

Abstract
The NF-κB transcription factor family has a crucial role in rapid responses to stress and pathogens. We show that the NF-κB subunit RelB is functionally associated with the aryl hydrocarbon receptor (AhR) and mediates transcription of chemokines such as Interleukin-8 (IL-8) via activation of AhR and protein kinase A (PKA). RelB physically interacts with AhR and binds to an unrecognized RelB/AhR responsive element (RelBAhRE) of the IL-8 promoter linking two signaling pathways to activate gene transcription. We found a time-dependent recruitment of AhR to the RelBAhRE site of IL-8 mediated by the AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) and via activation of PKA. Furthermore, NF-κB-binding sites that are preferentially recognized by RelB/p52 are a target for RelB/AhR complexes without addition of any stimuli implicating the endogenous function of the AhR. RelB/AhR complexes are also found to bind on Xenobiotic Responsive Elements (XRE), and RelB drastically increases the TCDD-induced XRE reporter activity. The interaction of RelB with AhR signaling, and AhR with NF-κB RelB signaling pathways represent a new mechanism of cross talk between the two nuclear receptor paradigms.
Keywords: Binding Sites, Cell Line, Tumor, Cyclic AMP-Dependent Protein Kinases, metabolism, Forskolin, pharmacology, Humans, Interleukin-8, genetics, metabolism, Molecular Sequence Data, NF-kappa B, metabolism, Promoter Regions (Genetics), Protein Binding, Receptors, Aryl Hydrocarbon, genetics, metabolism, Response Elements, Signal Transduction, drug effects, Tetrachlorodibenzodioxin, pharmacology, Transcription Factor RelB, genetics, metabolism, Transcription, Genetic, genetics
INTRODUCTION
The NF-kB/Rel transcription factors play critical roles in diverse cellular processes including adaptive and innate immunity, cell differentiation, proliferation, and apoptosis. Transcriptionally active NF-kB dimers are formed by combinatorial association of five subunits: p50, RelA (p65), p52, c-Rel, and RelB (1). The classic inducible NF-kB heterodimer consists of the p50 and RelA subunits, each contacting one half of the DNA binding site. The slight variations in the 10 base pair consensus sequence, 5′-GGGGYNNCCY-3′, confers a preference for selected Rel combinations (2). Compared to other members of the NF-kB family the biological mode of action of the RelB subunit has remained elusive. RelB does not express the functional properties common of the Rel family and no exclusive DNA binding activity had been discovered until recently. In vivo analysis revealed that the IκB Kinase (IKK)α activates an alternative NF-κB pathway based on processing of NF-κB2/p100 and release of RelB/p52 dimers in response to lymphotoxin β receptor (LTβR) trimers (3). Gene induction by IKKα depends on selective activation of RelB/p52 dimers, which recognize a unique type of NF-κB binding site (5′-NGGAGAYTTN-3′) regulating organogenic chemokines such as B lymphocyte chemoattractant (BLC) or the B cell-activating factor of the tumor necrosis factor family (BAFF) (4). Unlike p50 or RelA, which are expressed in virtually all cell types, RelB is predominantly present in lymphoid tissue and can be constitutively expressed in the nucleus (5).
The AhR is a member of basic helix-loop-helix (bHLH-PAS) transcription factors including Period (Per), AhR nuclear translocator (ARNT), and single minded (SIM) regulating hypoxia, circadian rhythm, and cellular processes like differentiation and apoptosis (6). The AhR is well described as a ligand-dependent activated transcription factor. About 15 years ago Hankinson and coworkers (7) identified the encoded protein ARNT which is required for ligand-dependent translocation of the AhR into the nucleus and its binding to XRE mediating induction of xenobiotic metabolizing enzymes (classical AhR/ARNT pathway). Numerous exogenous compounds (e.g. polycyclic aromatic hydrocarbons, benzimidazoles and flavonoids) with various binding affinities have been shown to bind to and activate the AhR (8), but the physiological ligand or function of the AhR remained a key question. However, the conservation of the receptor in a wide range of animal species (including humans) suggests a fundamental role in cellular physiology. The non-activated form of the AhR is complexed with HSP90 and XAP2 in the cytosol, but depending on cell type and physiological conditions the AhR is also located in the nucleus in absence of exogenous ligand (9). XAP2 may enhance the rate of nuclear translocation of the ligand-bound human AhR complex and modulates the sub-cellular localization of the mouse AhR (10, 11). The AhR has a critical role in development: AhR null mice show deficiencies in liver development, increased apoptosis in liver and decreased accumulation of lymphocytes in the spleen and lymph nodes (12). This further indicates that the AhR is also located in the nucleus to regulate these physiological processes in the absence of exogenous ligands. Nuclear localization and activity of the AhR during embryonic development has also been reported (13). Recently, a PKA-dependent activation and nuclear translocation of the AhR by forskolin (FSK)/cAMP has been reported (14). However, the PKA-activated form of AhR was found to be different from the ligand-activated AhR and does not dimerize with ARNT, although the dimerization partner of the PKA-activated AhR and its regulatory function remained undiscovered.
In recent reports we have shown that the induction of IL-8 in vitro (15) as well as the induction of KC (homolog of human IL-8) in mice (16) by TCDD requires a functional AhR. By analyzing the mechanism of the AhR-mediated induction of IL-8, this study demonstrates the physical and functional association of the AhR and the NF-κB subunit RelB, resulting in transcriptional activation of IL-8. IL-8 promoter studies with human macrophages U937 and human hepatoma cell line HepG2 revealed a novel RelB/AhR responsive element (RelBAhRE) required for transcriptional activation of IL-8 by FSK as well as by the prototype of AhR ligands, TCDD. Using electromobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) assays, we demonstrate the recruitment of AhR to the RelBAhRE region of the IL-8 promoter stimulated by FSK and TCDD. Furthermore, supershift analysis revealed clear binding activity of AhR in complex with RelB on a recently identified NF-κB-binding site located on promoter regions of chemokines like BLC and BAFF that are induced by FSK and TCDD.
RESULTS
Induction of IL-8 by FSK and TCDD is AhR-dependent
In the present study we found that activation of the AhR by FSK or TCDD leads to a sustained induction of the pro-inflammatory chemokine IL-8 in human macrophages in a time-dependent manner (Fig. 1A). FSK was included in our study as an alternative activator of the AhR and inducer of IL-8 since FSK has been reported to activate AhR through a PKA-dependent mechanism (14) and has been shown to increase IL-8 (20). The FSK- and TCDD-induced mRNA expression in macrophages correlated with elevated protein level and secretion of IL-8 (Fig. 1B and C). Results from transfection studies with short interfering RNA (siRNA) into U937 macrophages to target AhR suggest that TCDD as well as FSK activate IL-8 through an AhR-dependent mechanism (Fig. 1D and E).
Fig. 1.
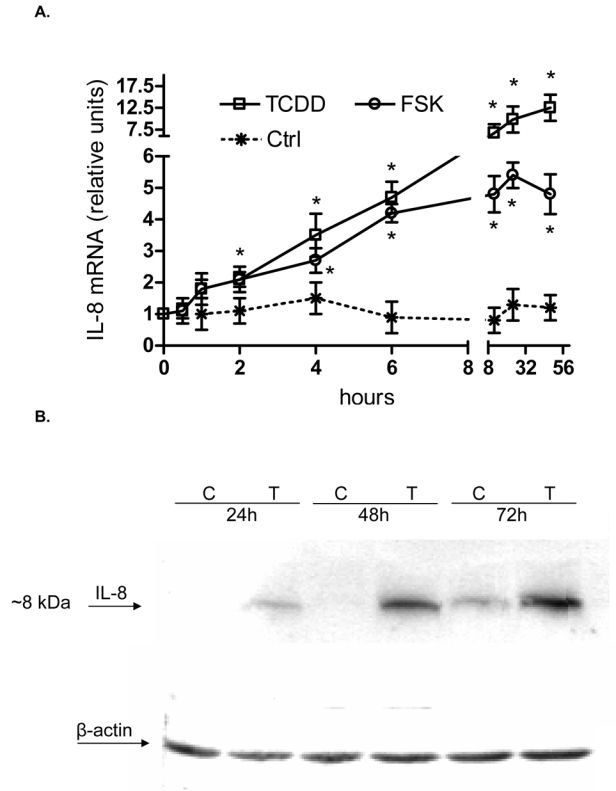
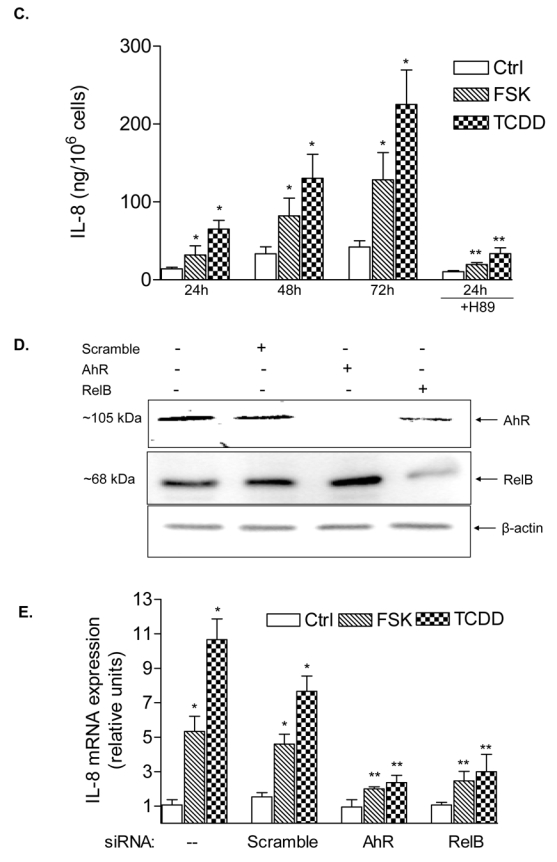
Induction of IL-8 is RelB- and AhR-dependent. A, Time-course study of IL-8 induction in U937 macrophages. Cells were treated for 0.5 to 48 h with 10 nM TCDD or 10 μM FSK. Control cells received only the vehicle solvent of 0.1% Me2SO or 0.1% PBS. Quantitative detection of IL-8 mRNA was performed using real time RT-PCR. Values for IL-8 mRNA expression are normalized to the expression of β-actin. *, significantly different from control (p<0.001). B, Time-dependent increase of IL-8 protein in U937 macrophages. The level of IL-8 protein in cell lysates from U937 macrophages after treatment with 10 nM TCDD (T) or 0.1 % Me2SO as vehicle control (C) as indicated were determined by Western blot analysis. C, Stimulated IL-8 secretion by activation of AhR in U937 macrophages. The level of IL-8 in the culture media of U937 macrophages was measured by ELISA. Results are expressed as ng IL-8 produced by 106 cells. *Values are the mean ± S.D. of three independent experiments and are significantly different from control (p<0.005). D, Western blot analysis of AhR and RelB protein levels 48h post-transfection with the indicated siRNAs. E, Quantitative IL-8 mRNA expression analyses after treatment with TCDD and FSK for 24 h as analyzed by real-time PCR. Total RNA was prepared 48 h post-transfection with either a scrambled siRNA or a specific siRNA targeted against AhR or RelB.
