

Drugs that at pico- to nanomolar concentration regulate ion channel function by high-affinity binding to their cognate receptor often have a “secondary pharmacology,” in which the same molecule at low micromolar concentrations regulates a diversity of membrane proteins in an apparently nonspecific manner. It has long been suspected that this promiscuous regulation of membrane protein function could be due to changes in the physical properties of the host lipid bilayer, but the underlying mechanisms have been poorly understood. Given that pharmacological research often involves drug concentrations that alter the physical properties of lipid bilayers, and that nonspecific drug effects are a major cause of attrition in drug discovery, this lack of understanding has been problematic. The present Perspective summarizes recent developments in the investigation of the bilayer-mediated mechanism that are transforming it into a subject of quantitative science. It is described how the hydrophobic interactions between a membrane protein and the host lipid bilayer provide the basis for a mechanism, whereby protein function is regulated by the bilayer physical properties. The use of gramicidin channels as single-molecule force transducers for measuring drug-induced changes in the bilayer physical properties (bilayer stiffness), and for predicting drug effects on membrane protein function, is described.
Introduction
When experimental evidence for the existence of ion channels started to emerge, the receptor concept had already reached maturity. It was established that drugs could regulate cellular function by high affinity, stoichiometric binding to specific receptors—and that receptor function, in turn, could be regulated by allosteric mechanisms, based on one receptor conformation having a higher affinity for a drug than another. The theories and technologies of ligand–receptor interactions provided a methodological framework, based on equilibrium energetic considerations, which allowed for quantitative predictions that could be experimentally tested (Colquhoun, 1998; Rang, 2006). Not surprisingly, the study of ion channel regulation by specific drug binding has been very successful. It has long been suspected that drugs regulate ion channel function also by another mechanism, namely by changing the physical properties of the host lipid bilayer (for recent summaries, see Lundbæk, 2006; Andersen and Koeppe, 2007). This mechanism has not had an easy life, however. The regulation of membrane protein function by changes in the bilayer physical properties, generally, has been poorly understood, and the hypothesis that the effects of a drug could be due to changes in bilayer physics has been based on a diagnosis of exclusion, i.e., being effects for which specific mechanisms have not been identified. Yet, pharmacological studies on ion channels often involve drug concentrations that alter the physical properties of lipid bilayers (compare Lundbæk, 2006), and nonspecific drug effects are a major cause of attrition in pharmaceutical development projects (Leeson and Springthorpe, 2007). Thus, the lack of understanding of the bilayer-mediated effects of drugs has been, and to a large extent still is, a problem in (and for) drug development.
The present Perspective describes recent studies on the bilayer-mediated mechanism that are transforming it into a diagnosis of inclusion and a subject of quantitative science. First, I summarize findings that suggest that drugs can regulate membrane protein function by altering the physical properties of the host lipid bilayer. Then, I describe the hydrophobic coupling mechanism, which provides a conceptual framework for understanding membrane protein regulation by the bilayer physical properties. Finally, I describe how the gramicidin channel can be used as a prototypical example of an ion channel that is regulated by the bilayer physical properties, as well as a molecular probe for characterizing drug-induced changes in these properties.
Promiscuous Regulation of Membrane Proteins
Amphiphilic drugs that at pico- to nanomolar concentrations regulate ion channel function by high-affinity binding to their cognate receptor often have a “secondary pharmacology,” in which the same molecule at low micromolar concentrations regulates a diversity of unrelated membrane proteins in an apparently nonspecific manner. A given compound may regulate a number of structurally unrelated proteins, and a given protein may be (similarly) regulated by a number of structurally dissimilar compounds (compare Lundbæk, 2006). It has long been suspected that amphiphiles could regulate membrane proteins by absorbing to the host lipid bilayer and thereby alter the bilayer physical properties, and that such a mechanism could be involved in the many nonspecific effects.
Amphiphile-induced Changes in Lipid Bilayer Physics
As reviewed by Seeman (1972), early studies noted that many lipophilic drugs modulate membrane protein function in an apparently nonspecific manner and prevent nerve cell excitation without appreciably altering the membrane resting potential. This was referred to as the drug's “membrane stabilizing” effect. It was later noted that such drugs protect red blood cells from osmotic hemolysis at the same concentrations where nerve excitation is blocked. This led to the use of the term “membrane stabilizing” also to describe the physical stabilization of cellular membranes (whatever the actual mechanism). It was proposed that the modulation of membrane protein function and cellular excitability could be due to the changes in cell membrane physical properties, but a possible causal relation/mechanism was not identified.
Later it was shown that amphiphiles, at the concentrations where membrane protein function promiscuously is regulated, may alter a number of parameters describing the physical properties of lipid bilayers (compare Lundbæk, 2006; Andersen and Koeppe 2007). A number of studies focused on the role of the bilayer “fluidity”, but the mechanisms whereby the fluidity of a bilayer might regulate membrane protein function have never been clear. Most importantly, the equilibrium distribution among conformational states of a membrane protein cannot be altered by a change in bilayer fluidity per se (Lee, 1991).
The Hydrophobic Coupling Mechanism
Beginning in the 1980s, it has become increasingly clear that membrane protein function can be regulated by changes in the host bilayer thickness or lipid intrinsic curvature (the curvature adopted by an isolated, unconstrained monolayer) (Andersen and Koeppe, 2007). These findings supported the notion that amphiphiles could regulate membrane protein function by altering the collective physical properties of the bilayer. They further provided the basis for development of a working model of protein–bilayer interactions that is based on equilibrium energetic considerations and allows for quantitative predictions that can be experimentally tested.
