

Abstract
Thrombospondin-1, an antiangiogenic matricellular protein, binds with high affinity to the angiogenic fibroblast growth factor-2, affecting its bioavailability and activity. The present work aimed at further locating the fibroblast growth factor-2 binding site of thrombospondin-1 and investigating its activity, using recombinant thrombospondin-1 proteins. Only recombinant constructs containing the thrombospondin-1 type III repeats bound fibroblast growth factor-2, whereas other domains, including the known anti-angiogenic type I repeats, were inactive. Binding was specific and inhibited by the anti thrombospondin-1 monoclonal antibody B5.2. Surface plasmon resonance analysis on BIAcore revealed a binding affinity (Kd) of 310 nM for the type III repeats and 11 nM for intact thrombospondin-1. Since the type III repeats bind calcium, the effect of calcium on thrombospondin-1 binding to fibroblast growth factor-2 was investigated. Binding was modulated by calcium, as thrombospondin-1 or the type III repeats bound to fibroblast growth factor-2 only in calcium concentrations <0.3 mM. The type III repeats inhibited binding of fibroblast growth factor-2 to endothelial cells, fibroblast growth factor-2-induced endothelial cell proliferation in vitro and angiogenesis in the chorioallantoic membrane assay in vivo, thus indicating the antiangiogenic activity of the domain. In conclusion, this study demonstrates that the fibroblast growth factor-2 binding site of thrombospondin-1 is located in the type III repeats. The finding that this domain is active in inhibiting angiogenesis indicates that the type III repeats represent a novel antiangiogenic domain of thrombospondin-1.
Keywords: Thrombospondin, FGF-2, angiogenesis inhibitors, calcium
INTRODUCTION
Thrombospondin-1 (TSP-1) is a matricellular protein (Bornstein, 2001). Through its modular structure, it binds simultaneously to cell surface receptors, matrix components, proteolytic enzymes, cytokines and growth factors, modulating the cell response to the environment (Bornstein & Sage, 2002). As with other matricellular proteins, TSP-1 is involved in complex biological processes including development, response to injury and several pathologies. It plays a complex role in physiological and pathological angiogenesis, particularly in tumor angiogenesis and consequently in tumor progression (Lawler & Detmar, 2004).
TSP-1 belongs to a family of 5 related vertebrate proteins (Adams & Lawler, 2004). It is a homotrimer. Each monomer consists of an N-terminal globular module, followed by the coiled-coil oligomerization domain, a procollagen module, three properdin-like type I repeats, the epidermal growth factor (EGF)-like type II repeats, the type III repeats, and the C-terminal globular domain. The type I repeats, which are present in TSP-1, TSP-2 and many other molecules, are responsible for most of the antiangiogenic activities (Lawler & Detmar, 2004).
Sequence identity/homology among the 5 TSPs is highest at the C-terminal end. Thus, the most conserved region among TSPs is the carboxy-terminal cassette - consisting of the type II and type III repeats and the C-terminal globular end - considered the hallmark of all TSPs (Adams, 2004). The type III, aspartate-rich repeats constitute the calcium-binding domain of TSPs. Several active sequences have been mapped in this domain (Adams, 2004), including the cell adhesion RGD sequence, the binding sites for neutrophil elastase and cathepsin G (Hogg, 1994), and attachments site for neutrophils (Majluf-Cruz et al., 2000) and sickle red blood cells (Watkins, Du, Scott, Ouwehand & Hillery, 2003).
TSP-1 directly binds to growth factors and angiogenesis regulatory factors, including fibroblast growth factor-2 (FGF-2) (Taraboletti et al., 1997), transforming growth factor beta (TGFβ), platelet derived growth factor (PDGF), vascular endothelial growth factor (VEGF), hepatocyte growth factor/scatter factor (HGF/SF), and Tat (reviewed in(Rusnati & Presta, 2006)). Binding of TSP-1 to growth factors affects their location, activation, and biological activity. We previously reported that binding of TSP-1 to FGF-2 affects the growth factor’s interaction with heparan sulfate proteoglycans (HSPG) on the cell surface and in the extracellular matrix, ultimately affecting FGF-2 bioavailability and activity (Margosio et al., 2003; Taraboletti et al., 1997).
Our previous studies indicated that the FGF-2 binding site is located in the 140-kDa carboxy-terminal fragment of TSP-1 (Taraboletti et al., 1997). In the present study we used recombinant portions of TSP-1 to further locate the FGF-2 binding site of TSP-1 and to investigate the activity of the identified domain in FGF-2-induced angiogenesis.
MATERIALS AND METHODS
Reagents
TSP-1 was purified from thrombin-stimulated human platelets (Taraboletti, Roberts, Liotta & Giavazzi, 1990). Human recombinant FGF-2 (R&D Systems, Minneapolis, MN) was obtained through the NCI-Biological Resources Branch (Frederick, MD). NeoMarkers anti TSP-1 monoclonal antibodies were from Lab Vision (Fremont, CA).
Expression of recombinant human TSP fragments
We used the vector pAcGP67.coco (COCO) to express the TSP-derived constructs (Mosher, Huwiler, Misenheimer & Annis, 2002). The COCO vector is a modified baculovirus transfer vector pAcGP67A (PharMingen, San Diego, CA) that has the signal peptide for GP67 and directs the recombinant protein into the secretory apparatus. DNA encoding for a six histidine-tag is immediately 3’ to the multiple cloning site. At the N-terminus, a 3 or 4 amino acid tail, dependent on the 5’ cloning site, precedes the start of the protein of interest after cleavage of the signal peptide. Modular segments of the cDNAs encoding human TSP-1 were cloned into pAcGP67.coco, virus was produced, and proteins were isolated and characterized as described (Hannah, Misenheimer, Annis & Mosher, 2003).
Biotin labeling
FGF-2, TSP-1 and the recombinant TSP-1 fragments were biotinylated as described (Margosio et al., 2003), except that the biotinylated TSP-1 fragments were isolated by chromatography on Micro Bio-Spin columns (Bio-Rad Lab, Milan, Italy).