FSK and TCDD mediate IL-8 activation via a RelB/AhR binding motif
The production of IL-8 is usually not constitutive and can be induced rapidly by a wide range of stimuli such as TNFα, IL-1β, LPS, metals, hypoxia, reactive oxygen species, or cellular stress (21, 22). Several studies have shown that the sequence spanning -1 to -133 bp within the 5′ upstream regulatory region of the IL-8 gene is essential for transcriptional regulation of the gene. Previous studies identified three promoter binding sites for transcription factors of the AP-1, Oct-1, and NF-κB family, which are involved in the transcriptional control of the IL-8 gene (23, 24). We were interested in the relevance of these binding sites and performed transfection experiments with deletion reporter constructs of the IL-8 promoter. Our data revealed that the region spanning -1 to -50 bp upstream the start site of the IL-8 promoter is sufficient to induce promoter activity of the IL-8 gene mediated by TCDD or FSK (Fig. 2A). Comparing this short promoter sequence with consensus binding elements we could identify an eight bp sequence which contains an AhR/ARNT- and NF-κB-like binding site (5′-GGGTGCAT-3′). Using EMSA we were interested in verifying changes in the binding activities of this XRE/NF-κB-like sequence as well as identifying corresponding binding proteins which may bind on XRE or NF-κB sites. We found that DNA binding activity to the XRE/NF-κB-like sequence was enhanced in nuclear extracts from TCDD as well as FSK treated U937 macrophages, compared to control cells (Fig. 2B and C). Supershift analysis in Fig. 2B revealed that AhR together with RelB are the dominant proteins binding to this unrecognized binding element of the IL-8 promoter: whereas p50 or RelA does not bind to the RelB/AhR responsive element, which is called RelBAhRE from here on. Furthermore, we could not observe any binding activity of ARNT (Fig. 2B) on the RelBAhRE site. ARNT is well described as the dimerization partner of the ligand-activated form of the AhR binding to XRE which is essential for TCDD-induced cytochrome P4501a1 (CYP1A1) activity (25). To determine the importance of the XRE-like component in the newly identified RelBAhRE sequence for the TCDD-mediated IL-8 activation, a T-to-C point mutation was introduced (5- GGGCGCAT -3′, M1) as shown in Fig. 2D. The T residue is a total requirement for the activity of XRE consensus elements, and a T-to-C mutation is known to fully eliminate binding of the AhR/ARNT complex (26). In contrast to AhR/ARNT complexes, binding activity of the RelB/AhR complex is not reduced but even further increased (Fig. 2E) by this point mutation. This was confirmed by an elevated promoter activity of the mutation construct M1 (Fig. 2F). Supershift analysis confirmed that M1 like the wild type (wt) RelBAhRE oligonucleotide binds RelB and AhR, but not p50, RelA, or ARNT (Fig. 2G). In order to investigate the importance of the first and second G, which are conserved in consensus NF-κB sites as well as in consensus XRE, two point mutations (5′-CGCTGCAT-3′, M2, Fig. 2D) were introduced similar to an earlier report (27). The two G-to-C point mutations drastically reduced the TCDD- and FSK-induced binding activity of RelBAhRE as well as activation of the IL-8 promoter (Fig. 2E and F).
Fig. 2.
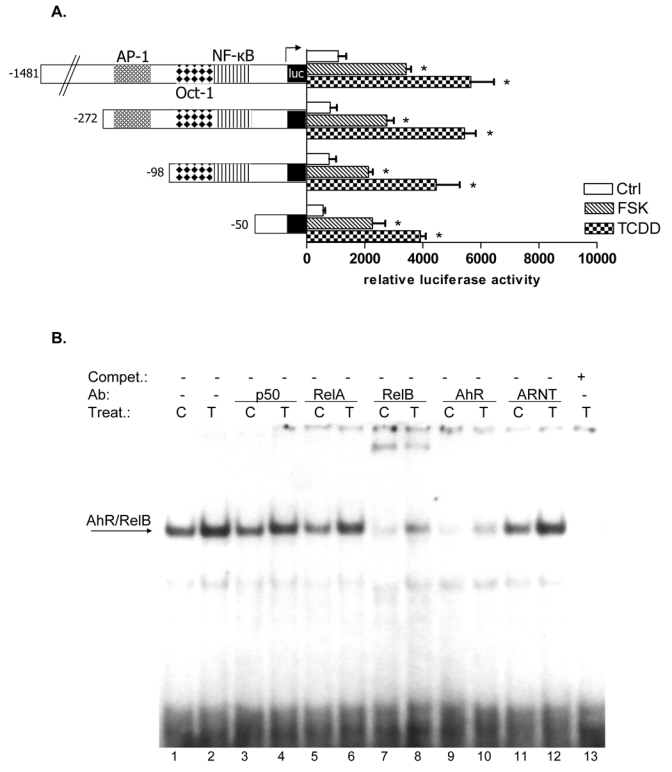
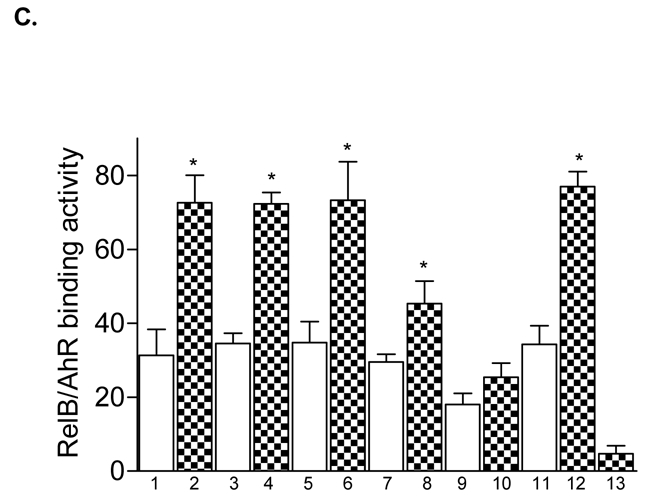
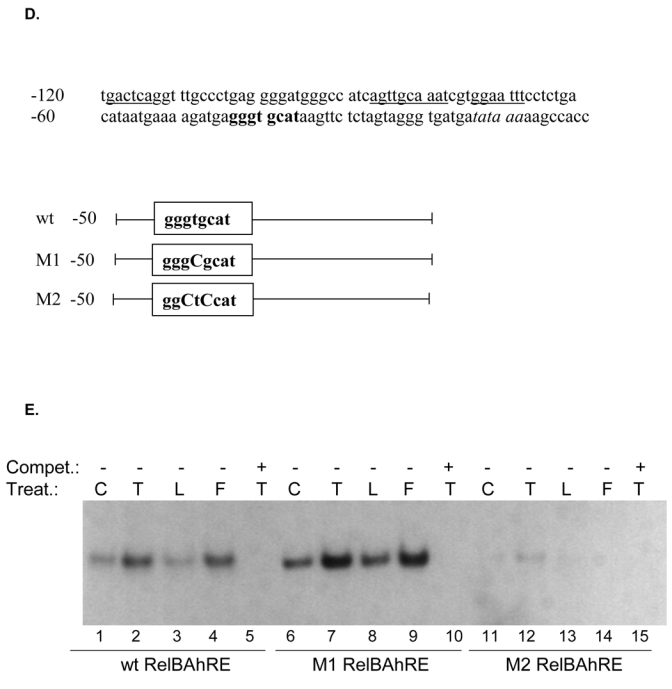
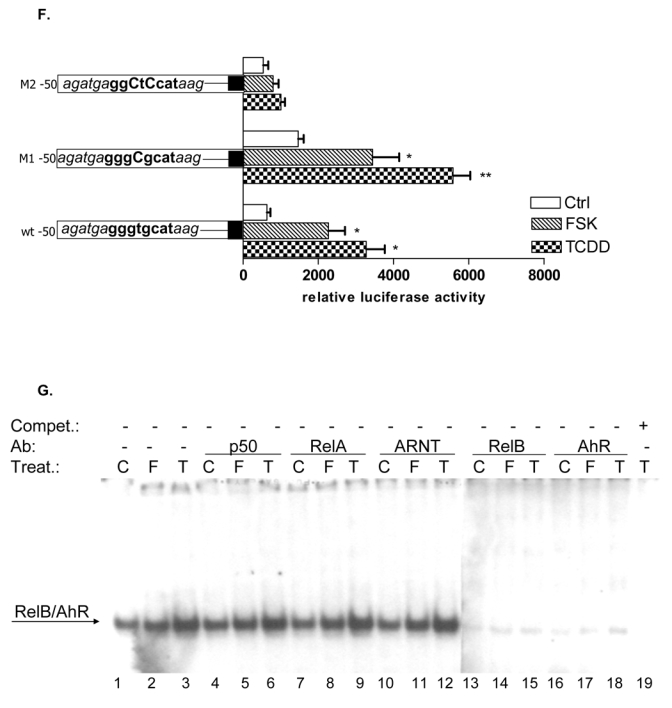
RelB and AhR mediate FSK- and TCDD-induced IL-8 activation. A, The AP-1, the Oct-1, and the NF-κB sites of the 5′-flanking region of the IL-8 gene are located 126 bp, 94 bp, and 80 bp, respectively, upstream of the start site of transcription in the IL-8 gene. U937 cells were transiently transfected with deletion constructs corresponding to the first 272, 137, 98, or 50 bp of the 5′-flanking region of the IL-8 gene and treated with 10 nM TCDD or 10 μM FSK for 24 h. Relative luciferase units are given as mean values of triplicates as a result of three independent experiments. *, significantly different from control (p<0.001). B, Supershift analyses with p50-, RelA-, RelB-, AhR-, and ARNT-specific antibodies were performed with a 32P-end-labeled oligonucleotide (5′-AGATGAGGGTGCATAAGTTC-3′) containing the RelBAhRE site of the IL-8 gene with nuclear extracts of untreated and TCDD-stimulated cells treated for 90 min. A 100-fold molar excess of unlabeled RelBAhRE was added. C, Densitometric evaluation of band intensities of the RelB/AhR complexes. Results of three independent experiments are shown as mean values ± S.D. *, significantly different from control cells (p<0.001). D, Nucleotide sequence of the wild-type (wt) -50 bp IL-8 construct corresponding to the 5′-flanking region of the first -120 bp upstream of the start site. The TATA box is in italic type, the AP-1, Oct-1, and NF-κB sites are underlined, the RelBAhRE site is shown in boldface letters. A one point mutation (M1) or two point mutations (M2) were introduced in the RelBAhRE site of the -50 bp construct. E, DNA binding of nuclear proteins from U937 macrophages to the RelBAhRE probe of the IL-8 promoter or RelBAhRE with two different point mutations M1 and M2. U937 macrophages were treated with 10 nM TCDD (T), 2 μg/ml LPS (L), 10 μM FSK (F), or received 0.1% Me2SO as vehicle control (C), and nuclear proteins were extracted at 90 min. A 100-fold molar excess of unlabeled wildtype oligonucleotides was added. F, U937 cells were transiently transfected with -50 wt IL-8 construct and the mutation constructs M1 or M2. After transfection, cells were treated with 10 nM TCDD or 10 μM FSK for 24 h. Relative luciferase activity units are given as mean values of triplicates as a result of three independent experiments. *, significantly different from control (p<0.005); **, significantly higher than only -50 wt transfected cells treated with TCDD or FSK (p<0.005). G, DNA binding of nuclear proteins from U937 macrophages to oligos containing a point mutation M1 of the RelBAhRE probe of the IL-8 promoter. U937 macrophages were treated with 10 μM FSK (F), 10 nM TCDD (T), or received 0.1% Me2SO as vehicle control (C), and nuclear proteins were extracted at 90 min. Supershift analyses with p50-, RelA-, ARNT-, RelB-, or AhR-specific antibodies were performed to identify proteins binding to the mutated M1 RelBAhRE sequence of IL-8. A 100-fold molar excess of unlabeled oligonucleotide was added.