Current descriptions of the structural arrangement of cell membrane lipid bilayers are based on the fluid mosaic model (FMM) (Singer and Nicolson, 1972). This model rests on the generalization that the gross structure of a lipid bilayer is organized by the need for optimizing hydrophobic interactions among the bilayer-forming molecules. The FMM describes the average structural organization of a lipid bilayer by the hydrophobic interactions among the bilayer-forming molecules. It does not consider the dynamic regulatory potential of these interactions.
Membrane protein function often involves conformational changes at the hydrophobic exterior of the protein transmembrane segment, as reviewed in Lundbæk (2006). Fig. 1 shows an example; the calcium-ATPase in a Ca2+-bound and a Ca2+-free state (Toyoshima and Nomura, 2002). As may be seen from the figure, the conformational changes at the protein–bilayer interface are substantial (and rather complex). Due to the protein–bilayer hydrophobic interactions, such conformational changes give rise to a local bilayer perturbation, with an associated energetic cost (Mouritsen and Bloom, 1984; Sackmann, 1984). The change in bilayer perturbation energy (ΔΔGbilayer) contributes to the total free energy difference between the conformational states (ΔGtotal). That is:
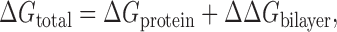 |
(1) |
where ΔGprotein is the free energy change intrinsic to the protein. The distinction between ΔGprotein and ΔΔGbilayer is not unambiguous, but Eq. 1 provides for a framework in which protein–bilayer interactions may be examined. Changes in the physical properties of the bilayer that affect the magnitude of ΔΔGbilayer will alter the protein conformational equilibrium and function. Thus the protein–bilayer hydrophobic interactions, through the ΔΔGbilayer contribution to ΔGtotal, provide for a mechanism whereby protein function is coupled to the bilayer physical properties (for recent reviews see Lundbæk, 2006; Andersen and Koeppe, 2007). Fig. 2 illustrates this hydrophobic coupling mechanism (HCM) in the case of an ion channel, where channel opening involves a decrease in hydrophobic length (length of the hydrophobic transmembrane segment), which causes a corresponding decrease in bilayer hydrophobic thickness (thickness of the hydrophobic core). Therefore the probability of channel opening depends on the energetic cost of locally thinning the bilayer.
Figure 1.

The calcium-ATPase in a Ca2+-bound and a Ca2+-free state (Toyoshima et al., 2000; Toyoshima and Nomura, 2002). Prepared with Pymol using PDB structures 1SU4 and 1IWO.
Figure 2.

Hydrophobic coupling between a membrane protein and the host lipid bilayer. A protein conformational change causes a local bilayer deformation. Modified from Andersen and Koeppe (2007).
For a membrane protein with two interconvertible states, the HCM may be described as
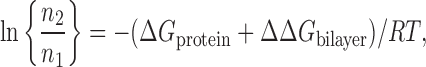 |
(2) |
where the ratio n2/n1 describes the equilibrium distribution between the number of molecules in each state, and R and T are the gas constant and temperature in Kelvin, respectively (Lundbæk et al., 2004). For the HCM to be operative in regulation of membrane protein function, ΔΔGbilayer should be larger than ∼1 kcal/mol, and changes in bilayer composition should alter ΔΔGbilayer by more than ∼0.5 kcal/mol.
What is the energetic cost of the bilayer perturbation associated with a membrane protein conformational change; and can changes in bilayer molecular composition considerably alter this cost? This has been investigated using gramicidin channels in planar lipid bilayers as a model system.
Gramicidin Channels as Molecular Force Transducers
The cation-selective gramicidin A (gA) channel, both structurally and functionally, is one of the most well-described ion channels. The channel structure has been determined at atomic resolution and channel function can be studied with single-molecule resolution in planar bilayers or cellular membranes (for recent reviews see Lundbæk, 2006; Andersen et al., 2007).
The ion-conducting gA channel is formed by transbilayer association of pentadecapeptide subunits residing in each monolayer of a lipid bilayer (Fig. 3 A). When the bilayer hydrophobic thickness (d0) exceeds the gA channel's hydrophobic length (l ∼2.2 nm) (Elliott et al., 1983), channel formation involves a local adjustment of the bilayer thickness to match the channel's hydrophobic exterior (Fig. 3 A). The ensuing bilayer deformation energy contributes to the energetic cost of channel formation (compare Eq. 1) and changes in the bilayer physical properties are reflected as changes in channel appearance rate (f), channel lifetime (τ), and channel dimerization constant (the number of conducting channels). Therefore gA channels can be used as molecular probes to measure the energetic cost of the bilayer deformation associated with a change in hydrophobic length of an embedded protein (Lundbæk, 2006; Andersen and Koeppe, 2007).
Figure 3.

(A) Gramicidin channel formation by the transbilayer association of two monomers causes a local bilayer deformation. Modified from Andersen and Koeppe (2007). (B) Relation between ln{τ} and the hydrophobic thickness of monoacylglyceride bilayers. Results obtained by Elliot et al. (1983) using squalene in the bilayer forming solution. Figure modified from Lundbæk and Andersen (1999). (C) Effects of lysophosphatidylcholine (LPC) on gA channel behavior in a diphytanoylphosphatidyl-choline/n-decane bilayer. Single channel current traces from the same lipid bilayer membrane before (top) and after (bottom) addition of LPC. Modified from Lundbæk and Andersen (1994).
The energetics of adjusting the thickness of a lipid bilayer to match the length of an embedded protein has been analyzed using the continuum elastic theory of liquid crystal deformations (Fig. 2 and Fig. 3 A) (Huang, 1986; Nielsen et al., 1998; Lundbæk and Andersen, 1999; Nielsen and Andersen, 2000). The major energetic contributions to the bilayer deformation energy are the cost of bilayer compression and monolayer bending (Nielsen et al., 1998; Nielsen and Andersen, 2000). The compression contribution is determined by the compression modulus and the change in bilayer thickness. The bending contribution, correspondingly, is determined by the bending modulus and the difference between the monolayer curvature and the lipid intrinsic curvature.