Binding of FGF-2 to immobilized TSP-1 or TSP-1 fragments
Microtiter plates (Probind, Falcon, Becton-Dickinson Co., Lincoln Park, NJ) were incubated for 2 h at 37°C or o.n. at 4°C with TSP-1 or recombinant TSP-1 fragments in 40 µl PBS. The plates were washed with PBS 0.1% BSA, and non-specific binding sites were saturated by a 30 min-incubation with 1% BSA in PBS (PBS-BSA). Biotin-labeled FGF-2 (2–4 ng/well, in 40 µl) was added in PBS-BSA, and incubated at room temperature for 3 h. Wells were washed with PBS 0.1% BSA and bound FGF-2 was quantified by peroxidase-conjugated ExtrAvidin (Sigma) followed by 1.2-phenylenediamine dihydrocloride (OPD; Dako, Glostrup, Denmark) as a chromogen. The reaction was stopped with 0.5M H2SO4 and absorbance at 490 nm was measured.
Binding of labeled TSP-1 or TSP-1 fragments to immobilized FGF-2
Plates were coated with FGF-2 (0.1 µg in 40 µl PBS/well) o.n. at 4°C; then treated with PBS-BSA as described above. Biotin-labeled TSP-1 or TSP recombinant fragments, with or without inhibitors, were added at the indicated concentration, in PBS-BSA. After 3 h incubation, wells were washed and binding quantified as above.
Analysis of the TSP-1 fragment / FGF-2 complex in solution
Labeled FGF-2 (5 nM) was added to a solution (20 µl) containing the TSP-1 fragment (1 µM) in PBS and incubated for 2 h at 4°C. Samples were analyzed by Western blot after resolving in non-reducing 10% polyacrylamide SDS-PAGE. Labeled species were detected using peroxidase-conjugated ExtrAvidin and a chemiluminescence detection kit (ECL; Amersham).
BIAcore binding assay
A BIAcore X apparatus (BIAcore Inc, Piscataway, NJ) was used. Surface plasmon resonance was exploited to measure changes in refractive index caused by the binding of FGF-2 to E123CaG-1. E123CaG-1 (25 µg/ml) or BSA (used as a negative control and for blank subtraction) in 100 mM sodium acetate pH 3.0 were allowed to react with a flow cell of a CM5 sensorchip, pre-activated with 50 ml 0.2 M N-ethayl-N’-(3-diethylaminopropyl)carbodiimide hydrochloride and 0.05 M N-hydroxysuccinimide, leading to the immobilization of 1700 RU (approximately 25 pmoles/mm2) of E123CaG-1. Increasing concentrations of FGF-2 in 10 mM HEPES, 150 mM NaCl, 3.4 mM EDTA, 0.005% surfactant P20, pH 7.4 (HBS) were injected over the E123CaG-1 or BSA surfaces for 4 min., and then washed until dissociation was observed. In competition experiments, binding of FGF-2 (330 nM) to the E123CaG-1 surface was evaluated in the presence of the TSP-1 fragments (1.5 µM). Alternatively, FGF-2 was immobilized to the CM5 sensorchip as described (Margosio et al., 2003) and increasing concentrations of the TSP-1 fragments in HBS were injected as above. Binding parameters were calculated by the non-linear curve fitting software package BIAevaluation 3.2 using a single site model with correction for mass transfer. Only sensorgrams whose fitting gave values of X2 close to 100 were used (Khalifa, Choulier, Lortat-Jacob, Altschuh & Vernet, 2001). Dissociation constant (Kd) was derived from the dissociation rate (koff)/association rate (kon) ratio (kinetics).
Binding of FGF-2 to endothelial cells
Subconfluent cultures of bovine aortic endothelial cells (BAEC) in 96 well plates were incubated for 30 min at 4°C in serum-free DMEM with 0.15% gelatin and 25 mM HEPES (DMEM-gelatin). The medium was then replaced with cold DMEM-gelatin containing labeled FGF-2 (final concentration 10 ng/ml) with or without the indicated concentrations of recombinant TSP-1 fragments. The plate was incubated for 2 h at 4°C, washed with cold DMEM-gelatin and bound FGF-2 was measured as described above for the binding of FGF-2 to TSP-1.
Proliferation assay
BAEC (2500 cells/well) were plated in 96-well plates in DMEM 1.5% FCS. After 24 h, FGF-2 (5 ng/ml) was added, with or without the TSP-1 fragments, and incubated for 72 h. Plates were then stained and analyzed as described (Taraboletti et al., 1997).
Chorioallantoic membrane (CAM) assay
Fertilized White Leghorn chicken eggs were incubated at 37°C at constant humidity. On day 3, a square window was opened in the shell, 2 to 3 ml of albumen was removed to allow detachment of the developing CAM, and the window was sealed with a glass. On day 8, 1 mm3 sterilized gelatin sponges (Gelfoam Upjohn, Kalamazoo, MI) were placed on the CAM as described (Ribatti, Nico, Vacca & Presta, 2006). Sponges were loaded with: 1 µl of PBS or FGF-2 (0.5 µg), with or without E3Ca-1 (5 µg). At day 12, CAMs were photographed in ovo under a stereomicroscope (Olympus), and blood vessels entering the sponge within the focal plane of the CAM were counted by two observers in a double blind fashion.
RESULTS
Localization of the FGF-2 binding domain in the E3CaG-1 portion of TSP-1
We previously reported that the FGF-2 binding site is located in the 140 kDa proteolytic fragment of TSP-1 (Taraboletti et al., 1997). This trimeric fragment, contains several functional binding sites. The type I repeats (P in Figure 1) contain the know antiangiogenic sequences of TSP-1 and bind to CD36, integrins, TGFβ and extracellular matrix components; the EGF-like repeats (E in Figure 1) bind to integrins; the type III repeats (Ca in Figure 1) bind integrins and proteolytic enzymes; and the lectin-like module (G in Figure 1) binds CD47 (Adams, 2004; Calzada et al., 2004; Hogg, 1994; Majluf-Cruz et al., 2000; Short et al., 2005; Sid et al., 2004). Therefore all of these domains could be candidate binding sites for FGF-2. As a strategy to identify which domain bound FGF-2 we used recombinant portions of the 140 kDa fragment, spanning the entire region with partial overlaps. The three recombinant proteins were: i) CP123-1 (28.3 kDa), containing the procollagen and the 3 type I repeats; ii) P3E123-1 (23.8 kDa), containing the third type I repeat and the 3 type II, EGF-like repeats; iii) E3CaG-1 (60.2 kDa), with the third EGF-like domain, the type III calcium-binding repeats, and the globular carboxyterminal end of TSP-1 (Figure 1).
Figure 1.