PKA-dependent activation of IL-8 by FSK and TCDD is mediated through RelB and AhR
Cotransfection with siRNA specific for AhR and RelB notably decreased the TCDD- as well as FSK–mediated activation of the IL-8 promoter (Fig. 3A) and induction of IL-8 mRNA expression (Fig. 1E), thus underlining the requirement of AhR and RelB to mediate the activation of IL-8 by TCDD or FSK. These results are supported by over-expression of AhR and RelB which enhanced the activation of the IL-8 promoter in a dose-dependent manner (Fig. 3B and C). To verify the specificity of RelB and AhR, cells were transfected with an ARNT expression plasmid which did not significantly changed the IL-8 promoter activity (Fig. 3D). Nuclear proteins from cells transfected with siAhR or siRelB showed significant decreased binding activity and no effect of TCDD treatment on RelBAhRE in EMSA, which supports the role of AhR (Fig. 3E). To determine whether activation of IL-8 by FSK and TCDD is PKA-dependent, U937 macrophages were transfected with the IL-8 reporter in presence or absence of a PKA wild type, a PKA mutant expression plasmid, and the PKA inhibitor H89. The requirement of PKA for TCDD and FSK to activate the IL-8 promoter was evident (data not shown), which is supported by EMSA showing decreased binding activity of RelBAhRE in cells pretreated with H89 (Fig. 3F).
Fig. 3.

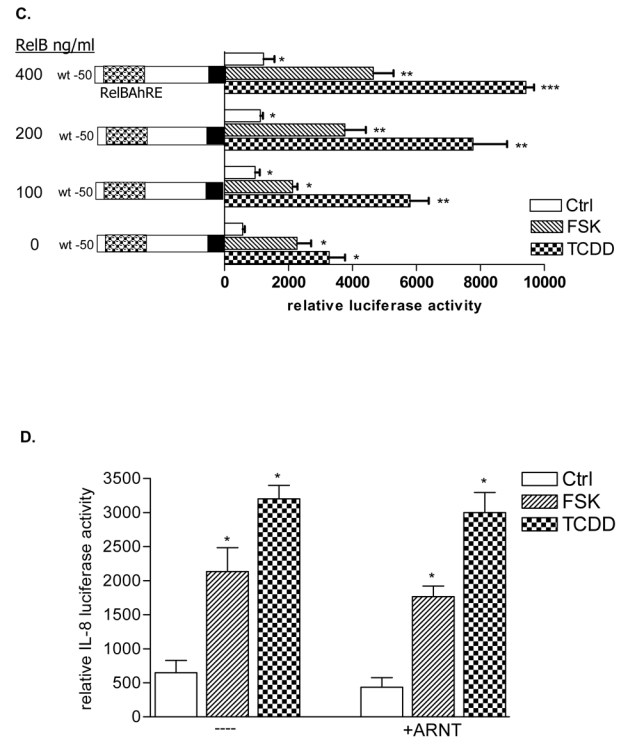
Requirement of PKA in AhR- and RelB-mediated IL-8 promoter and RelBAhRE binding activity induced by FSK and TCDD. A, Transfection of siRNA targeted against AhR or RelB mRNA prevents TCDD- and FSK-mediated induction of IL-8 promoter activity in U937 macrophages. Cells were transfected with either a scrambled siRNA or a specific siRNA targeted against AhR or RelB and the -50 wt IL-8 construct. After 24 h cells were treated with 10 nM TCDD or 10 μM FSK for 24 h. B, Cotransfection with an AhR C, RelB or D, ARNT expression plasmid. U937 macrophages were transiently transfected with wildtype -50 bp IL-8 construct and cotransfected with increasing amounts (100–400 ng/ml) of an AhR (B) RelB (C) or 200 ng/ml ARNT (D) expression plasmid. After transfection for 24 h, cells were treated with 10 nM TCDD, 10 μM FSK, or 0.1% Me2SO for 24 h. Relative luciferase activity units are given as mean values of triplicates as a result of three independent experiments. *, significantly different from control (p<0.005). **, significantly higher than cells treated with TCDD or FSK transfected with only -50 wt IL-8 construct (p<0.005). ***, significantly higher than cells treated with TCDD or FSK cotransfected with -50 wt IL-8 and 100 ng RelB or AhR (p<0.005). E, DNA binding of nuclear proteins from U937 macrophages to RelBAhRE of the IL-8 promoter requires AhR and RelB. U937 macrophages were transfected with either a scrambled siRNA or a specific siRNA targeted against AhR or RelB treated with 10 nM TCDD (T) or received 0.1% Me2SO as vehicle control (C) and nuclear proteins were extracted at 90 min. A 100-fold molar excess of unlabeled wildtype oligonucleotides was added. F, DNA binding of nuclear proteins from U937 macrophages to RelBAhRE of the IL-8 promoter depends on PKA. U937 macrophages were treated with 1 μM H89 (H), 1 μM H89 plus 10 μM FSK (H+F), 10 μM FSK (F), 1 μM H89 plus 10 nM TCDD (H+T), 10 nM TCDD (T), or received 0.1% Me2SO as vehicle control (C) and nuclear proteins were extracted at 90 min. A 100-fold molar excess of unlabeled wildtype oligonucleotides was added.
Physical association of AhR and RelB
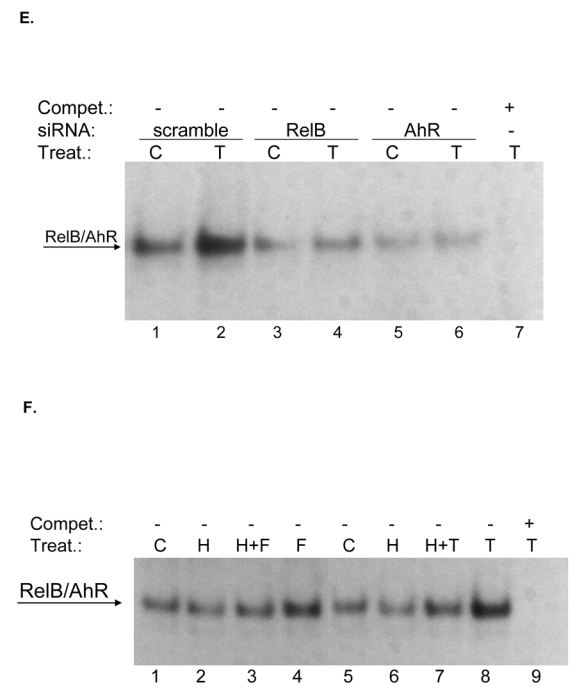
In order to investigate the physical association between RelB and AhR, co-immunoprecipitation studies were performed. The results show that AhR and RelB proteins are interacting in control as well as TCDD-treated cells (Fig. 4A and B). To verify the effect of the vehicle Me2SO, we compared the vehicle controls with medium controls (untreated cells). No significant effect of Me2SO at a concentration of 0.1% was observed on the interaction or binding activity of RelB and AhR on a RelBAhRE oligonucleotide (data not shown). Although TCDD did not affect the apparent association between AhR and RelB, the functional activity of this complex has been clearly stimulated by TCDD or FSK as shown in EMSA and transient transfection studies (Fig. 2A to F). Since ligand-activated AhR is known to dimerize with ARNT in the nucleus, we tested the possible interaction of ARNT with RelB. ARNT was found complexed with AhR in TCDD-treated cells as expected and no interaction of ARNT and RelB could be detected (Fig. 4C), which is supported by results obtained from gel shift studies (Fig. 2B and Fig. 5E). No association of AhR with NF-κB proteins p50 or RelA could be detected by co-immunoprecipitation (data not shown) or EMSA (Fig. 5A) studies.
Fig. 4.