A lipid bilayer, in response to the deformation change induced by gA channel formation, exerts a disjoining force (F) on the channel. To a first approximation, F may be expressed as:
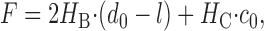 |
(3) |
where HB and HC, are elastic coefficients reflecting the bilayer compression and bending moduli, and c0 is the lipid intrinsic curvature (Lundbæk et al., 2005). Based on Eq. 3, F should be increased by (a) larger values of the channel–bilayer mismatch; (b) larger values of the elastic moduli; (c) more negative values of c0 (as the coefficient Hc is negative). An increase in F, in turn, will decrease f and τ.
The relation between gA channel lifetime and channel–bilayer mismatch has been studied in monoacylglyceride bilayers of varying thickness (Kolb and Bamberg, 1977; Elliott et al., 1983; Huang, 1986; Lundbæk and Andersen, 1999). As shown in Fig. 3 B, ln{τ} is linearly decreasing function of d0. This is what would be expected if the bilayer disjoining force on the channel is given by Eq. 3 (Lundbæk and Andersen, 1999). The slope of the ln{τ} vs. d0 relation provides a measure of the bilayer deformation energy associated with channel formation. For a bilayer with a d0 of 3 nm, this value is ∼10 kcal/mol, a value that agrees surprisingly well with that estimated using the continuum theory of elastic bilayer deformations (Lundbæk and Andersen, 1999). The bilayer elastic energy should scale as a linear function of the channel radius (Nielsen and Andersen, 2000), and the corresponding value for a typical membrane protein with a radius of 2–3 nm (rather than 1 nm for gA) would be 20–30 kcal/mol. For comparison, the energy released by hydrolysis of one molecule of ATP is ∼9 kcal/mol (Veech et al., 1979). The bilayer deformation energy, associated with a protein conformational change, can be of a considerable magnitude.
The modulation of gA channel function by changes in lipid intrinsic curvature of the bilayer-forming lipid similarly conforms to the expectations of the continuum theory of elastic liquid crystal deformations. When c0 of dioleoylphosphatidylserine/n-decane bilayers is varied by Ca2+-induced changes in electrostatic repulsion among the negatively charged lipid head groups, a more negative c0 increases F, and thus decreases f and τ (compare Eq. 3) (Lundbæk et al., 1997). The presence of 100 μM Ca2+ increases the energetic cost of channel formation by 2.5 kcal/mol. The decrease in f is much larger than that in τ. This finding is expected because f reflects the energetic cost of a local change in bilayer thickness, which is much larger than that reflected in τ. Separation of the channel-forming subunits to the distance where channel conductance is lost involves a local increase in bilayer thickness of ∼0.16 nm (Durkin et al., 1993; Miloshevsky and Jordan, 2004). In contrast, adjusting the nonperturbed bilayer thickness (∼5 nm) to match the length of the monomers separated by ∼0.16 nm, involves a decrease of ∼2.7 nm.
Overall, studies of gA channels in a wealth of different bilayers have led to the conclusion that the hydrophobic coupling between a lipid bilayer and an embedded protein provide for an energetically important mechanism, whereby protein function can be regulated (Lundbæk, 2006).
Amphiphile-induced Changes in Bilayer Stiffness
The use of gA channels as molecular force transducers has been developed into a technology for measuring the effects of changes in bilayer molecular composition on the bilayer elastic response to a change in hydrophobic length of an embedded protein. A change in the bilayer physical properties that for a given bilayer deformation alters the bilayer disjoining force on a gA channel is operationally defined as a change in bilayer stiffness (Lundbæk et al., 1996, 2004). A decrease in stiffness reduces the disjoining force, which increases f and τ (and vice versa). A change in stiffness further should cause a larger change in f than in τ (see Lundbæk et al., 2004).
The effects of amphiphiles on the bilayer stiffness measured using gA channels will be illustrated with a few examples. Lysophospholipids (LPLs) at low nanomolar concentrations may regulate membrane protein function by specific mechanisms (Ishii et al., 2004). At low micromolar concentrations, however, LPLs regulate a plethora of unrelated proteins in a seemingly nonspecific manner (Lundbæk and Andersen, 1994). LPLs are micelle-forming compounds that promote a positive-going change in c0 (Fuller and Rand, 2001). Moreover, LPLs, as water-soluble amphiphiles generally, should decrease the elastic moduli of a lipid bilayer due to their reversible absorption to the bilayer (Evans et al., 1995). The LPL lysophospatidylcholine (LPC), in accordance, decreases the compression and bending moduli of phospholipid bilayers in an aqueous solution (Zhelev, 1998; McIntosh et al., 1995). These effects of LPLs should all decrease the bilayer disjoining force on a gA channel (compare Eq. 3). Fig. 3 C shows the effects of LPC, at a nominal concentration of 1 μM, on gA channel behavior in a lipid bilayer (Lundbæk and Andersen, 1994). (The aqueous LPC concentration is likely to be lower than 1 μM, see Fig. 5.) Both f and τ are increased, and there is a larger increase in f than in τ (for the purpose of visual clarity, a current trace with a modest increase in f is shown). At a nominal concentration of 2 μM, LPC causes an ∼400-fold increase in the number of conducting channels. This corresponds to a decrease in the energetic cost of channel formation by ∼3.6 kcal/mol. The effects of LPC are not due to specific interactions with the gA channel; similar changes in channel function are observed using several other lysolipids or synthetic micelle-forming amphiphiles, as well as by using different sequence-modified gA analogues (Lundbæk and Andersen, 1994; Lundbæk et al., 2004). Therefore, LPC decreases lipid bilayer stiffness.