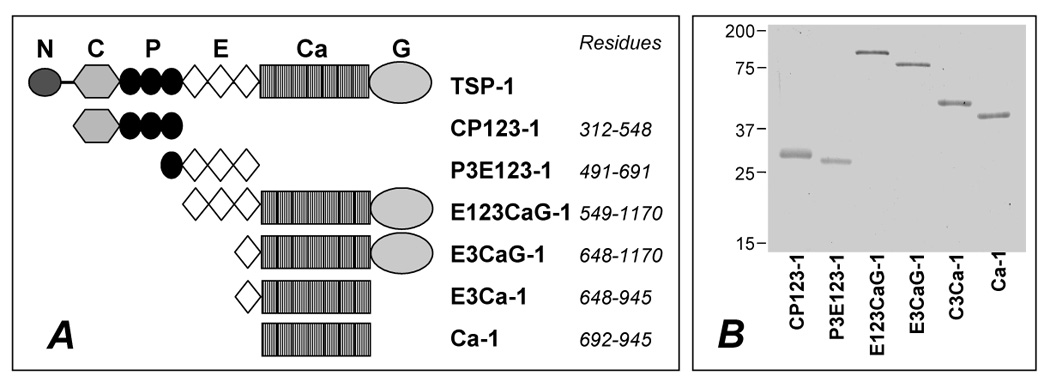
A) Schematic representations of the human TSP-1 monomer and the recombinant fragments used in this study. Abbreviation for the structural domains are: N, amino-terminal domain; C, procollagen like domain; P, properdin like, type I repeat; E, EGF-like, type II repeat; Ca, calcium-binding, type III repeats; G, globular carboxy-terminal domain. Residues are related to the initiating methionine. B) Purified recombinant proteins were resolved by SDS-PAGE and stained with Coomassie blue. Molecular weight markers are indicated in kDa. The type III repeats-containing domains migrated more slowly than predicted by their molecular weight, as reported (Anilkumar, Annis, Mosher & Adams, 2002; Hannah, Misenheimer, Annis & Mosher, 2003).
We first analyzed the binding of biotin-labeled FGF-2 to increasing amounts of immobilized proteins. FGF-2 bound to immobilized E3CaG-1, although at a lesser extent compared to whole TSP-1, whereas binding to equimolar coatings of CP123-1 or P3E123-1 was negligible (Figure 2A). The affinity of FGF-2 for immobilized E3CaG-1 was lower compared to TSP-1 (Figure 2A), suggesting a possible cooperative involvement of sequences in distant sites of the molecule. To test this possibility, binding of FGF-2 to combinations of the three fragments was tested. No combination improved the binding compared to E3CaG-1 alone (not shown), indicating that sequences in CP123-1 or P3E123-1 did not contribute to FGF-2 binding. Likewise, the longer segment E123CaG-1 had the same FGF-2-binding ability of E3CaG-1 (not shown).
Figure 2.
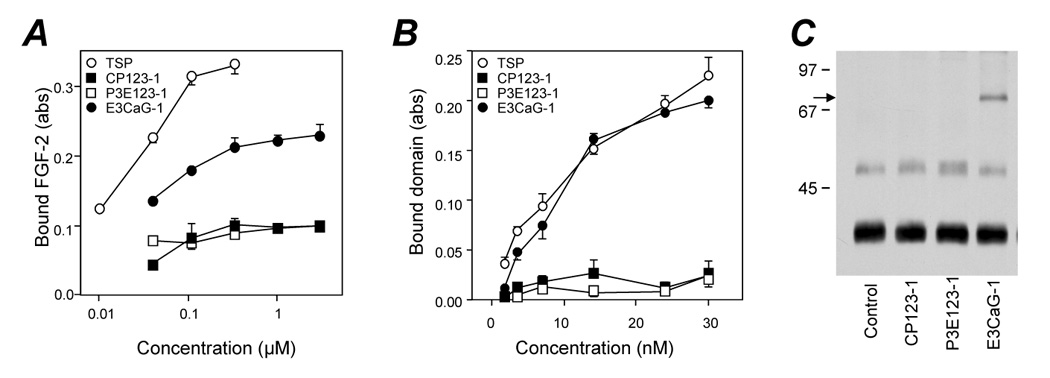
FGF-binding ability of CP123-1, P3E123-1, and E3CaG-1. A) Binding of labeled FGF-2 to plastic coated with increasing concentrations of TSP-1 or TSP-1 fragment. B) Binding of labeled TSP-1 or fragments, at the indicated concentrations, to immobilized FGF-2. Data are the amount of bound FGF-2, as absorbance (Abs), mean and SD of triplicates. C) Formation of the complex between FGF-2 and the indicated TSP-1 fragment in solution. Samples were analyzed in Western blot. Arrow indicates the putative FGF-2/E3CaG-1 complex. Molecular weight markers (in kDa) are on the left. Experiments in A were done in calcium-free buffers, in B calcium concentration was < 0.1 mM.
We also tested the reverse condition, namely the binding of free labeled recombinant TSP-1 fragments to immobilized FGF-2. Again, only E3CaG-1, but not CP123-1 nor P3E123-1, significantly bound FGF-2 (Figure 2B). In this case, free E3CaG-1 and TSP-1 bound in a similar way to immobilized FGF-2, suggesting that, in the process of immobilization, E3CaG may bind to plastic in conformations unfavorable to FGF binding.
Next, we analyzed the formation of the complex between labeled FGF-2 and each TSP-1 recombinant fragment in solution. After incubation of the two reagents in solution, the samples were resolved by SDS-PAGE under non-reducing conditions, followed by Western blot analysis. All samples showed major and minor bands corresponding to monomeric and dimeric FGF-2, respectively. The sample containing E3CaG-1 showed an additional high molecular weight band, compatible with a complex between FGF-2 and E3CaG-1 (Figure 2C). Noteworthy, the complex was visualized in the absence of a cross-linking agent, indicating the strength of binding, resistant to SDS and boiling. No band was observed when the construct was incubated in the absence of FGF-2 (not shown).
These findings indicated that the FGF-2 binding site is located in a region of approximately 60 kDa, comprising the third EGF-like domain through the C-terminus.
Localization of the FGF-2 binding domain in the type III repeats of TSP-1
We next investigated which subpart of the E3CaG-1 region retained the FGF-2 binding ability. To this purpose we used two smaller recombinant fragments: i) E3Ca-1 (34.1 kDa), containing the third type II repeat and the type III repeats but lacking the globular domain; ii) Ca-1 (29.9 kDa), containing only the type III repeats (Figure 1). Labeled FGF-2 bound to both immobilized E3Ca-1 and Ca-1 to a degree comparable to E3CaG-1 (Figure 3A). Moreover, both labeled fragments bound immobilized FGF-2 (Figure 3B).