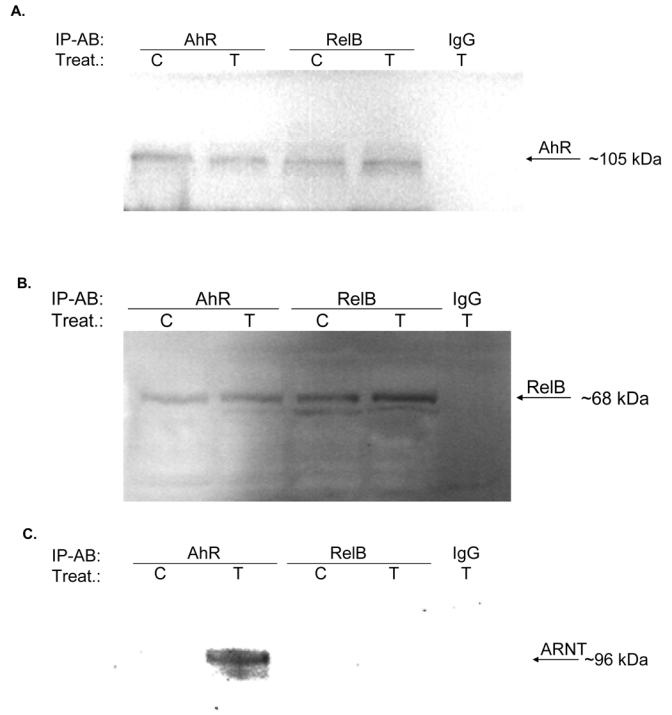
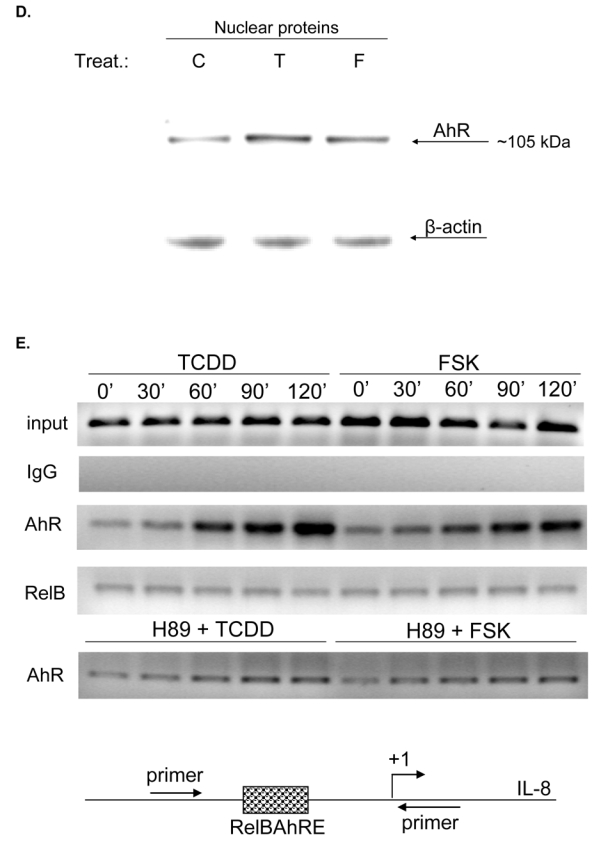
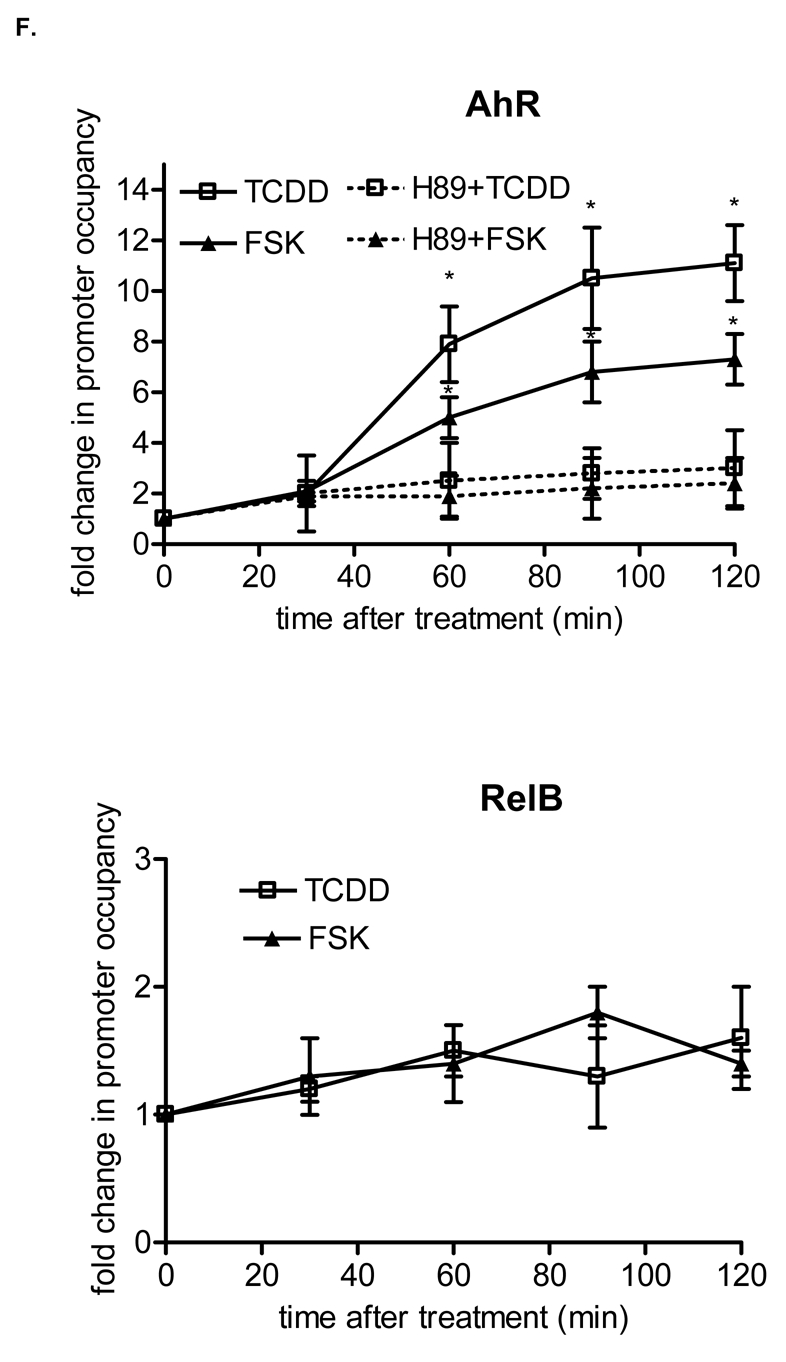
Physical association of AhR and RelB. U937 macrophages were treated with TCDD (10 nM) or Me2SO (0.1%) and after 90 min nuclear proteins were prepared for nuclear complex co-immunoprecipitation followed by Western blot analysis to detect specific association of AhR and RelB. A, immunoprecipitation of AhR with antibody against RelB. Samples of total cell lysates were incubated with rabbit lgG as the negative control, anti-AhR antibody (positive control), and antibody against RelB. The blot was probed with antibody against AhR protein after Western transfer. B, immunoprecipitation of RelB with antibody against AhR. The cell lysates were incubated with rabbit IgG, antibody against RelB (positive control), and antibody against AhR. After Western transfer, the blot was stained with antibody against RelB. C, ARNT is not associated with RelB. The cell lysates were incubated with antibody against RelB, AHR (positive control), and rabbit IgG. The Western blot was stained with an antibody against the ARNT protein. D, Increased nuclear accumulation of AhR protein. The level of AhR in nuclei from U937 macrophages 90 min after treatment with 10 nM TCDD (T) or 10 μM FSK (F), or 0.1% Me2SO (C) as indicated were determined by Western blot analysis using a AhR-specific antibody. E, FSK and TCDD stimulate the recruitment of AhR to the RelBAhRE region of the IL-8 promoter. U937 macrophages were treated with 10 nM TCDD and 10 μM FSK in presence or absence of 1 μM H89, or Me2SO (0.1%) for the indicated amounts of time. ChIP assays with antibodies to AhR and RelB proteins were analyzed by PCR using primer pairs covering the specified RelBAhRE region of human IL-8. Genomic DNA and the sonicated input DNA were separated by agarose gel electrophoresis and visualized by ethidium bromide staining. Arrows, position of primers used to test the recruitment of AhR or RelB to the IL-8 promoter region flanking the RelBAhRE region. F, ChIP assay samples were analyzed by real-time PCR as described in Materials and Methods and the results were normalized to time zero (no AhR activation by TCDD or FSK). *, significantly different from control cells (p<0.001)
Fig. 5.
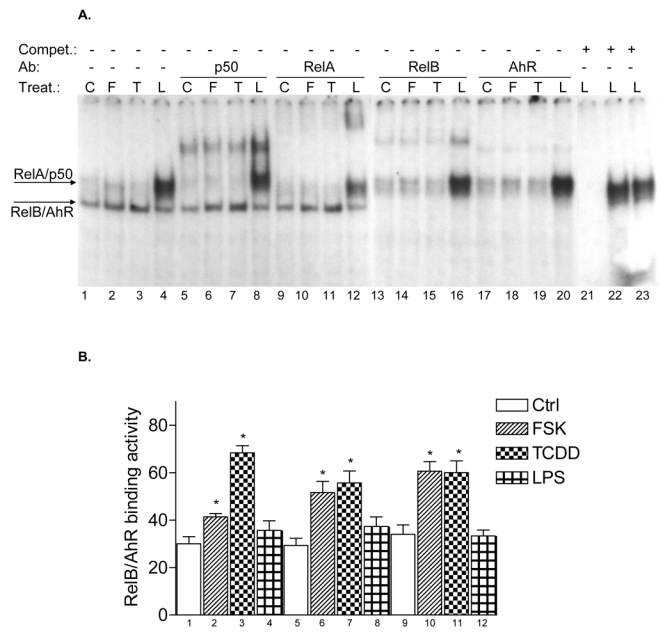
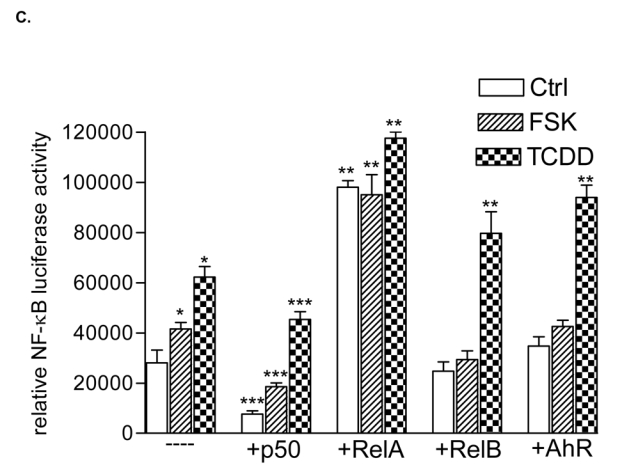
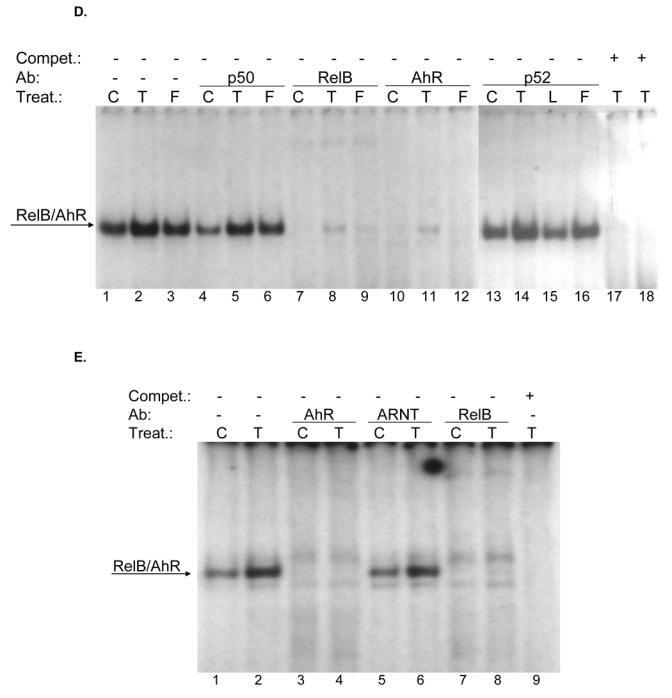
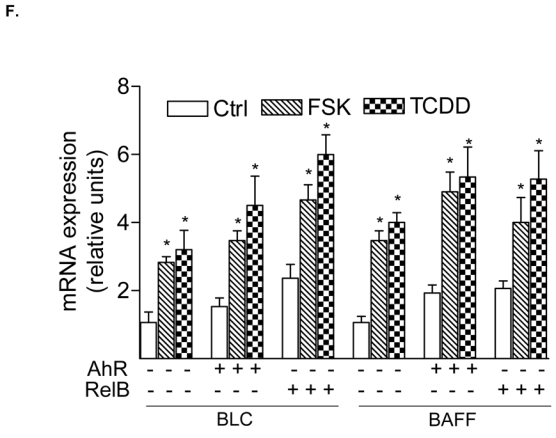
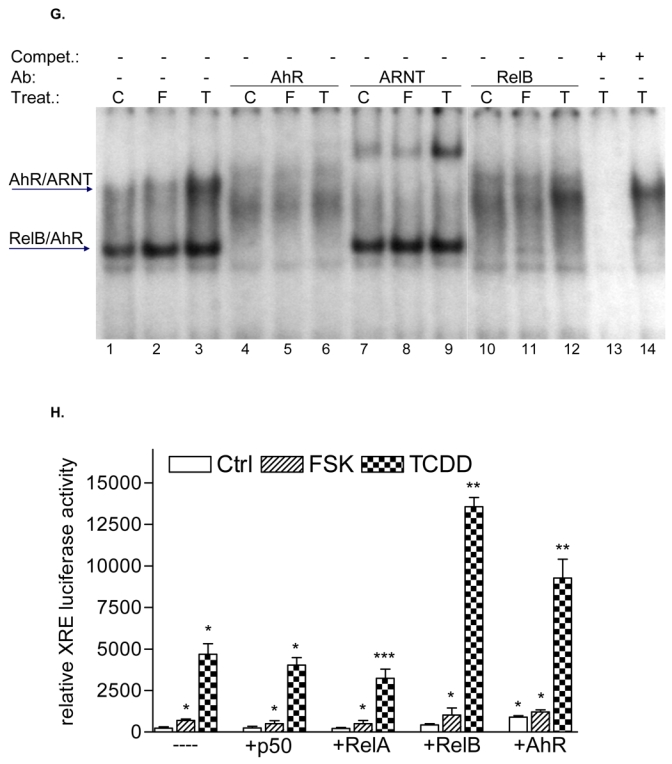
Effect of FSK, TCDD or LPS on NF-κB and XRE activity. A, U937 macrophages were treated with 10 μM FSK (F), 10 nM TCDD (T), 2 μg/ml LPS (L) or received 0.1% Me2SO as vehicle control (C), and nuclear proteins were extracted at 90 min. Nuclear protein extracts of non-stimulated, FSK-, TCDD-, or LPS-stimulated U937 macrophages were incubated with a NF-κB consensus probe. NF-κB proteins, present in nuclear extracts of 90 min treated U937 macrophages binding to the NF-κB site were identified by supershift analyses using p50-, RelA-, RelB-, or AhR-specific antibodies. Competition with a 100-fold excess of unlabeled NF-κB consensus, XRE-consensus, or RelBAhRE oligonucleotide from the IL-8 promoter confirms specificity of the complex. B, Densitometric evaluation of the band intensity of the lower band of the NF-κB complex. Results of three independent experiments are shown as mean values ± S.D. *, significantly different from control cells (p<0.001). C, Effect of overexpression of various Rel proteins and AhR on FSK- and TCDD-induced NF-κB reporter activity. NF-κB activity was evaluated in U937 macrophages by transient transfection of the corresponding reporter plasmid with or without cotransfection of 200 ng/ml of a p50, RelA, RelB or AhR expression plasmid. Transfected cells were incubated with 0.1% Me2SO, 10 μM FSK, or 10 nM TCDD for 24 h. Relative luciferase activity units are given as mean values of triplicates as a result of three independent experiments. *, significantly different from control (p<0.005) **, significantly higher than only NF-κB reporter plasmid transfected cells (p<0.005) ***, significantly lower than only NF-κB reporter plasmid transfected cells (p<0.005). D, Nuclear protein extracts of control (C), TCDD- (T), or FSK-stimulated (F) U937 macrophages were incubated with the RelB/p52 consensus oligo of the BLC promoter. A possible binding of p50, RelB, AhR, and p52 was identified by supershift analyses. To confirm specificity a 100-fold excess of unlabeled RelB/p52 consensus or RelBAhRE oligonucleotide from the IL-8 promoter was added. E, Nuclear protein extracts of control (C) or TCDD- (T) stimulated U937 macrophages were incubated with an oligonucleotide containing the RelB/p52 of the BAFF promoter. A possible binding of AhR, ARNT, and RelB was identified by supershift analyses. To confirm specificity a 100-fold excess of unlabeled RelB/p52 oligonucleotide from the BAFF promoter was added. F, Induction of BLC and BAFF is increased in AhR and RelB overexpressing cells. Cells were transiently transfected with 200 ng/ml of AhR or RelB expression plasmid. Control cells were transfected