Figure 5.

A note on amphiphile concentrations. It is difficult to quantitatively compare the effects of amphiphiles on membrane protein function in different experimental setups. Amphiphiles adsorb to all hydrophobic surfaces and the aqueous concentration, generally, will be lower than the nominal concentration. First, adsorption to the walls of experimental containers (e.g., pipettes) may be considerable, and increasing with the surface area/volume ratio. For LPC, with a molecular area ∼0.7 nm2, 80% of the molecules in 5 μl of a 10 μM solution would be needed to cover the surface of a hydrophobic cube with that volume. Second, the adsorption to lipid bilayers may be considerable. A recent study, measuring the effects of free fatty acids on lipid bilayer stiffness, found that in the absence of a lipid bilayer 30% of a fatty acid adsorbed to the Teflon experimental chamber; when fatty acid was added to a chamber containing a bilayer, however, the aqueous concentration was about two orders of magnitude lower than the nominal concentration! (Bruno et al., 2007). Because the lipid/water partition coefficient for amphiphiles is high, the bilayer amphiphile concentration will be affected by the ratio between lipid volume and total volume in the experimental setup. In binding experiments using membrane fragments, the membrane volume (of which the majority is lipids) may constitute 2% of the total volume (Søgaard et al., 2006). In bilayer stiffness measurements the bilayer-forming lipid solution constitutes only ∼0.1% of the total volume. Moreover, in whole cell voltage clamp experiments using continuously flowing experimental solution, the lipid volume may be considered as infinitely small compared with the total volume. As a consequence, a given nominal amphiphile concentration will result in bilayer concentrations that are related as: whole cell voltage clamp > bilayer stiffness measurements > binding experiments.
The gA channel becomes particularly useful when amphiphile-induced changes in the parameters used to describe the bilayer the physical properties, in isolation, would be expected to have opposite effects on the bilayer deformation energy associated with a protein conformational change. Capsaicin, the pungent ingredient in pepper, at high nanomolar concentrations, interacts with the TRPV1 receptor (Caterina et al., 1997); at low micromolar concentrations, capsaicin alters the function of a wide range of membrane proteins (Lundbæk et al., 2005). Being a water-soluble amphiphile, capsaicin would be expected to decrease the bilayer elastic moduli and thus the bilayer disjoining force on a gA channel. Capsaicin also promotes a negative-going change in c0 (Aranda et al., 1995), however, which should increase the disjoining force. So what is the net effect on gA channels? At the concentrations where membrane protein is promiscuously regulated (≥10 μM), capsaicin increases both f and τ, that is, the bilayer stiffness is decreased (Lundbæk et al., 2005). Apparently the decrease in the bilayer elastic moduli trumps the negative-going change in c0.
Bilayer Stiffness and Ion Channels in Living Cells
To what extent are amphiphile-induced changes in bilayer stiffness, measured using gA channels in planar lipid bilayers, relevant for the function of complex membrane proteins in the heterogeneous cell membranes of living cells? This question has been addressed in studies of voltage-dependent sodium channels (VDSCs) in mammalian cells (Lundbæk et al., 2004, 2005). Capsaicin reversibly promotes inactivation of VDSCs, but does not affect channel activation. Triton X-100 and three other synthetic micelle-forming amphiphiles that decrease bilayer stiffnes, but are structurally dissimilar from capsaicin, have the same effects. Cholesterol, which increases bilayer stiffness, in contrast, prevents inactivation. That is, cholesterol depletion promotes inactivation and cholesterol addition to depleted cells prevents inactivation (this effect of cholesterol saturates above the normal cholesterol content, however). Capsaicin and the micelle-forming amphiphiles all cause a hyperpolarizing shift in the potential causing 50% channel inactivation (Vin). Fig. 4 compares the magnitude of this shift with the effect on bilayer stiffness, as characterized by the change in gA channel lifetime. At the lowest concentrations used, the quantitative relations for the different compounds are remarkably similar, suggesting that the amphiphile-induced regulation of VDSC function involves changes in bilayer physical properties that are (quantitatively) reflected in bilayer stiffness. At higher concentrations, the correlations deviate considerably (Fig. 4, insert).
Figure 4.

Relation between amphiphile-induced shifts in the potential causing 50% inactivation of VDSCs in HEK293 cells (Vin − Vin, contr) and change in gA channel lifetime in dioleoylphosphatidylcholine/n-decane bilayers (ln{τ/τcontr}). β-Octyl glucoside (βOG), capsaicin, Genapol X-100 (GX100), Triton X-100 (TX100), and reduced Triton X-100 (rTX100) were used at concentrations of 0.3, 1 mM; 30 μM; 4.5 μM; 10, 30 μM; 10, 30 μM. Cells were depolarized to +20 mV from 300-ms prepulses to potentials varying from −130 to +50 mV. The insert shows the relation between the effects on VDSCs and on gA channels including higher amphiphile concentrations at which the correlation between the effects on the two channel types breaks down. Modified from Lundbæk et al. (2005).
Table I shows results for all amphiphiles that have been characterized with respect to both effects on bilayer stiffness and VDSC function (Lundbæk et al., 2005). The nominal concentrations used in the bilayer stiffness measurements are lower than, or equal to, those affecting VDSCs (and a number of other membrane proteins). There is an almost one-to-one mapping of the changes in bilayer stiffness and VDSC function. Some of the amphiphiles shown in Table I, at submicromolar concentrations, regulate protein function by specific interactions, and one cannot exclude that such effects contribute to the results shown. The correlation between the changes in bilayer stiffness and VDSC function is quite general, however. Only one group of amphiphiles does not seem to conform to this correlation: long-chain alcohols decrease gA channel lifetime (Elliott et al., 1985) but promote VDSC inactivation (Armstrong and Binstock, 1964).