Figure 3.
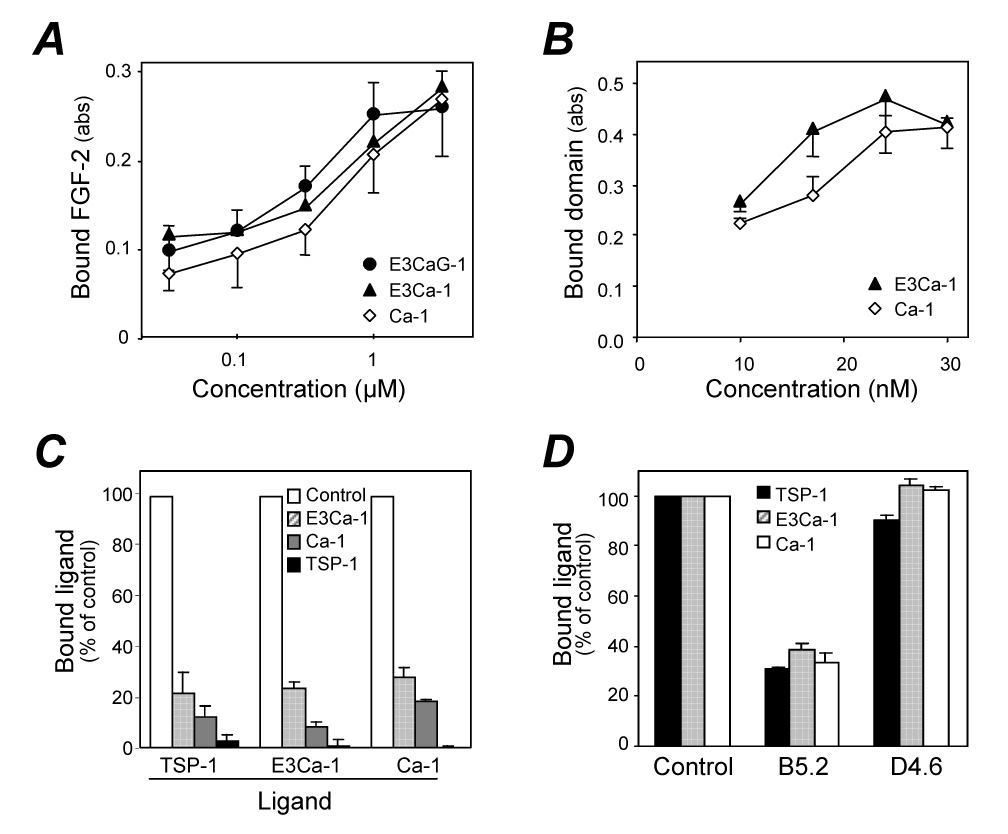
Localization of the FGF-binding site in the type III repeats of TSP-1. A) Binding of labeled FGF-2 to plastic coated with increasing concentrations of TSP-1 fragments. Data are the amount of bound FGF-2, as absorbance, mean and SD of triplicates. B) Binding of labeled E3Ca-1 or Ca-1, at the indicated concentrations, to immobilized FGF-2. C) Competition assay: binding of labeled TSP-1, E3Ca-1 and Ca-1 to immobilized FGF-2 in the presence of 2.5 µM unlabeled E3Ca-1, Ca-1, or TSP-1 used as competitors (see legend). Data are the percentage of control binding, in the absence of competitors. D) Effect of the monoclonal anti-TSP-1 antibodies B5.2 and D4.6 (50 µg/ml) on the binding of TSP-1, E3Ca-1, or Ca-1 to immobilized FGF-2. Binding is the percentage of control, in the absence of antibodies. Experiments were done in calcium-free buffers.
Binding of biotinylated E3Ca-1 and Ca-1 to FGF-2 was inhibited by unlabeled TSP-1, E3Ca-1 or Ca-1 used as competitors (Figure 3C). Among different anti TSP-1 monoclonal antibodies, binding was inhibited by the antibody B5.2 (recognizing a region comprised between the third type II repeat and the first type III repeat), but not by D4.6 (recognizing an unknown epitope in the type III repeats, Figure 3D), nor by isotype-matched control antibodies (not shown). In accordance with our previous findings (Taraboletti et al., 1997), heparin (100 µg/ml) blocked the binding of TSP-1 and Ca-1 to FGF-2 (88 and 99% inhibition, respectively, not shown).
Real time surface plasmon resonance studies
To calculate the binding parameters of the E123CaG-1/FGF-2 interaction, increasing concentrations of FGF-2 were injected over Biacore sensorchips coated with E123CaG-1 or BSA, as control. FGF-2 dose-dependently bound E123CaG-1 (Figure 4A), with an association rate constant (kon) of 9.9 × 104 M-1s-1, a dissociation rate constant (koff) of 1.4 × 10-2 s-1, and hence a dissociation constant (Kd) = koff/kon of 140 nM.
Figure 4.
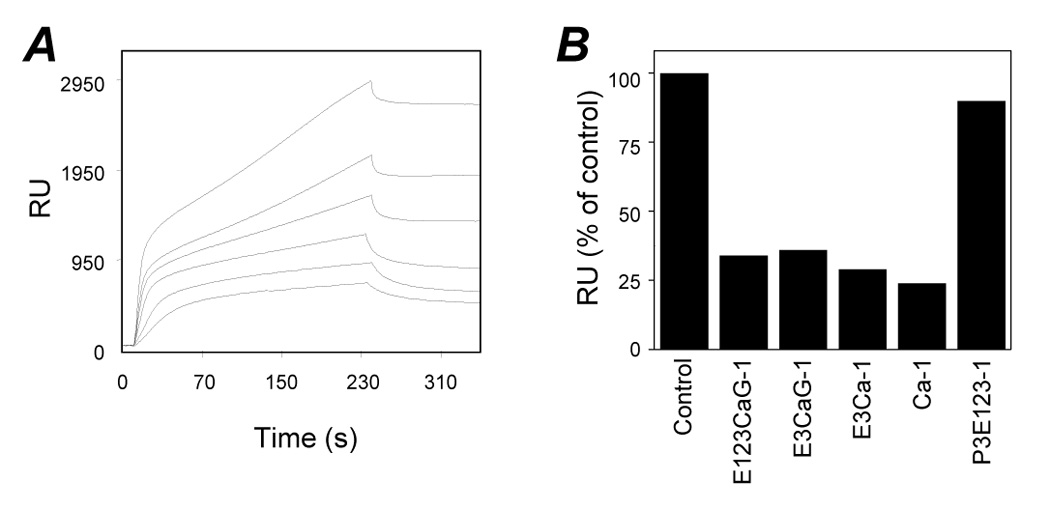
Real time biomolecular studies of FGF-2 interaction with TSP-derived peptides. A) Overlay of blank-subtracted sensograms showing the binding of FGF-2 (1400, 1120, 840, 560, 280 and 140 nM from top to bottom) to immobilized E123CaG-1. B) Competition assay. FGF-2 was loaded onto the BIAcore CM5 sensorchip containing immobilized E123CaG-1 in the absence or in the presence of the indicated TSP-1-fragment (1.5 µM). Data are the percentage of FGF-2 bound to the sensorchip at the equilibrium in the absence of any competitor. Experiments were done in calcium-free buffers.