with an empty control vector. After 72 h cells were treated with 10 nM TCDD or 10 μM FSK for 24 h. Expression of BLC and BAFF mRNA was analyzed by real-time PCR as described above. *, significantly different from control cells (p<0.01); **, significantly higher than control, TCDD or FSK treated cells (p<0.01) G, Nuclear protein extracts of control (C, lane 1), FSK- (F, lane 2), or TCDD-stimulated (T, lane 3) U937 macrophages were incubated with 32P-labeled oligonucleotide containing a XRE consensus element of the CYP1A1 promoter. A possible binding of AhR, ARNT, and RelB was identified by supershift analyses using AhR-, ARNT-, or RelB-specific antibodies. To confirm specificity a 100-fold excess of unlabeled XRE consensus or RelBAhRE oligonucleotide from the IL-8 promoter was added. H, Effect of overexpression of various Rel proteins and AhR on FSK- and TCDD-induced XRE reporter activity. XRE activity was evaluated in U937 macrophages by transient transfection of the corresponding reporter plasmid with or without cotransfection of 200 ng/ml of a p50, RelA, RelB or AhR expression plasmid. Transfected cells were incubated with 0.1% Me2SO, 10 μM FSK, or 10 nM TCDD for 24 h. Relative luciferase activity units are given as mean values of triplicates as a result of three independent experiments. *, significantly different from control (p<0.005) **, significantly higher than only XRE reporter plasmid transfected cells (p<0.005) ***, significantly lower than only XRE reporter plasmid transfected cells (p<0.005).
Enhanced recruitment of AhR to a novel RelBAhRE binding site of the IL-8 promoter
Binding activity of RelBAhRE of the IL-8 promoter was elevated by TCDD as well as FSK due to an increased nuclear localization of AhR which is indicated by increased protein levels of AhR in nuclear extracts of TCDD and FSK treated U937 macrophages (Fig. 4D). These results are confirmed by chromatin immunoprecipitation (ChIP) assays with U937 human macrophages to study the recruitment of AhR and RelB proteins to the RelBAhRE element of the IL-8 promoter (Fig. 4E). For quantification, the ChIP samples were analyzed by real-time PCR; their relative enrichment levels are shown in Fig. 4F. Our data demonstrate the enhanced recruitment of AhR to the RelBAhRE element of IL-8 stimulated by TCDD and FSK. Increased occupancy of the RelBAhRE promoter region by AhR was evident after 30 min, peaking at approximately 90 min and sustained thereafter during the course of the treatment. The ChIP analysis demonstrate a higher increase of AhR binding at RelBAhRE compared with results from EMSA, which might be due to the fact that ChIP includes the chromatin context and the dynamic of the cell which is not the case in EMSA. No apparent significant kinetic differences were observed in the occupancy of the RelBAhRE region by RelB under these conditions. The critical role of PKA to recruit AhR was verified by significantly less occupancy of the RelBAhRE region by AhR in cells treated with TCDD or FSK in presence of H89, which is in line with EMSA showing a lower binding activity of RelBAhRE by pretreatment with H89 (Fig. 3F). No recruitment of ARNT was observed to the RelBAhRE region (data not shown) within the IL-8 promoter, demonstrating the specific binding of RelB and recruitment of AhR to this promoter region of IL-8 (Fig. 4E).
FSK and TCDD signaling induce binding of RelB/AhR complexes to NF-κB binding sites
Since RelB is a subunit of the NF-κB family which binds to NF-κB consensus sequences we were interested in the possible coexistence of RelB and AhR complex binding to a NF-κB consensus element. EMSA in Fig. 5A shows that TCDD and FSK stimulate binding activity of the lower NF-κB complex. Supershift analyses with AhR-specific antibodies revealed that AhR indeed binds to a NF-κB consensus element present in the lower complex of the classical TNFα- or LPS-activated NF-κB complex which also contains the NF-κB subunits RelB and p50. However, a physical interaction of AhR with RelA, which forms RelA homodimers or heterodimers with p50 after treatment with LPS has not been observed (Fig. 5A). A 100-fold excess of cold NF-κB oligonucleotide completely abolished formation of both NF-κB complexes, whereas excess of cold XRE consensus or RelBAhRE oligonucleotide abolished specifically the lower complex. The LPS-induced upper complex formed by RelA and p50 was not affected indicating the specific binding of AhR and RelB on these DNA binding sequences (Fig. 5A). In contrast to the classical activation pathway of NF-κB and inflammatory signaling by TNFα or LPS through their respective receptors TNFR1/2 and TLR/IL-1R, TCDD obviously activates NF-κB through an enhanced recruitment of AhR which is complexed with RelB and binds to NF-κB response elements. This suggestion is supported by the TCDD-induced binding activity of the lower complex containing AhR and RelB (Fig. 5B). As expected, RelA increased the constitutive NF-κB-reporter activity drastically whereas p50 over-expression inhibited NF-κB activity. RelB and AhR increased the TCDD- but not FSK-stimulated NF-κB-activity (Fig. 5C).