TABLE I.
Effects of Amphiphiles on Bilayer Stiffness and VDSC Function
| gA
|
VDSC
|
|||
|---|---|---|---|---|
| Amphiphile | Bilayer stiffness | Activation | Inactivation | |
| Positive c0 | LPC | ↓(a) | ↓(b) | ↑(b) |
| Triton X-100 | ↓(c) | 0(c) | ↑(c) | |
| Reduced Triton X-100 | ↓(c) | 0(c) | ↑(c) | |
| Genapol X-100 | ↓(c) | 0(c) | ↑(c) | |
| β-Octyl Glucoside | ↓(c) | 0(c) | ↑(c) | |
| Pentobarbital | ↓(d) | 0(e) | ↑(e) | |
| Valproate | ↓(d) | 0(f) | ↑(f) | |
| Diazepam | ↓(d) | 0(g) | ↑(g,h) | |
| Capsazepine | ↓(i) | 0(i) | ↑(i) | |
| Genistein | ↓(j) | 0↓(k,r) | ↑(k,r) | |
| Negative c0 | Capsaicin | ↓(i) | 0↓(i) | ↑(i) |
| Docosahexaenoic acid | ↓(n) | 0(o) | ↑(o) | |
| Arachidonic acid | ↓(q,l) | 0(s) | ↑(s) | |
| Chlorpromazine | ↓(d) | 0(m,g) | ↑(m,g) | |
| Oleic acid | 0(n) | 0(o) | 0(o) | |
| Cholesterol | ↑(c,p) | ↓(c) | ↓(c) | |
(a) Lundbæk and Andersen, 1994, (b) Shander et al., 1996, (c) Lundbæk et al., 2004, (d) Sun, Y., V. Jogini, and O.S. Andersen. 2002. Biophys. J. 82:550a, (e) Rehberg et al., 1995, (f) Schauf, 1987, (g) Wakamori et al., 1989, (h) Backus et al., 1991, (i) Lundbæk et al., 2005, (j) Hwang et al., 2003, (k) Wang et al., 2003, (l) Bruno et al., 2006, (m) Ogata and Narahashi, 1989, (n)Bruno et al., 2007, (o)Vreugdenhil et al., 1996, (p) Lundbæk et al., 1996, (q) Bruno et al., 2005, (r) Paillart et al., 1997, (s) Lee et al., 2002.
It is possible to extend this argument more generally. Table II shows the effects of a number of amphiphiles on the function of gA channels and of five other channel types, all together representing three protein superfamilies. There is a near perfect correlation between the changes in bilayer stiffness and the function of the different channel types. As in Table I, one cannot exclude that specific interactions contribute to the effects shown, but the correlation seems too general to be due to specific mechanisms.
TABLE II.
Effects of Amphiphiles on Bilayer Stiffness and Ion Channel Function
| Superfamily
|
Prokaryotic channel
|
Voltage dependent channels
|
Cys-loop receptors
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Channel type
|
gA
|
VDSC
|
N-type Ca2+
|
BKCa
|
nAChR
|
GABAA
|
|||
| Function | Bilayer stiffness | Activation | Inactivation | Activation | Inactivation | Activation | Desensitization | Muscimol binding |
|
| Positive c0 | Triton X-100 | ↓(a) | 0(a) | ↑(a) | 0(b) | ↑(b) | ↑(c,d) | ↑(e) | |
| β-Octyl glucoside | ↓(a) | 0(a) | ↑(a) | 0(b) | ↑(b) | ↑(d) | ↑(e) | ||
| Pentobarbital | ↓(v) | 0(w) | ↑(w) | 0(x) | ↑(x) | ↑(y) | ↑(m) | ||
| Negative c0 | Capsaicin | ↓(f) | 0↓(f) | ↑(f) | ↑(g) | ↑(e) | |||
| Docosahexaenoic acid | ↓(h) | 0(i) | ↑(i) | ↑(j) | ↑(k) | ↑(l,e) | |||
| Arachidonic acid | ↓(n,o) | 0(u) | ↑(u) | ↑(p) | ↑(p | ↑(q) | ↑(k) | ↑(l) | |
| Cholesterol | ↑(a,r) | ↓(a) | ↓(a) | 0(b) | ↓(b) | ↓(s) | ↓(t) | ↓(e) | |
The effects of structurally different amphiphiles on bilayer stiffness measured using gA channels and on; voltage-dependent sodium channels (VDSC); N-type calcium channels (N-type Ca2+); calcium activated potassium channels (BKCa); nicotinic acetylcholine receptors (nAChR), and GABAA receptors. (a) Lundbæk et al., 2004, (b) Lundbæk et al., 1996, (c) Anwyl and Narahashi, 1980, (d) McCarthy and Moore, 1992, (e) Søgaard et al., 2006, (f) Lundbæk et al., 2005, (g) Ellis et al., 1997, (h) Bruno et al., 2007, (i) Vreugdenhil et al., 1996, (j) Ye et al., 2002, (k) Bouzat and Barrantes, 1993, (l) Witt et al., 1999, (m) Quast and Brenner, 1983, (n) Bruno et al., 2005, (o) Bruno et al., 2006, (p) Liu et al., 2001, (q) Denson et al., 2000, (r) Lundbæk et al., 1996, (s) Chang et al., 1995, (t) Baenziger et al., 2000, (u) Lee et al., 2002, (v) Sun, Y., V. Jogini, and O.S. Andersen. 2002. Biophys. J. 82:550a, (w) Rehberg et al., 1995, (x) Gundersen et al., 1988, (y) Firestone et al., 1994. Table modified from Søgaard et al. (2006).