To evaluate the FGF-2-binding ability of shorter TSP-1 segments, competition-binding assays were performed. FGF-2 was injected over the E123CaG-1 surface in the absence or presence of each fragment. Binding of FGF-2 to E123CaG-1 was prevented by all the Ca-containing fragments, but not by the P3E123 fragment, which does not bind FGF-2 (Figure 4B).
All the Ca-containing TSP-1 segments bound to immobilized FGF-2 with Kd spanning from 314 nM to 1400 nM (not shown), thus in the range of the values calculated for the interaction of immobilized E123CaG-1 with free FGF2. In these same experimental conditions, intact TSP-1 bound FGF-2 with a Kd of 11 nM (Margosio et al., 2003), confirming that the domains have a lower affinity for FGF-2 compared to intact TSP-1.
FGF-2/TSP-1 binding is calcium-dependent
The type III repeats of TSP-1 bind calcium, that affects the physical dimension of TSP, sensitivity to proteolysis, and cell attachment properties (reviewed in (Adams, 2004)). We therefore investigated the role of calcium in the binding of TSP-1, E3CaG-1 and Ca-1 to FGF-2.
Binding of labeled TSP-1, E3CaG-1 or Ca-1 to immobilized FGF-2 was measured in buffers containing increasing concentrations of calcium. In all cases, binding occurred only in low calcium concentrations but was inhibited in the presence of calcium concentrations higher than 0.3 mM, with remarkable concordance between TSP-1 and the two domains (Figure 5A,B). Calcium (1mM) also inhibited the formation of the E3CaG-1/FGF-2 complex in solution (Figure 5B, inset).
Figure 5.
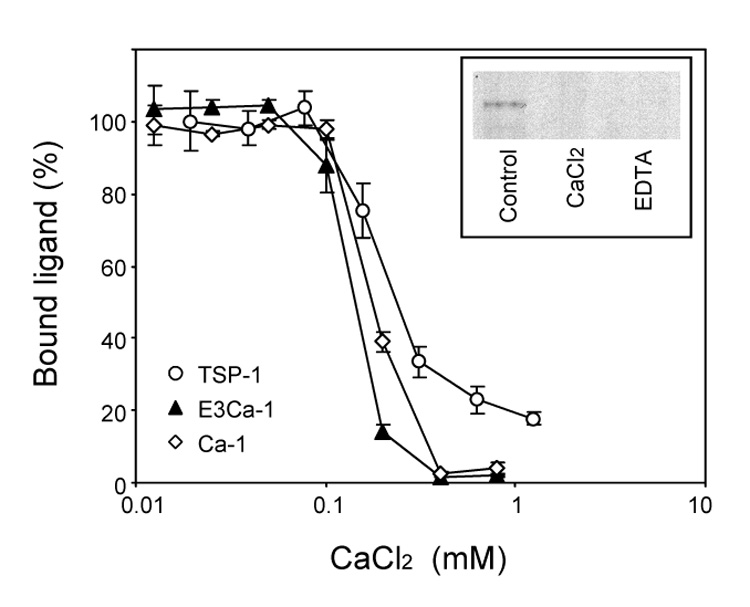
Effect of calcium on the interaction of TSP-1 and its fragments with FGF-2. Binding of 20 nM labeled TSP-1, E3Ca-1 or Ca-1 to immobilized FGF-2 was tested in the presence of the indicated concentration of CaCl2. Data are the amount of bound labeled protein, as the percentage of maximal binding (in calcium-free buffer), mean and SD of triplicates, from one experiment representative of 4–6. Inset: labeled FGF-2 and E3CaG-1 in solution were incubated in calcium-free buffer (control) or in the presence of CaCl2 (1 mM) or EDTA (5 mM). The samples were then analyzed by Western blot.
Calcium ions inhibited the binding of FGF-2 to E3Ca-1 and Ca-1with an IC50 of 0.095 ± 0.02 and 0.15 ± 0.01 mM, respectively (mean and SE of 4–6 experiments). Both transitions were steep and occurred over a 2.5-fold concentration range. The effects of calcium on the binding of the domains to FGF-2 (both IC50 value and steep dose dependency) closely parallel those on the induction of changes in the secondary structure of E3Ca-1 and Ca-1 as monitored by far ultraviolet circular dichroism (EC50 of 0.15 and 0.21 mM, Hill coefficient of 5.1 and 4.6 for E3Ca-1 and Ca-1, respectively) (Hannah, Misenheimer, Annis & Mosher, 2003), strongly indicative of an association between backbone structure detected by circular dichroism and ability to bind FGF-2.
EDTA treatment leads to profound alterations of the calcium-binding domain and shape change of the C-terminal region (Kvansakul, Adams & Hohenester, 2004; Misenheimer, Hannah, Annis & Mosher, 2003). Addition of EDTA (5 mM) abolished the binding of TSP-1 to FGF-2 (93% inhibition, not shown) and prevented the formation of the E3CaG-1/FGF-2 complex in solution (Figure 5B, inset).