To address whether a recently identified κB-binding site (5′-GGGAGATTTG-3′) located on the promoter of chemokines like BLC and BAFF that is preferentially recognized by RelB/p52 dimers and not RelA/p50 dimers (4) is also a target for AhR- and RelB-containing dimers we performed EMSA with the specific κB-binding site located on promoters of BLC at position -115 bp and BAFF at position -71 bp. Both probes exhibited strong binding activity to nuclear extracts of U937 macrophages which was further increased using nuclear extracts of FSK- or TCDD-stimulated cells (Fig. 5D and E). Supershift analysis revealed clear binding activity of AhR in complex with RelB. The presence of ARNT, p50, or p52 subunits dimerized with RelB or AhR could not be detected in FSK-, TCDD-, or LPS-stimulated cells. This result agrees with previous studies showing that binding of p52/RelB requires the degradation of the inhibitory p52 precursor, p100, which is mediated by LTβR signaling and IKKα, but not by TNFα or LPS and IKKβ or IKKγ (28). Migration and binding activity of the RelB/p52 probes were very similar to the RelBAhRE probe of the IL-8 promoter. In all cases, the detected protein-DNA complexes were specific, as indicated by competition experiments with RelB/p52 and RelBAhRE probes (Fig. 5D). These results suggest that the RelB/AhR complex is also involved in the regulation of other chemokines like BLC or BAFF containing the specific RelB/p52 κB-binding site on their promoter. As suspected, treatment with FSK or TCDD led to induction of BLC and BAFF mRNA in U937 macrophages and overexpression of AhR and RelB further increased the level of BLC and BAFF in control, FSK-, and TCDD-stimulated cells (Fig. 5F). These results and previous EMSA strongly suggest that BLC and BAFF gene induction is not only mediated via RelB/p52 but also RelB/AhR complexes.
RelB binds on a XRE consensus site of the CYP1A1 promoter and increases XRE-activity
Using EMSA we investigated the possible interaction of RelB with an XRE consensus element of a CYP1A1 promoter sequence. Supershift analyses with RelB specific antibodies revealed clear binding activity of RelB in complex with AhR in nuclear extracts of U937 macrophages (Fig. 5G). Excess of cold XRE oligonucleotide completely abolished formation of both XRE complexes, whereas excess of unlabeled NF-κB consensus or RelBAhRE oligonucleotide abolished specifically the lower complex which does not contain ARNT protein complexed with AhR (Fig. 5G). A very similar pattern of AhR/ARNT and AhR/RelB binding on consensus XRE was found in the human and mouse hepatoma cell lines HepG2 and Hepa1c1c7, respectively (unpublished data). The data are indicating the specific binding of AhR and RelB on DNA binding sequences which do not involve binding of AhR/ARNT complexes. Since the ligand-activated AhR preferably forms heterodimers with ARNT in the nucleus as shown in EMSA of TCDD-treated cells (Fig. 5G) RelB might bind to a different active form of the AhR located in the nucleus. The existence of a non-ligand activated form of the AhR in the nucleus has been described earlier (9, 14, 29). Overexpression of RelA had an inhibitory effect on TCDD-induced XRE activity, whereas over-expression of RelB like AhR drastically increased the TCDD-induced activity of the XRE reporter construct (Fig. 5H). Compared to TCDD, FSK had only a small but statistically significant effect on XRE activity, which was further increased by overexpression of RelB. These results underline the cross-talk between RelB and AhR and indicate the supportive action of RelB on AhR-mediated transcriptional activation.
DISCUSSION
Our present analysis reveals that an activated AhR (through TCDD or FSK) associated with RelB mediates IL-8 gene transcription via an unrecognized cis-acting element (RelBAhRE). Interestingly, a T-to-C mutation of the XRE-like component (3′-GTGCAT-5′) of the RelBAhRE sequence led to enhanced binding activity of RelB/AhR and increased IL-8 promoter activity. These findings indicate that the binding site of the RelB/AhR complex is distinctly different from the typical XRE (3′-GCGTG-5′) of the ligand activated AhR/ARNT complex, where the T and the third and fifth G are a strict requirement to bind AhR/ARNT dimers (26). The T-to-C mutation in the RelBAhRE site seems to reflect more characteristics of a NF-κB binding site than the original RelBAhRE site. The enhanced recruitment of AhR and binding of RelB/AhR dimers to RelBAhRE induced by FSK or TCDD require PKA activity for the full induction of IL-8. In line with these findings an increased FSK/cAMP-stimulated activation followed by nuclear localization of AhR, which does not dimerize with ARNT has been reported earlier (14), although the dimerization partner of the PKA-mediated nuclear AhR could not be revealed by these authors. In a previous report we could show that TCDD treatment is associated with an early increase of PKA activity leading to the induction of C/EBPβ (18). Thus, it seems reasonable that TCDD induces nuclear translocation of cytosolic AhR through an elevation of PKA activity, besides the classical well described ligand-dependent activation and nuclear translocation of the AhR, which forms heterodimers with ARNT (30). These data imply that increased AhR nuclear localization and DNA binding activity induced by FSK or TCDD is not due to increased AhR synthesis but rather a direct phosphorylation of the AhR protein. Although, ligand binding is an important mechanism of nuclear receptor activation, other receptors, including ERα and ERβ, can be activated by specific kinases as well (31).
Furthermore, we observed an enhanced binding activity of RelB/AhR complexes on the RelB/p52 consensus element of the BLC and BAFF promoter by TCDD or FSK. The enhanced binding activity was associated with an increased expression of BLC and BAFF mRNA, which was further elevated in AhR and RelB overexpressing cells. The chemokine BLC and the B cell activating factor BAFF are known to contain RelB/p52 responsive sites on their promoter and have been shown to be regulated through the alternative NF-κB pathway (4). Thus, our study establishes an example of how an activated AhR pathway connects to the NF-κB subunit RelB (alternative AhR/RelB pathway, Fig. 6) in order to cooperatively regulate inflammatory gene expression.
Fig. 6.
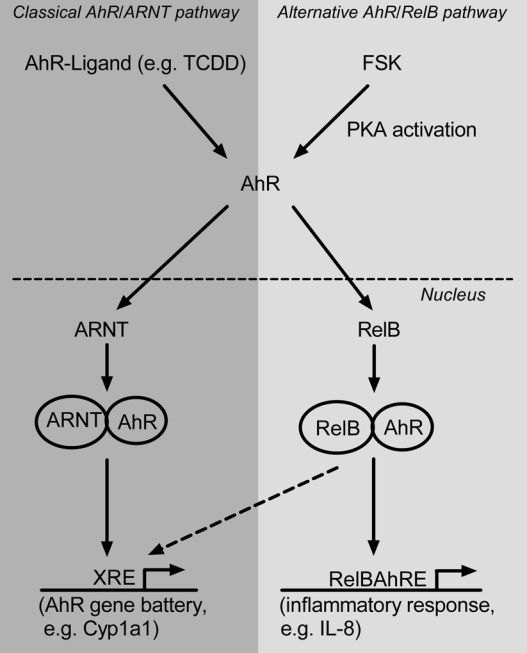
Model of the new mechanism of cross talk between AhR and RelB. Ligand-activated or unliganded AhR activated by PKA translocates into the nucleus and interacts with RelB to occupy RelBAhRE-responsive promoters as in the case of IL-8 (alternative AhR/RelB pathway). AhR agonists induce the recruitment of AhR/ARNT complexes to XRE-responsive promoters such as CYP1A1 (classical AhR/ARNT pathway).
As our above data suggest the recruitment of AhR and RelB may have important consequences on NF-κB and AhR signaling, and also reveals an important role for AhR in NF-κB-dependent transcription, as well as for RelB in AhR-dependent transcription through XRE sites. There are conflicting reports on the effect of NF-κB activation through TNFα or LPS on TCDD-induced expression of AhR target genes. Some studies report an inhibition (32), whereas other groups (33–35) and own results show an activation of NF-κB activity. Other investigators observed that TCDD leads to induction of the pro-inflammatory gene IL-1β (36) and a sustained induction of NF-κB binding activity (37). These differences may be due to different cell types, culture conditions, serum lots, and treatment regimes. Puga et al. (37) concluded that an increased formation of p50/p50 complexes might be responsible for the TCDD-mediated effect on NF-κB reporter activity. Our results collectively demonstrate that the AhR interacts with RelB and that activation of the AhR leads to an increased NF-κB activity. Current data also show that the increased binding activity of the lower complex of the NF-κB element mediated by TCDD or FSK is obviously due to an increased nuclear accumulation of AhR complexed with RelB rather than increased binding of p50/p50 homodimers, which are believed to repress NF-κB activity (38). This hypothesis is in line with other reports showing an increased NF-κB binding in mouse hepatoma cells (Hepa1c1c7) transfected with an AhR expression plasmid (32). In the case of the chemokines IL-8, BLC, and BAFF the interaction of AhR and the NF-κB member RelB enhances the gene activity by TCDD or FSK. The supportive action of RelB on AhR signaling is also clearly indicated by a distinct increase of TCDD-mediated XRE-Luc reporter activity through overexpression of RelB and the binding of RelB on a XRE consensus sequence. Similar results were received from parallel IL-8- and XRE-Luc reporter studies with the human hepatoma cell line HepG2 (unpublished data) indicating that the observed mechanism of RelB and AhR interaction is not limited to macrophages and exists in other cell types as well. Recently we could show that TCDD induces KC (homolog of human IL-8) in various tissues of mice of C57Bl/6 mice (16). Two structurally distinct κB sequence motifs have been identified for mouse KC (39) and the second κB motif (3′-GGGTGT-5′) of KC shows sequence homology to RelBAhRE. Results from AhRnls mice show that the induction of KC by TCDD depends on the nuclear translocation of the AhR. The induction of KC in liver of C57Bl/6 wild type mice was associated with an increased expression of F4/80 indicating the infiltration of macrophages which suggests the physiological relevance and biological consequence of the AhR/RelB pathway (16).
Despite the present study our understanding of the molecular mechanisms of AhR and RelB crosstalk is far from complete. For instance the PKA-mediated signal(s) that stimulates nuclear translocation of AhR and complex formation with RelB are unknown, and may include the activation of other signaling pathways and kinases. We believe that the PKA-stimulated association of AhR with RelB represents an important mechanism mediating crosstalk between NF-κB and AhR signaling pathways, but also a new mechanism of RelB and AhR action. Because AhR associates not only with ARNT (classical AhR/ARNT pathway) but also with RelB, we propose a model of an alternative AhR/RelB pathway in which AhR and RelB regulates inflammatory genes like IL-8, BLC, or BAFF.
Some of the major future questions are: how is the functional separation between the two AhR signaling pathways regulated? The alternative AhR/RelB pathway obviously overlaps with the alternative NF-κB pathway and has a regulatory function on the expression of especially chemokines. Organogenic chemokines like BLC and BAFF are regulated by the alternative NF-κB pathway and are required for the recruitment of macrophages, T cells, and B cells to secondary lymphoid organs (3). Studies with AhR-deficient mice are showing a distinct decrease of lymphocytes in spleen and lymph nodes (12) suggesting the critical role of AhR in the alternative NF-κB pathway. By contrast, the classical AhR/ARNT pathway is mostly responsible for rapid responses to xenobiotics and activation of genes encoding xenobiotic metabolizing enzymes including CYP1A1, CYP1A2, and CYP1B1 through XRE sites. Another open question is whether the identified RelBAhRE binding site of IL-8 is a unique sequence that is selectively recognized by RelB/AhR dimers and not by RelB/p52 dimers, the ubiquitous target of the alternative NF-κB pathway, and the possible existence on promoters of other target genes. Current data indicate that AhR influences NF-κB signaling and RelB regulates AhR signaling and XRE activity. Thus, it will also be important to determine if RelB/AhR complexes are recruited to other AhR target genes or restricted strictly to consensus XREs of CYP1A1 promoters. Even with the differences in the mechanisms and type of NF-κB and AhR activation, our model suggests that the role of PKA-dependent phosphorylation in assembling a RelB/AhR signaling complex is a conserved strategy that has evolved to regulate genes in response to environmental stressors and inflammatory signals.
