Nunez BA, Walker MK. Characterizing the role of endothelin-1 in the progression of cardiac hypertrophy in aryl hydrocarbon receptor (AhR) null mice. Toxicol Appl Pharmacol. 2006;212(2):127–135. doi: 10.1016/j.taap.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Mahon MJ, Gasiewicz TA. Chelatable metal ions are not required for aryl hydrocarbon receptor transformation to a DNA binding form: phenanthrolines are possible competitive antagonists of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch Biochem Biophys. 1992;297(1):1–8. doi: 10.1016/0003-9861(92)90633-8. [DOI] [PubMed] [Google Scholar]
- Puga A, Tomlinson CR, Xia Y. Ah receptor signals cross-talk with multiple developmental pathways. Biochem Pharmacol. 2005;69(2):199–207. doi: 10.1016/j.bcp.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Ramadass P, Meerarani P, Toborek M, Robertson LW, Hennig B. Dietary flavonoids modulate PCB-induced oxidative stress, CYP1A1 induction, and AhR-DNA binding activity in vascular endothelial cells. Toxicol Sci. 2003;76(1):212–219. doi: 10.1093/toxsci/kfg227. [DOI] [PubMed] [Google Scholar]
- Reiterer G, Toborek M, Hennig B. Peroxisome proliferator activated receptors alpha and gamma require zinc for their anti-inflammatory properties in porcine vascular endothelial cells. J Nutr. 2004;134(7):1711–1715. doi: 10.1093/jn/134.7.1711. [DOI] [PubMed] [Google Scholar]
- Reiterer G, Toborek M, Hennig B. Zinc and cell signaling during inflammation: implications in atherosclerosis. Current Nutrititon and Food Science. 2006;2:23–28. [Google Scholar]
- Saatcioglu F, Perry DJ, Pasco DS, Fagan JB. Aryl hydrocarbon (Ah) receptor DNA-binding activity. Sequence specificity and Zn2+ requirement. J Biol Chem. 1990;265(16):9251–9258. [PubMed] [Google Scholar]
- Savouret JF, Berdeaux A, Casper RF. The aryl hydrocarbon receptor and its xenobiotic ligands: a fundamental trigger for cardiovascular diseases. Nutr Metab Cardiovasc Dis. 2003;13(2):104–113. doi: 10.1016/s0939-4753(03)80026-1. [DOI] [PubMed] [Google Scholar]
- Shi Y, Berg JM. Specific DNA-RNA hybrid binding by zinc finger proteins. Science. 1995;268(5208):282–284. doi: 10.1126/science.7536342. [DOI] [PubMed] [Google Scholar]
- Slim R, Toborek M, Robertson LW, Hennig B. Antioxidant protection against PCB-mediated endothelial cell activation. Toxicol Sci. 1999;52(2):232–239. doi: 10.1093/toxsci/52.2.232. [DOI] [PubMed] [Google Scholar]
- Stegeman JJ, Hahn ME, Weisbrod R, Woodin BR, Joy JS, Najibi S, Cohen RA. Induction of cytochrome P4501A1 by aryl hydrocarbon receptor agonists in porcine aorta endothelial cells in culture and cytochrome P4501A1 activity in intact cells. Mol Pharmacol. 1995;47(2):296–306. [PubMed] [Google Scholar]
- Suske G. The Sp-family of transcription factors. Gene. 1999;238(2):291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- Thiesen HJ, Bach C. Transition metals modulate DNA-protein interactions of SP1 zinc finger domains with its cognate target site. Biochem Biophys Res Commun. 1991;176(2):551–557. doi: 10.1016/s0006-291x(05)80219-0. [DOI] [PubMed] [Google Scholar]
- Wu X, Bishopric NH, Discher DJ, Murphy BJ, Webster KA. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol Cell Biol. 1996;16(3):1035–1046. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Davis CA, Molkentin JD, Lien CL, Duncan SA, Richardson JA, Olson EN. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci U S A. 2006;103(30):11189–11194. doi: 10.1073/pnas.0604604103. [DOI] [PMC free article] [PubMed] [Google Scholar]