Antiangiogenic activity of the type III repeats
To learn whether the FGF-2 binding domain of TSP-1, like intact TSP-1 (Taraboletti, Roberts, Liotta & Giavazzi, 1990; Taraboletti et al., 1997), inhibits FGF-2 activity, the recombinant fragments were tested in assays of FGF-2 binding to endothelial cells, induction of endothelial cell proliferation in vitro and angiogenesis in vivo. Both E3Ca-1 and Ca-1 inhibited the binding of labeled FGF-2 to endothelial cells (Figure 6A). This assay measures total binding, to both low affinity and high affinity receptors. Since low affinity receptors are the vast majority of the FGF-2 binding sites, this finding is indicative of an effect of the type III repeats on FGF-2 binding to cell surface heparan sulfates. E3Ca-1 and Ca-1 inhibited FGF-2-induced endothelial cell proliferation, with only marginal activity on baseline proliferation. In keeping with the presence of different antiproliferative sites in the TSP-1 molecule, the isolated type III repeats were less potent then entire TSP-1 in inhibiting endothelial cell proliferation (IC50 15±1 nM, 2.5±0.3 µM and 1.5±0.5 µM for TSP-1, E3Ca-1 and Ca-1, respectively). Nonetheless, we found that the B5.2 antibody which recognizes the type III repeats reduced the antiproliferative activity of whole TSP-1 (38±2% reduction, not shown), indicating that this domain relevantly contributes to the antiangiogenic activity of the intact molecule. The differences in domain concentrations active in inhibiting FGF-2 binding and cell proliferation were presumably due to the differences in the experimental conditions, since also TSP-1 showed activity at different concentrations in the two assays.
Figure 6.
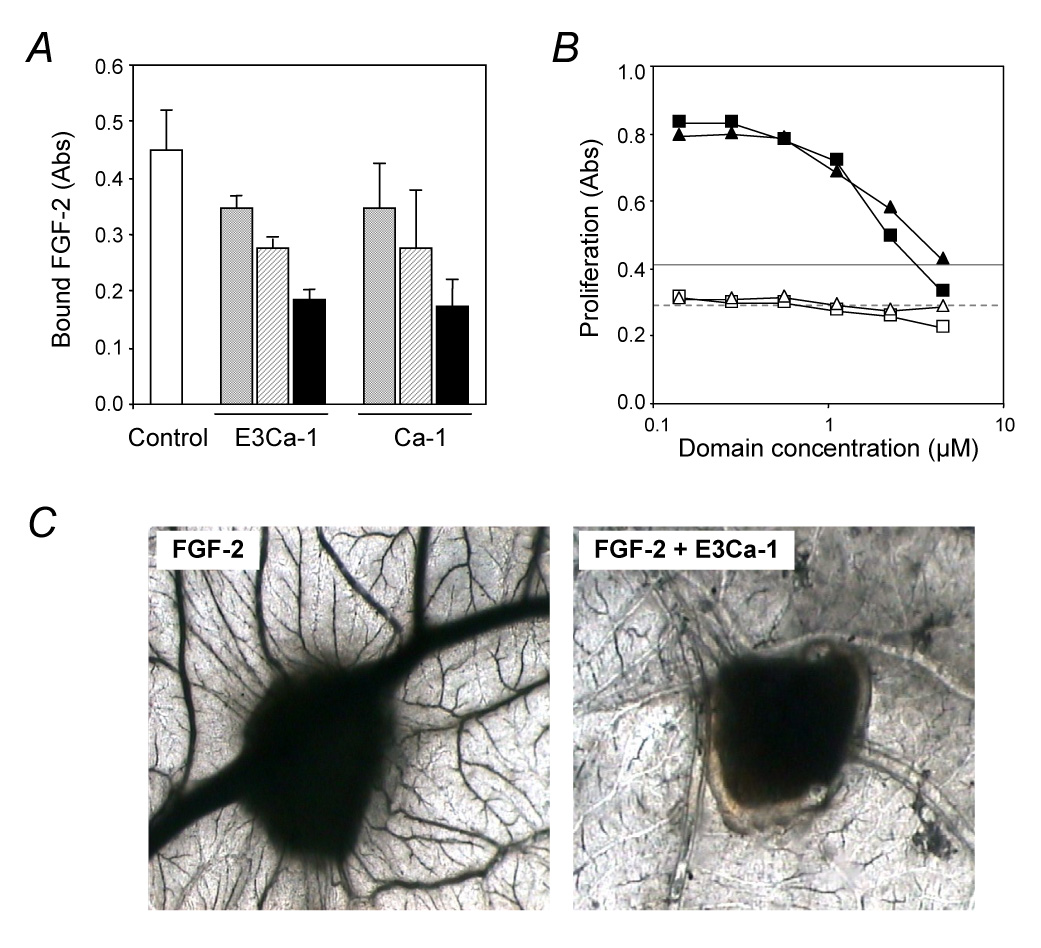
Antiangiogenic activity of the TSP-1-type III repeats. A) Binding of FGF-2 to endothelial cells. BAEC were incubated with labeled FGF-2 in the absence (control, white column) or presence of the indicated recombinant fragment (7.5 µM grey columns, 15 µM striped columns, 30 µM black columns). The amount of cell-bound FGF-2 is expressed as absorbance. B) Endothelial cell proliferation. BAEC were exposed to the indicated concentration of E3Ca-1 (triangles), Ca-1 (squares), or TSP-1 (25 µg/ml, lines) with (black symbols, solid line) or without (white symbols, dashed line) 5 ng/ml FGF-2 and incubated for 3 days. Proliferation is expressed as absorbance. C) FGF-2-induced angiogenesis in the CAM assay. FGF-2 was administered in the absence or presence of E3Ca-1 on day 8 and pictures were taken 4 days later (n=10). Original magnification: 50 x.
In vivo, macroscopic observations of the CAM showed that E3Ca-1 alone did not affect physiological angiogenesis, presenting few allantoic vessels arranged around the sponge similarly to PBS-loaded sponges (not shown). Sponges adsorbed with FGF-2 were surrounded by allantoic vessels developing radially towards the implant in a “spoked-wheel” pattern (Figure 6C). When sponges were loaded with FGF-2 and E3Ca-1, a significant reduction of FGF-2-induced angiogenic response was appreciable macroscopically: mean number of blood vessels entering the sponge was 28±4 (FGF-2 alone) versus 3±1 (FGF-2+E3Cal-1, p < 0.001).
DISCUSSION
TSP-1 directly binds to growth factors, including FGF-2 (Margosio et al., 2003; Taraboletti et al., 1997). In this study, by using recombinant portions of TSP-1, we have shown that the FGF-2-binding site is located in the type III repeats, and that this domain is active in inhibiting FGF-2 angiogenic activity. The major antiangiogenic site of TSP-1 has been localized in the type I repeats. Our findings therefore provide evidence of the presence of a different, novel antiangiogenic site of TSP-1, located on a different domain.