MATERIALS AND METHODS
Reagents and Antibodies
Dimethylsulfoxide (Me2SO), Phorbol-12-myristate-13-acetate (TPA), Tumor Necrosis Factor (TNF)α and lipopolysaccharide (LPS) were obtained from SIGMA (St. Louis, MO). [y-32P] ATP (6000 Ci/mmol) was purchased from ICN (Costa Mesa, CA). FSK and N-{2-[(p-bromocinamyl)amino]ethyl}-5-isoquinolinesulfonamide·2HCl (H89) was purchased from Calbiochem (San Diego, CA). TCDD (>99% purity) was originally obtained from Dow Chemicals Co (Midland, MI). Other molecular biological reagents were purchased from Qiagen (Valencia, CA) and Roche (Indianapolis, IN). Monoclonal ARNT, polyclonal RelA, p52 (Santa Cruz Biotechnology, Santa Cruz, CA), NF-κB member p50, RelB, c-Rel (Active Motif, Carlsbad, CA), and polyclonal AhR (Novus Biologicals, Littleton, CO) antibodies were used for Western blot analyses, Supershift in EMSA, and ChIP assays.
Plasmids and Site-directed mutagenesis
Details concerning the cloning of a 1.5 kb IL-8 promoter fragment, deletion and muation constructs into pGL3 Basic are described elsewhere (17). Mutation of RelBAhRE sequences of human IL-8 (GGGTGCAT to M1, GGGCGCAT or M2, GGCTCCAT) was carried out by site-directed mutagenesis (Stratagene, La Jolla, CA) using the following primers synthesized by Integrated DNA Technologies Inc. (Coralville, IA): M1-50-Mutant, 5′-GATGAGGGCGCATAAGTTCTCTAG-3′ and M2-50-Mutant, 5′-GATGAGGCTCCATAAGTTCTCTAG-3′. Insertion of mutations was confirmed by direct sequencing. The NF-κB luciferase reporter was from Clontech (Mountain View, CA) and XRE luciferase reporter was kindly provided by J. Abel (University of Duesseldorf, Germany). The AhR and ARNT expression plasmid was a kind gift of C. Bradfield (McArdle Laboratory for Cancer Research, Madison, WI). Expression vectors for p50 and RelA were kindly provided by W. Greene (J. David Gladstone Institute, San Francisco, CA). The RelB expression plasmid was kindly provided by U. Siebenlist (NIH, Bethesda, MD).
Cell Culture, Transfection Experiments, and Luciferase Assay
Human U937 monocytic cells were obtained from A.T.C.C. (Manassas, VA) and maintained in RPMI 1640 medium containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA) supplemented with 4.5 g/l glucose, 1 mM sodium pyruvate, and 10 mM HEPES. Cell culture was maintained at a cell concentration between 2 × 105 and 2 × 106 cells/ml. HepG2 cells from A.T.T.C. were maintained in MEM medium containing 10% fetal bovine serum (FBS). Hepa1c1c7 cells were a kind gift from O. Hankinson (University of California, Los Angeles) and maintained in α-minimum essential medium (Invitrogen). For transient transfection of U937 macrophages, cells were plated in RPMI with 10% FBS and 0.5 μg/ml TPA, which promotes differentiation into macrophages after 2 days. Transfection of plasmid DNA or siRNA into U937 macrophages was performed via Nucleofector technology. Briefly, 106 U937 macrophages were resuspended in 100 μl Nucleofector Solution V (Amaxa GmbH, Köln, Germany) and nucleofected with 1.0 μg plasmid DNA or 1.5 μg of the corresponding siRNA using program V-001, which is preprogrammed into the Nucleofector device (Amaxa GmbH). Following nucleofection, the cells were immediately mixed with 500 μl of prewarmed RPMI 1640 medium and transferred into 6-well plates containing 1.5 ml RPMI 1640 medium per well. 24h after transfection cells were treated with 10 nM TCDD, 10 μM FSK, or 0.1% Me2SO (control) for 24h. In case of siRNA transfection, the reduction of the target RNA and protein was detected by quantitative real-time RT-PCR and Western blot. siRNA to target human AhR (catalog no. M-004990) was designed and synthesized by Dharmacon (Lafayette, CO). siRNA to target human RelB (5′-GGAUUUGCCGAAUUAACAA-3′) and a negative control siRNA (catalog no. 10272280) were synthesized by Qiagen (Valencia, CA). For transient transfection experiments in HepG2, cells were plated in 24-well plates (1 × 105 cells/well) and transfected using jetPEI™ (PolyTransfection, Qbiogene, Irvine, CA), according to the manufacturer′s instructions. Briefly, 0.3 μg of the IL-8 construct was suspended in 25 μl of 150 mM sterile NaCl solution. Also 0.3 μl of jetPEI™ solution was suspended in 25 μl of 150 mM sterile NaCl solution. The jetPEI™/NaCl solution was then added to the DNA/NaCl solution and incubated at room temperature for 30 min. The medium in the wells was changed to fresh medium, and 50 μl of the DNA/jetPEI™ was added to each well. The transfection was allowed to proceed for 6 h and cells were treated with 10 nM TCDD, 10 μM FSK, or 0.1% Me2SO (control) for 24 h. To control the transfection efficiency cells were cotransfected with 0.1 μg per well β-galactosidase reporter construct. Luciferase activities were measured with the Luciferase Reporter Assay System (Promega, Madison, MI) using a luminometer (Berthold Lumat LB 9501/16, Pittsburgh, PA). Relative light units are normalized to β-galactosidase activity and to protein concentration, using Bradford dye assay (Bio-Rad, Hercules, CA).
IL-8 ELISA
The IL-8 concentration in culture supernatants was determined by ELISA as recommended by the manufacturer. Briefly, samples were added to 96-well microtiter plates, which were coated with monoclonal anti-IL-8 antibody (MAB-208, R&D Systems, Minneapolis, MN). After 2 h, the wells were washed four times and biotinylated anti-IL-8 antibody was added. After 1 h of incubation, the plates were washed three times, and streptavidin-HRP conjugate (RPN1231, Amersham, Buckinghamshire, UK) supplied, and the plates incubated for 20 min. Plates were washed again and chromogen substrate (Sigma Fast OPD, Sigma, St Louis, MO, USA) added. The plates were read at 450 nm.
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from U937 macrophages using a high pure RNA isolation kit (Qiagen) and cDNA synthesis was carried out as previously described (18). Quantitative detection of β-actin and IL-8 was performed with a LightCycler Instrument (Roche Diagnostics, Mannheim, Germany) using the QuantiTect SYBR Green PCR Kit (Qiagen, Valencia, CA) according to the manufacturer′s instructions. DNA-free total RNA (1.0 μg) was reverse-transcribed using 4 U Omniscript reverse transcriptase (Qiagen, Valencia, CA) and 1 μg oligo(dT)15 in a final volume of 40 μl. The primers for each gene were designed on the basis of the respective cDNA or mRNA sequences using OLIGO primer analysis software, provided by Steve Rosen and Whitehead Institute/MIT Center for Genome Research. The following primer sequences for human β-actin (forward primer 5′-GGACTTCGAGCAAGAGATGG-3′, reverse primer 5′-AGCACTGTGTTGGCGTACAG-3′), human IL-8 (forward primer 5′-CTGCGCCAACACAGAAATTA-3′, reverse primer 5′-ATTGCATCTGGCAACCCTAC-3′), human BAFF (forward primer 5′-CGTTCAGGGTCCAGAAGAAA-3′, reverse primer 5′-GTCCCATGGCGTAGGTCTTA-3′), and human BLC (forward primer 5′-GAGGCAGATGGAACTTGAGC-3′, reverse primer 5′-CTGGGGATCTTCGAATGCTA-3′) were used. All PCR assays were performed in triplicate. The intra-assay variability was <7%. For quantification data were analyzed with the LightCycler analysis software according to the manufacturer’s instructions. The variables were examined for one-sided Student’s t test. The results are given as the mean ± the standard error of the mean.
ChIP
U937 macrophages were seeded in 150-mm dishes and cultured in RPMI medium containing 10% FBS. FSK, TCDD, and H89 were added for the indicated times, and protein-DNA complexes were cross-linked with 1% formaldehyde for 10 min. Cells were washed with phosphate-buffered saline, harvested, and resuspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate) containing protease inhibitors (Roche, Mannheim, Germany) and sonicated with 5 sets of 10-sec pulses. The soluble chromatin was collected by centrifugation, and an aliquot of the chromatin was put aside and represented the input fraction. The supernatants were incubated with 30 μl of protein A/G Sepharose (50% slurry; Pharmacia) under gentle agitation for 2 h at 4°C. The supernatant was transferred to a new microcentrifuge tube, and 1 μg of antibody was added and incubated overnight at 4°C. Protein A/G-Sepharose (20 μl of a 50% slurry) was then added and incubated for 1.5 h. The pellets were successively washed for 10 min in 1 ml of buffer 1 (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS]), 1 ml of buffer 2 (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), 1 ml of LiCl buffer (20 mM Tris-HCl [pH 8.0], 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% Na-deoxycholate), and 2 × 1 ml of TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). Protein-DNA complexes were eluted in 120 μl of elution buffer (TE, 1% SDS) for 30 min, and the cross-links were reversed by overnight incubation at 65°C. DNA was purified using a PCR purification kit (QIAGEN) and eluted in 50 μl. ChIP DNA (5 μl) was amplified by real-time PCR with primers 5′-AATGAAAAGATGAGGGTGCAT-3′ and 5′-GCCAGCTTGGAAGTCATGTT-3′ covering the specified region RelBAhRE of IL-8. For real-time PCR, SYBR green qPCR supermix (Qiagen) was used to amplify a 182 bp fragment of the IL-8 promoter.
Nuclear complex co-immunoprecipitation assay and Western blot analyses
Preparation of nuclear extracts and co-immunoprecipitation was performed according to the manufacturer’s protocol (Active Motif). To analyze level of AhR and RelB protein in nuclei, nuclear protein extracts (15 μg) were separated on a 10% SDS-polyacrylamide gel and blotted onto a PVDF membrane (Immuno-Blot, BioRad, Herkules, CA). The antigen-antibody complexes were visualized using the chemoluminescence substrate SuperSignal®, West Pico (Pierce, Rockford, IL) as recommended by the manufacturer.