Perspective
The remarkable correlation between the effects of amphiphiles on bilayer stiffness, as measured using gA channels in planar lipid bilayers, and on ion channel function in living cells is somewhat surprising, and likely to break down at some point. Nevertheless, the correlations shown in Fig. 4 and in Tables I and II strongly suggest that gA-based measurements of amphiphile-induced changes in bilayer stiffness detect changes in the physical properties of lipid bilayers that are important for membrane protein function generally. Maybe this is because these measurements report net changes in the bilayer elastic response to a change in protein hydrophobic length, as experienced by a bilayer-embedded protein.
The results shown in Tables I and II also illustrate the fact that pharmacological research on ion channels often involves drug concentrations that are so high that the drug may alter the physical properties of lipid bilayers (as the stiffness measurements involve nominal concentrations that are, at least, as low as those used on the different ion channels.) Moreover, orally available drugs generally are amphiphiles (Lipinski et al., 1997), and in drug development the nonspecific effects of drugs that absorb to lipid bilayers are a major, and rising, problem. It seems suitable to end this Perspective by citing a recent review that describes this predicament: “The consequences of the marked increase in lipophilicity—the most important drug-like physical property—include a greater likelihood of lack of selectivity and attrition in drug development. Tackling the threat of compound-related toxicological attrition needs to move to the mainstream of medicinal chemistry decision making.” (Leeson and Springthorpe, 2007). The use of gA-based measurements of drug-induced changes in bilayer stiffness, or test of whether the effects of a drug can be reproduced by a structurally different compound known to alter bilayer stiffness (e.g., Triton X; Matta et al., 2007), may provide suitable tools in this endeavor.
Acknowledgments
I thank Olaf S. Andersen, Shemille Collingwood, Helgi I. Ingolfsson, Ruchi Kapoor, Jung Kim, and Subhi J. Al' Aref for stimulating discussions.
The present work was supported by a grant from the Danish Medical Research Council and National Institutes of Health Grant GM021342.
Abbreviations used in this paper: gA, gramicidin A; HCM, hydrophobic coupling mechanism; LPC, lysophospatidylcholine; LPL, lysophospholipid; VDSC, voltage-dependent sodium channel.
References
- Andersen, O.S., and R.E. Koeppe II. 2007. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 36:107–130. [DOI] [PubMed] [Google Scholar]
- Andersen, O.S., M.J. Bruno, H. Sun, and R.E. Koeppe II. 2007. Single-molecule methods for monitoring changes in bilayer elastic properties. Methods Mol. Biol. 400:543–570. [DOI] [PubMed] [Google Scholar]
- Anwyl, R., and T. Narahashi. 1980. Comparison of desensitization and time-dependent block of the acetylcholine receptor responses by chlorpromazine, cytochalasin B, Triton X-100 and other agents. Br. J. Pharmacol. 69:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda, F.J., J. Villalain, and J.C. Gomez-Fernandez. 1995. Capsaicin affects the structure and phase organization of phospholipid membranes. Biochim. Biophys. Acta. 1234:225–234. [DOI] [PubMed] [Google Scholar]
- Armstrong, C.M., and L. Binstock. 1964. The effects of several alcohols on the properties of the squid giant axon. J. Gen. Physiol. 48:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus, K.H., P. Pflimlin, and G. Trube. 1991. Action of diazepam on the voltage-dependent Na+ current. Comparison with the effects of phenytoin, carbamazepine, lidocaine and flumazenil. Brain Res. 548:41–49. [DOI] [PubMed] [Google Scholar]
- Baenziger, J.E., M.L. Morris, T.E. Darsaut, and S.E. Ryan. 2000. Effect of membrane lipid composition on the conformational equilibria of the nicotinic acetylcholine receptor. J. Biol. Chem. 275:777–784. [DOI] [PubMed] [Google Scholar]
- Bouzat, C.B., and F.J. Barrantes. 1993. Effects of long-chain fatty acids on the channel activity of the nicotinic acetylcholine receptor. Receptors Channels. 1:251–258. [PubMed] [Google Scholar]
- Bruno, M.J., R.E. Koeppe, and O.S. Andersen. 2005. Modification of gramicidin channel function by PUFAs depends on double-bond structure. Biophys. J. 88:575. [Google Scholar]
- Bruno, M.J., R.E. Koeppe, and O. Andersen. 2006. Polyunsaturated fatty acids alter lipid bilayer elasticity. Biophys. J. 90:1769. [Google Scholar]
- Bruno, M.J., R.E. Koeppe II, and O.S. Andersen. 2007. Docosahexaenoic acid alters bilayer elastic properties. Proc. Natl. Acad. Sci. USA. 104:9638–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina, M.J., M.A. Schumacher, M. Tominaga, T.A. Rosen, J.D. Levine, and D. Julius. 1997. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 389:816–824. [DOI] [PubMed] [Google Scholar]
- Chang, H.M., R. Reitstetter, R.P. Mason, and R. Gruener. 1995. Attenuation of channel kinetics and conductance by cholesterol: an interpretation using structural stress as a unifying concept. J. Membr. Biol. 143:51–63. [DOI] [PubMed] [Google Scholar]
- Colquhoun, D. 1998. Binding, gating, affinity and efficacy: the interpretation of structure–activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacology. 125:924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson, D.D., X. Wang, R.T. Worrell, and D.C. Eaton. 2000. Effects of fatty acids on BK channels in GH(3) cells. Am. J. Physiol. Cell Physiol. 279:C1211–C1219. [DOI] [PubMed] [Google Scholar]
- Durkin, J.T., L.L. Providence, R.E. Koeppe II, and O.S. Andersen. 1993. Energetics of heterodimer formation among gramicidin analogues with an NH2-terminal addition or deletion. Consequences of a missing residue at the join in channel. J. Mol. Biol. 231:1102–1121. [DOI] [PubMed] [Google Scholar]
- Elliott, J.R., D. Needham, J.P. Dilger, and D.A. Haydon. 1983. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Biophys. Acta. 735:95–103. [DOI] [PubMed] [Google Scholar]
- Elliott, J.R., D. Needham, J.P. Dilger, O. Brandt, and D.A. Haydon. 1985. A quantitative explanation of the effects of some alcohols on gramicidin single-channel lifetime. Biochim. Biophys. Acta. 814:401–404. [DOI] [PubMed] [Google Scholar]
- Ellis, J.L., J.S. Sham, and B.J. Undem. 1997. Tachykinin-independent effects of capsaicin on smooth muscle in human isolated bronchi. Am. J. Respir. Crit. Care Med. 155:751–755. [DOI] [PubMed] [Google Scholar]
- Evans, E., W. Rawicz, and A.F. Hofmann. 1995. Lipid bilayer expansion and mechanical disruption in solutions of water-soluble bile acid. In Bile Acids in Gastroenterology Basic and Clinical Advances, A. F. Hofmann, G. Paumgartner, and A. Stiehl, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 59–68.