The type III repeats, together with the EGF-like type II repeats and the carboxy terminal globular end, constitutes the most conserved portion of TSPs. The most remarkable activity discerned for the repeats to date has been cooperative binding of calcium ions (Adams, 2004). Analysis of the crystal structure of type III repeat-containing proteins has revealed a peculiar protein “wire” structure organized around a metal core (Carlson et al., 2005; Kvansakul, Adams & Hohenester, 2004). Up to 30 calcium ions bind each TSP-1 or TSP-2 subunit (Hannah, Misenheimer, Pranghofer & Mosher, 2004; Kvansakul, Adams & Hohenester, 2004). Some of the bound calciums are not readily exchangeable in equilibrium dialysis experiments (Misenheimer, Hannah, Annis & Mosher, 2003). We found that calcium-saturated TSP-1, E3Ca-1 and Ca-1 did not bind to FGF-2. The finding that the transitions for binding of E3Ca-1 and Ca-1 to FGF-2 and alterations of secondary structure occur over the same ranges of calcium concentration indicates that TSP-1/FGF-2 binding requires the type III repeats to be in a calcium-depleted structure. Nevertheless, the presence of EDTA, which presumably removes all bound calcium and causes maximal loss of structure, abolished binding of FGF-2 to TSP-1 or E3Ca-1. This finding implies that TSP-1/FGF-2 binding is controlled by a structural change of TSP-1 that is induced by reduction in calcium ion concentration in the environment but requires the presence of residual bound calcium.
Other activities of TSP type III repeats are regulated by calcium, including attachment of neutrophils (Majluf-Cruz et al., 2000) and binding to collagen (Galvin, Vance, Dixit, Fink & Frazier, 1987), neutrophil elastase and cathepsin G (Hogg, 1994). In most cases, the induced activity is ascribed to a calcium-dependent structural modification that allows the exposure of the active site (Kvansakul, Adams & Hohenester, 2004). Similarly, it is conceivable that the FGF-2 binding site is cryptic in calcium-saturated TSP-1, and becomes exposed in low calcium concentrations, supporting the concept that the readily exchangeable calcium ions have a critical role in controlling TSP structure and functions (Adams, 2004).
In experimental settings, calcium loading of TSP-1 is modulated by varying the calcium concentration in the environment. Little is known about the physiological mechanisms that regulate calcium loading of TSP in vivo. The local concentration of extracellular calcium undergoes dynamic fluctuations under physiological conditions, and in highly specialized tissues calcium concentrations transiently drop to levels compatible with the TSP/FGF binding (Hofer, 2005). However, the actual relevance of local calcium fluctuation (for example in the tumor environment) in determining the ability of TSP-1 to bind to FGF-2 still remains to be elucidated. Besides calcium, other factors can induce the exposure of active TSP sequences, by altering the tertiary structure of the type III repeats. The status of disulphide bonds between consecutive type III repeats regulates the exposure of the RGD sequence (Kvansakul, Adams & Hohenester, 2004) and the binding sites for neutrophil elastase and cathepsin G (Hogg, 1994). The conformation of TSP type III repeats is also affected by the binding of certain molecules to TSP, even at sites distant from this domain (Dardik & Lahav, 1999; Watkins, Du, Scott, Ouwehand & Hillery, 2003). Altogether, these considerations suggest that the exposure of the FGF-2 binding site is achieved through both calcium-dependent and calcium-independent mechanisms.
Binding of TSP-1 to FGF-2 prevents the growth factor interaction with HSPG on the cell surface and in the extracellular matrix (Margosio et al., 2003; Taraboletti et al., 1997). Since HSPG modulate the activity of heparin-binding factors (Iozzo, 2005) the binding of TSP-1 to FGF-2 might have important biological consequences. Indeed we previously found that the interaction of TSP-1 with FGF-2 prevented FGF-2 binding to endothelial cells, internalization, interaction with the extracellular matrix and proliferative activity (Margosio et al., 2003; Taraboletti et al., 1997). In agreement, in this study we have found that the FGF-2 binding type III repeat inhibit FGF-2 binding to endothelial cells and FGF-2-induced endothelial cell proliferation, suggesting that this domain has the potential to affect the growth factor bioavailability and activity. The finding that the type III repeat prevent FGF-2-induced angiogenesis in the CAM further point to a role for this domain in angiogenesis.
This study further indicates that the activity of TSP-1 depends on the environmental conditions. Local fluctuations of calcium concentration, the presence of TSP-1-degrading proteases, TSP-1 ligands or receptors might modulate the bioavailability of functional sites of TSP-1, dictating the final effect of TSP-1 on cell invasion, adhesion and angiogenesis. The complex role of TSP-1 in angiogenesis - through the proangiogenic N-terminal domain or the anti-angiogenic type I and type III repeats - has been analyzed in several experimental models, including angiogenesis assays in the CAM and cornea (this study and (Taraboletti et al., 2000)). The isolated, recombinant domains provide a useful tool to dissect the role of each domain in various experimental conditions, and will help clarify the impact of TSP-1 on angiogenesis in different settings, including tumor angiogenesis.
In conclusion, this study demonstrates the presence of a novel FGF-2-binding antiangiogenic site in the TSP-1 type III repeats. Binding to FGF-2 is affected by local conformational changes induced by calcium concentration. Moreover, we have found that this domain is active in inhibiting FGF-2-induced angiogenesis. Additional investigations are warranted to fully clarify the relevance of the FGF-2 /type III repeats interaction in the physiological and pathological roles of thrombospondin.
ACKNOWLEDGEMENTS
We thank F. Sangalli and A. Silini for helpful assistance.