Mice
AhRnls mice were generated and kindly provided by Christopher Bradfield as described earlier (19). AhRnls mice carrying the lower affinity AhRd allele were backcrossed to C57BL/6 and injected once with 100 μg/kg TCDD. C57BL/6J wildtype mice carrying the high-affinity AhRb allele received a single dose 15 μg/kg TCDD. Male C57BL/6J mice, 8 weeks old, were purchased from Jackson Laboratory (Sacramento, CA). The animals had free access to water and food according to the guidelines set by the University of California Davis. When TCDD was used, animals were injected once intraperitoneally with TCDD. After 7 days animals were weighed and sacrificed, and organs were immediately removed for total RNA extraction.
EMSA
Nuclear extracts were isolated from U937 cells as described previously (18). In brief, 5 ×106 cells were treated with 10 nM TCDD, 10 μM FSK, or 2 μg/ml LPS for 90 min unless noted otherwise in the figure legends, and harvested in Dulbecco’s PBS containing 1 mM PMSF and 0.05 μg/μl of aprotinin. After centrifugation the cell pellets were gently resuspended in 1 ml of hypotonic buffer (20 mM HEPES, 20 mM NaF, 1 mM Na3VO4, 1mM Na4P2O7, 1 mM EDTA, 1 mM EGTA, 0.5 mM PMSF, 0.13 μM okadaic acid, 1 mM dithiothreitol, pH 7.9, and 1 μg/ml each leupeptin, aprotinin, and pepstatin). The cells were allowed to swell on ice for 15 min and then homogenized by 25 strokes of a Dounce-homogenizer. After centrifugation for 1 min at 16,000 × g nuclear pellets were resuspended in 300 μl ice-cold high-salt buffer (hypotonic buffer with 420 mM NaCl, and 20% glycerol). The samples were passed through a 21-gauge needle and stirred for 30 min at 4°C. The nuclear lysates were microcentrifuged at 16,000 × g for 20 min, aliquoted and stored at −80°C. Protein concentrations were determined by the method of Bradford. Sequences for double-stranded oligonucleotides used in EMSA are shown in Table 1. DNA-protein binding reactions were carried out in a total volume of 15 μl containing 10 μg nuclear protein, 60,000 cpm of DNA oligonucleotide, 25 mM Tris buffer (pH 7.5), 50 mM NaCl, 1 mM EDTA, 0.5 mM dithiothreitol, 5% glycerol, and 1 μg poly (dI-dC). The samples were incubated at room temperature for 20 min. Supershift analysis were performed by adding 2 μg of monoclonal ARNT, polyclonal RelA (Santa Cruz Biotechnology, Santa Cruz, CA), NF-κB member p50, RelB, c-Rel (Active Motif, Carlsbad, CA), or polyclonal AhR (Novus Biologicals, Littleton, CO) antibodies to the reaction mixtures. Competition experiments were performed in the presence of a 100-fold molar excess of unlabeled DNA fragments. Protein-DNA complexes were resolved on a 4% nondenaturating polyacrylamide gel and visualized by exposure of the dehydrated gels to X-ray films. For quantitative analysis, respective bands were quantified using a ChemiImager™4400 (Alpha Innotech Corporation, San Leandro, CA).
Table 1.
EMSA oligonucleotide sequences.
| RelBAhRE | 5′-AGATGAGGGTGCATAAGTTC-3′ |
| M1 RelBAhRE | 5′-AGATGAGGGCGCATAAGTTC-3′ |
| M2 RelBAhRE | 5-AGATGAGGCTCCATAAGTTC-3′ |
| XRE consensus | 5′-GCCCCGGAGTTGCGTGAGAAGAGCCTGG-3′ |
| NF-κB consensus | 5′-AGTTGAGGGGACTTTCCCAGGC-3′ |
| RelB/p52 binding site | 5′-GATGAGGGAGATTTGGTTCTCTAG-3′ |
Note. Binding sequences are underlined. Point mutations are in bold italic letters.
Statistics
All data were obtained from at least three independent experiments performed in duplicate, and the results are given as the mean ± the standard error of the mean. To demonstrate statistical significance, the variables were examined for one-sided Student’s t test. The level of significance was p<0.05.
Acknowledgments
We thank Chris Bradfield and Ed Glover (McArdle laboratory for Cancer Research at the University of Wisconsin) for generously providing a breeding pair of AhRnls mice. We thank Thomas Haarmann-Stemmann and Josef Abel for excellent technical support, Roland Schmidt, Gisela Degen, and Gille Salbert for critical reading of this article.
This work was supported by research grant R01-ES005233 and core center grant, P30-ES05707 from the National Institute of Environmental Health Sciences.
Footnotes
“This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.”
References
- 1.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–403. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 2.Kunsch C, Ruben SM, Rosen CA. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol Cell Biol. 1992;10:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;4:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 4.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 2004;21:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;8:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 6.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 7.Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- 8.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 9.Singh SS, Hord NG, Perdew GH. Characterization of the activated form of the aryl hydrocarbon receptor in the nucleus of HeLa cells in the absence of exogenous ligand. Arch Biochem Biophys. 1996;329:47–55. doi: 10.1006/abbi.1996.0190. [DOI] [PubMed] [Google Scholar]
- 10.Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Divergent roles of hepatitis B virus X-associated protein 2 (XAP2) in human versus mouse Ah receptor complexes. Biochemistry. 2004;43:700–709. doi: 10.1021/bi035827v. [DOI] [PubMed] [Google Scholar]
- 11.Petrulis JR, Hord NG, Perdew GH. Subcellular localization of the aryl hydrocarbon receptor is modulated by the immunophilin homolog hepatitis B virus X-associated protein 2. J Biol Chem. 2000;275:37448–37453. doi: 10.1074/jbc.M006873200. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 13.Walisser JA, Bunger MK, Glover E, Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci USA. 2004;101:16677–16682. doi: 10.1073/pnas.0404379101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oesch-Bartlomowicz B, Huelster A, Wiss O, Antoniou-Lipfert P, Dietrich C, Arand M, Weiss C, Bockamp E, Oesch F. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: divergent signaling pathways. Proc Natl Acad Sci USA. 2005;102:9218–9223. doi: 10.1073/pnas.0503488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumura F, Vogel CF. Evidence supporting the hypothesis that one of the main functions of the aryl hydrocarbon receptor is mediation of cell stress responses. Biol Chem. 2006;387:1189–1194. doi: 10.1515/BC.2006.146. [DOI] [PubMed] [Google Scholar]
- 16.Vogel CF, Nishimura N, Sciullo E, Li W, Wong P, Matsumura F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice. Arch Biochem Biophys. 2007 doi: 10.1016/j.abb.2007.01.015. In press. [DOI] [PubMed] [Google Scholar]
- 17.Freund A, Jolivel V, Durand S, Kersual N, Chalbos D, Chavey C, Vignon F, Lazennec GA. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene. 2004;23:6105–6114. doi: 10.1038/sj.onc.1207815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel CF, Sciullo E, Park S, Liedtke C, Trautwein C, Matsumura F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J Biol Chem. 2004;279:8886–8894. doi: 10.1074/jbc.M310190200. [DOI] [PubMed] [Google Scholar]
- 19.Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem. 2003;278:17767–17774. doi: 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- 20.Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, Labow MA. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leuk Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- 22.Roebuck KA. Regulation of interleukin-8 gene expression. J Interferon Cytokine Res. 1999;5:429–438. doi: 10.1089/107999099313866. [DOI] [PubMed] [Google Scholar]
- 23.Mukaida N, Mahe Y, Matsushima K. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J Biol Chem. 1990;265:21128–21133. [PubMed] [Google Scholar]
- 24.Harant H, de Martin R, Andrew PJ, Foglar E, Dittrich C, Lindley IJ. Synergistic activation of interleukin-8 gene transcription by all-trans-retinoic acid and tumor necrosis factor-alpha involves the transcription factor NF-kappaB. J Biol Chem. 1996;271:26954–26961. doi: 10.1074/jbc.271.43.26954. [DOI] [PubMed] [Google Scholar]
- 25.Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol. 2006;72:267–279. doi: 10.1016/j.bcp.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Shen ES, Whitlock JP., Jr Protein-DNA interactions at a dioxin-responsive enhancer. Mutational analysis of the DNA-binding site for the liganded Ah receptor. J Biol Chem. 1992;267:6815–6819. [PubMed] [Google Scholar]
- 27.Sogawa K, Numayama-Tsuruta K, Takahashi T, Matsushita N, Miura C, Nikawa J, Gotoh O, Kikuchi Y, Fujii-Kuriyama Y. A novel induction mechanism of the rat CYP1A2 gene mediated by Ah receptor-Arnt heterodimer. Biochem Biophys Res Commun. 2004;318:746–755. doi: 10.1016/j.bbrc.2004.04.090. [DOI] [PubMed] [Google Scholar]
- 28.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A forth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petrulis JR, Hord NG, Perdew GH. Subcellular localization of the aryl hydrocarbon receptor is modulated by the immunophilin homolog hepatitis B virus X-associated protein 2. J Biol Chem. 2000;275:37448–37453. doi: 10.1074/jbc.M006873200. [DOI] [PubMed] [Google Scholar]
- 30.Nebert DW, Puga A, Vasiliou V. Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann N Y Acad Sci. 1993;685:624–640. doi: 10.1111/j.1749-6632.1993.tb35928.x. [DOI] [PubMed] [Google Scholar]
- 31.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 32.Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274:510–515. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- 33.Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene. 2000;19:5498–5506. doi: 10.1038/sj.onc.1203945. [DOI] [PubMed] [Google Scholar]
- 34.Baba T, Mimura J, Gradin K, Kuroiwa A, Watanabe T, Matsuda Y, Inazawa J, Sogawa K, Fujii-Kuriyama Y. Structure and expression of the Ah receptor repressor gene. J Biol Chem. 2001;276:33101–33110. doi: 10.1074/jbc.M011497200. [DOI] [PubMed] [Google Scholar]
- 35.Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS. Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J Immunol. 2005;175:90–103. doi: 10.4049/jimmunol.175.1.90. [DOI] [PubMed] [Google Scholar]
- 36.Sutter TR, Guzman K, Dold KM, Greenlee WF. Targets for dioxin: genes for plasminogen activator inhibitor-2 and interleukin-1 beta. Science. 1991;254:415–418. doi: 10.1126/science.1925598. [DOI] [PubMed] [Google Scholar]
- 37.Puga A, Barnes SJ, Chang C, Zhu H, Nephew KP, Khan SA, Shertzer HG. Activation of transcription factors activator protein-1 and nuclear factor-kappaB by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Pharmacol. 2000;59:997–1005. doi: 10.1016/s0006-2952(99)00406-2. [DOI] [PubMed] [Google Scholar]
- 38.Kang SM, Tran AC, Grilli M, Lenardo MJ. NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science. 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 39.Ohmori Y, Fukumoto S, Hamilton TA. Two structurally distinct kappa B sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. J Immunol. 1995;155:3593–3600. [PubMed] [Google Scholar]