- Firestone, L.L., J.K. Alifimoff, and K.W. Miller. 1994. Does general anesthetic-induced desensitization of the Torpedo acetylcholine receptor correlate with lipid disordering? Mol. Pharmacol. 46:508–515. [PubMed] [Google Scholar]
- Fuller, N., and R.P. Rand. 2001. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 81:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen, C.B., J.A. Umbach, and B.E. Swartz. 1988. Barbiturates depress currents through human brain calcium channels studied in Xenopus oocytes. J. Pharmacol. Exp. Ther. 247:824–829. [PubMed] [Google Scholar]
- Huang, H.W. 1986. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 50:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, T.C., R.E. Koeppe II, and O.S. Andersen. 2003. Genistein can modulate channel function by a phosphorylation-independent mechanism: importance of hydrophobic mismatch and bilayer mechanics. Biochemistry. 42:13646–13658. [DOI] [PubMed] [Google Scholar]
- Ishii, I., N. Fukushima, X. Ye, and J. Chun. 2004. Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 73:321–354. [DOI] [PubMed] [Google Scholar]
- Kolb, H.A., and E. Bamberg. 1977. Influence of membrane thickness and ion concentration on the properties of the gramicidin a channel. Autocorrelation, spectral power density, relaxation and single-channel studies. Biochim. Biophys. Acta. 464:127–141. [DOI] [PubMed] [Google Scholar]
- Lee, A.G. 1991. Lipids and their effects on membrane proteins: evidence against a role for fluidity. Prog. Lipid Res. 30:323–348. [DOI] [PubMed] [Google Scholar]
- Lee, G.Y., Y.K. Shin, C.S. Lee, and J.H. Song. 2002. Effects of arachidonic acid on sodium currents in rat dorsal root ganglion neurons. Brain Res. 950:95–102. [DOI] [PubMed] [Google Scholar]
- Leeson, P.D., and B. Springthorpe. 2007. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 6:881–890. [DOI] [PubMed] [Google Scholar]
- Lipinski, C.A., F. Lombardo, B.W. Dominy, and P.J. Feeney. 1997. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23:3–25. [DOI] [PubMed] [Google Scholar]
- Liu, L., C.F. Barrett, and A.R. Rittenhouse. 2001. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am. J. Physiol. Cell Physiol. 280:C1293–C1305. [DOI] [PubMed] [Google Scholar]
- Lundbæk, J.A. 2006. Regulation of membrane protein function by lipid bilayer elasticity-a single molecule technology to measure the bilayer properties experienced by an embedded protein. Jounal of Physics - Condensed Matter. 18:S1305–S1344. [DOI] [PubMed] [Google Scholar]
- Lundbæk, J.A., and O.S. Andersen. 1994. Lysophospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J. Gen. Physiol. 104:645–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbæk, J.A., and O.S. Andersen. 1999. Spring constants for channel-induced lipid bilayer deformations-estimates using gramicidin channels. Biophys. J. 76:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbæk, J.A., P. Birn, J. Girshman, A.J. Hansen, and O.S. Andersen. 1996. Membrane stiffness and channel function. Biochemistry. 35:3825–3830. [DOI] [PubMed] [Google Scholar]
- Lundbæk, J.A., A.M. Maer, and O.S. Andersen. 1997. Lipid bilayer electrostatic energy, curvature stress, and assembly of gramicidin channels. Biochemistry. 36:5695–5701. [DOI] [PubMed] [Google Scholar]
- Lundbæk, J.A., P.H.A.J. Birn, R. Søgaard, C. Nielsen, J. Girshman, M.J. Bruno, S.E. Tape, J. Egebjerg, D.V. Greathouse, G.L. Mattice, et al. 2004. Regulation of sodium channel function by bilayer elasticity the importance of hydrophobic coupling: effects of micelle-forming amphiphiles and cholesterol. J. Gen. Physiol. 123:599–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbæk, J.A., P. Birn, S.E. Tape, G.E. Toombes, R. Søgaard, R.E. Koeppe II, S.M. Gruner, A.J. Hansen, and O.S. Andersen. 2005. Capsaicin regulates voltage-dependent ssodium channels by altering lipid bilayer elasticity. Mol. Pharmacol. 68:680–689. [DOI] [PubMed] [Google Scholar]
- Matta, J.A., R.L. Miyares, and G.P. Ahern. 2007. TRPV1 is a novel target for omega-3 polyunsaturated fatty acids. J. Physiol. 578:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, M.P., and M.A. Moore. 1992. Effects of lipids and detergents on the conformation of the nicotinic acetylcholine receptor from Torpedo californica.J. Biol. Chem. 267:7655–7663. [PubMed] [Google Scholar]
- McIntosh, T.J., S. Advani, R.E. Burton, D.V. Zhelev, D. Needham, and S.A. Simon. 1995. Experimental tests for protrusion and undulation pressures in phospholipid bilayers. Biochemistry. 34:8520–8532. [DOI] [PubMed] [Google Scholar]