Supported by grants from the Italian Association for Cancer Research (AIRC), the European Union FPS/6 LSHC-CT-2003-503233, the NIH (HL54462), and Ministero dell’Unversità e della Ricerca (MIUR, FIRB2001 and PRIN2005). B.M. received fellowships from the Italian Foundation for Cancer Research (FIRC), and successively from Emilio Mussini Fund; B.A.H. from the American Heart Association (AHA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams JC. Functions of the conserved thrombospondin carboxy-terminal cassette in cellextracellular matrix interactions and signaling. Int J Biochem Cell Biol. 2004;36:1102–1114. doi: 10.1016/j.biocel.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar N, Annis DS, Mosher DF, Adams JC. Trimeric assembly of the C-terminal region of thrombospondin-1 or thrombospondin-2 is necessary for cell spreading and fascin spike organisation. J Cell Sci. 2002;115:2357–2366. doi: 10.1242/jcs.115.11.2357. [DOI] [PubMed] [Google Scholar]
- Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- Calzada MJ, Annis DS, Zeng B, Marcinkiewicz C, Banas B, Lawler J, et al. Identification of novel beta1 integrin binding sites in the type 1 and type 2 repeats of thrombospondin-1. J Biol Chem. 2004;279:41734–41743. doi: 10.1074/jbc.M406267200. [DOI] [PubMed] [Google Scholar]
- Carlson CB, Bernstein DA, Annis DS, Misenheimer TM, Hannah BL, Mosher DF, et al. Structure of the calcium-rich signature domain of human thrombospondin-2. Nat Struct Mol Biol. 2005;12:910–914. doi: 10.1038/nsmb997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardik R, Lahav J. Functional changes in the conformation of thrombospondin-1 during complexation with fibronectin or heparin. Exp Cell Res. 1999;248:407–414. doi: 10.1006/excr.1999.4415. [DOI] [PubMed] [Google Scholar]
- Galvin NJ, Vance PM, Dixit VM, Fink B, Frazier WA. Interaction of human thrombospondin with types I-V collagen: direct binding and electron microscopy. J Cell Biol. 1987;104:1413–1422. doi: 10.1083/jcb.104.5.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah BL, Misenheimer TM, Annis DS, Mosher DF. A polymorphism in thrombospondin-1 associated with familial premature coronary heart disease causes a local change in conformation of the Ca2+-binding repeats. J Biol Chem. 2003;278:8929–8934. doi: 10.1074/jbc.m211185200. [DOI] [PubMed] [Google Scholar]
- Hannah BL, Misenheimer TM, Pranghofer MM, Mosher DF. A polymorphism in thrombospondin-1 associated with familial premature coronary artery disease alters Ca2+ binding. J Biol Chem. 2004;279:51915–51922. doi: 10.1074/jbc.M409632200. [DOI] [PubMed] [Google Scholar]
- Hofer AM. Another dimension to calcium signaling: a look at extracellular calcium. J Cell Sci. 2005;118:855–862. doi: 10.1242/jcs.01705. [DOI] [PubMed] [Google Scholar]
- Hogg PJ. Thrombospondin 1 as an enzyme inhibitor. Thrombosis and Haemostasis. 1994;72:787–792. [PubMed] [Google Scholar]
- Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646–656. doi: 10.1038/nrm1702. [DOI] [PubMed] [Google Scholar]
- Khalifa MB, Choulier L, Lortat-Jacob H, Altschuh D, Vernet T. BIACORE data processing: an evaluation of the global fitting procedure. Anal Biochem. 2001;293:194–203. doi: 10.1006/abio.2001.5119. [DOI] [PubMed] [Google Scholar]
- Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. Embo J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler J, Detmar M. Tumor progression: the effects of thrombospondin-1 and -2. Int J Biochem Cell Biol. 2004;36:1038–1045. doi: 10.1016/j.biocel.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Majluf-Cruz A, Manns JM, Uknis AB, Yang X, Colman RW, Harris RB, et al. Residues F16-G33 and A784-N823 within platelet thrombospondin-1 play a major role in binding human neutrophils: evaluation by two novel binding assays. J Lab Clin Med. 2000;136:292–302. doi: 10.1067/mlc.2000.109407. [DOI] [PubMed] [Google Scholar]
- Margosio B, Marchetti D, Vergani V, Giavazzi R, Rusnati M, Presta M, et al. Thrombospondin 1 as a scavenger for matrix-associated fibroblast growth factor 2. Blood. 2003;102:4399–4406. doi: 10.1182/blood-2003-03-0893. [DOI] [PubMed] [Google Scholar]
- Misenheimer TM, Hannah BL, Annis DS, Mosher DF. Interactions among the three structural motifs of the C-terminal region of human thrombospondin-2. Biochemistry. 2003;42:5125–5132. doi: 10.1021/bi026983p. [DOI] [PubMed] [Google Scholar]
- Mosher DF, Huwiler KG, Misenheimer TM, Annis DS. Expression of recombinant matrix components using baculoviruses. Methods Cell Biol. 2002;69:69–81. doi: 10.1016/s0091-679x(02)69008-9. [DOI] [PubMed] [Google Scholar]
- Ribatti D, Nico B, Vacca A, Presta M. The gelatin sponge-chorioallantoic membrane assay. Nature Protocols. 2006;1:85–91. doi: 10.1038/nprot.2006.13. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Presta M. Extracellular angiogenic growth factor interactions: an angiogenesis interactome survey. Endothelium. 2006;13:93–111. doi: 10.1080/10623320600698011. [DOI] [PubMed] [Google Scholar]
- Short SM, Derrien A, Narsimhan RP, Lawler J, Ingber DE, Zetter BR. Inhibition of endothelial cell migration by thrombospondin-1 type-1 repeats is mediated by beta1 integrins. J Cell Biol. 2005;168:643–653. doi: 10.1083/jcb.200407060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sid B, Sartelet H, Bellon G, El Btaouri H, Rath G, Delorme N, et al. Thrombospondin 1: a multifunctional protein implicated in the regulation of tumor growth. Crit Rev Oncol Hematol. 2004;49:245–258. doi: 10.1016/j.critrevonc.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Taraboletti G, Roberts D, Liotta LA, Giavazzi R. Platelet thrombospondin modulates endothelial cell adhesion, motility, and growth: a potential angiogenesis regulatory factor. J Cell Biol. 1990;111:765–772. doi: 10.1083/jcb.111.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboletti G, Belotti D, Borsotti P, Vergani V, Rusnati M, Presta M, et al. The 140-kilodalton antiangiogenic fragment of thrombospondin-1 binds to basic fibroblast growth factor. Cell Growth Differ. 1997;8:471–479. [PubMed] [Google Scholar]
- Taraboletti G, Morbidelli L, Donnini S, Parenti A, Granger HJ, Giavazzi R, et al. The heparin binding 25 kDa fragment of thrombospondin-1 promotes angiogenesis and modulates gelatinase and TIMP-2 production in endothelial cells. Faseb J. 2000;14:1674–1676. doi: 10.1096/fj.99-0931fje. [DOI] [PubMed] [Google Scholar]
- Watkins NA, Du LM, Scott JP, Ouwehand WH, Hillery CA. Single-chain antibody fragments derived from a human synthetic phage-display library bind thrombospondin and inhibit sickle cell adhesion. Blood. 2003;102:718–724. doi: 10.1182/blood-2002-11-3497. [DOI] [PubMed] [Google Scholar]