- Miloshevsky, G.V., and P.C. Jordan. 2004. Gating gramicidin channels in lipid bilayers: reaction coordinates and the mechanism of dissociation. Biophys. J. 86:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouritsen, O.G., and M. Bloom. 1984. Mattress model of lipid-protein interactions in membranes. Biophys. J. 46:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, C., M. Goulian, and O.S. Andersen. 1998. Energetics of inclusion-induced bilayer deformations. Biophys. J. 74:1966–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, C., and O.S. Andersen. 2000. Inclusion-induced bilayer deformations: effects of monolayer equilibrium curvature. Biophys. J. 79:2583–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata, N., and T. Narahashi. 1989. Block of sodium channels by psychotropic drugs in single guinea-pig cardiac myocytes. Br. J. Pharmacol. 97:905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart, C., E. Carlier, D. Guedin, B. Dargent, and F. Couraud. 1997. Direct block of voltage-sensitive sodium channels by genistein, a tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther. 280:521–526. [PubMed] [Google Scholar]
- Quast, U., and O. Brenner. 1983. Modulation of [3H]muscimol binding in rat cerebellar and cerebral cortical membranes by picrotoxin, pentobarbitone, and etomidate. J. Neurochem. 41:418–425. [DOI] [PubMed] [Google Scholar]
- Rang, H.P. 2006. The receptor concept: pharmacology's big idea. Br. J. Pharmacol. 147(Suppl 1):S9–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehberg, B., E. Bennett, Y.H. Xiao, S.R. Levinson, and D.S. Duch. 1995. Voltage- and frequency-dependent pentobarbital suppression of brain and muscle sodium channels expressed in a mammalian cell line. Mol. Pharmacol. 48:89–97. [PubMed] [Google Scholar]
- Sackmann, E. 1984. Physical basis of trigger processes and membrane structures. In Biological Membranes. D. Chapman, editor. Academic Press Inc., Ltd., London. 105–143.
- Schauf, C.L. 1987. Anticonvulsants modify inactivation but not activation processes of sodium channels in Myxicola axons. Can. J. Physiol. Pharmacol. 65:1220–1225. [DOI] [PubMed] [Google Scholar]
- Seeman, P. 1972. The membrane actions of anesthetics and tranquilizers. Pharmacol. Rev. 24:583–655. [PubMed] [Google Scholar]
- Shander, G.S., A.I. Undrovinas, and J.C. Makielski. 1996. Rapid onset of lysophosphatidylcholine-induced modification of whole cell cardiac sodium current kinetics. J. Mol. Cell. Cardiol. 28:743–753. [DOI] [PubMed] [Google Scholar]
- Singer, S.J., and G.L. Nicolson. 1972. The fluid mosaic model of the structure of cell membranes. Science. 175:720–731. [DOI] [PubMed] [Google Scholar]
- Søgaard, R., T.M. Werge, C. Bertelsen, C. Lundbye, K.L. Madsen, C.H. Nielsen, and J.A. Lundbæk. 2006. GABA(A) receptor function is regulated by lipid bilayer elasticity. Biochemistry. 45:13118–13129. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., and H. Nomura. 2002. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 418:605–611. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., M. Nakasako, H. Nomura, and H. Ogawa. 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 405:647–655. [DOI] [PubMed] [Google Scholar]
- Veech, R.L., J.W.R. Lawson, N.W. Cornell, and H.A. Krebs. 1979. Cytosolic phosphorylation potential. J. Biol. Chem. 254:6538–6547. [PubMed] [Google Scholar]
- Vreugdenhil, M., C. Bruehl, R.A. Voskuyl, J.X. Kang, A. Leaf, and W.J. Wadman. 1996. Polyunsaturated fatty acids modulate sodium and calcium currents in CA1 neurons. Proc. Natl. Acad. Sci. USA. 93:12559–12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamori, M., M. Kaneda, Y. Oyama, and N. Akaike. 1989. Effects of chlordiazepoxide, chlorpromazine, diazepam, diphenylhydantoin, flunitrazepam and haloperidol on the voltage-dependent sodium current of isolated mammalian brain neurons. Brain Res. 494:374–378. [DOI] [PubMed] [Google Scholar]
- Wang, Y., M.B. Wagner, R. Kumar, J. Cheng, and R.W. Joyner. 2003. Inhibition of fast sodium current in rabbit ventricular myocytes by protein tyrosine kinase inhibitors. Pflugers Arch. 446:485–491. [DOI] [PubMed] [Google Scholar]
- Witt, M.R., C.F. Poulsen, B. Lukensmejer, S.E. Westh-Hansen, J. Nabekura, N. Akaike, and M. Nielsen. 1999. Structural requirements for the interaction of unsaturated free fatty acids with recombinant human GABAA receptor complexes. Ann. N. Y. Acad. Sci. 868:697–700. [DOI] [PubMed] [Google Scholar]
- Ye, D., D. Zhang, C. Oltman, K. Dellsperger, H.C. Lee, and M. VanRollins. 2002. Cytochrome p-450 epoxygenase metabolites of docosahexaenoate potently dilate coronary arterioles by activating large-conductance calcium-activated potassium channels. J. Pharmacol. Exp. Ther. 303:768–776. [DOI] [PubMed] [Google Scholar]
- Zhelev, D.V. 1998. Material property characteristics for lipid bilayers containing lysolipid. Biophys. J. 75:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]