

Abstract
Studies using protein synthesis inhibitors have provided key support for the prevalent view that memory formation requires the initiation of protein synthesis as a primary element of the molecular biology of memory. However, many other interpretations of the amnesia data have received far less attention. These include: a) Protein synthesis may play a constitutive role in memory formation, providing proteins prior to an experience that can be activated by training; b) Protein synthesis may be needed to replace proteins available prior to learning but ‘consumed’ by learning; c) Inhibition of protein synthesis impairs the well-being of neurons, leading to an inability to deliver resources needed for memory formation; and d) Inhibition of protein synthesis results in abnormal neural functions that interfere with memory. One of these, abnormal release of neurotransmitters after inhibition of protein synthesis, is detailed here, along with a review of many circumstances in which it appears that protein synthesis at the time of training is not required for the formation of new memories.
Evidence of activation of cell signaling molecules and transcription factors is another form of support for a role of training-initiated protein synthesis in memory. However, recent findings suggest that many of these molecules are activated by training and remain activated for days after training, i.e. activated for times well beyond those typically invoked for memory consolidation processes. Reviewing these results, this paper suggests that the long-lasting molecular changes may be the basis of a form of intra-cellular memory, one responsible for up-regulating the probability that a neuron, once activated in this manner, will engage in future plasticity. This view melds ideas of modulation of memory with those of consolidation of memory.
Keywords: anisomycin, memory consolidation, memory modulation, memory, molecular bases
1. INTRODUCTION
Many reports begin with a well-accepted statement that there are at least two stages of memory. As generally proposed, memory is maintained soon after experience, by a short-lived temporary process that is dependent on modifications of existing proteins (e.g., Kandel and Schwartz, 1982). As this memory mechanism decays, mechanisms responsible for permanent memory storage supplant the temporary process. These mechanisms are generally believed to be dependent on new protein synthesis and to form the basis for cellular memory consolidation at a cellular level (Squire, 1987; Dudai, 2002; Kandel, 2001).
Of several ways in which new protein synthesis may be important to memory (Glassman, 1969), one is the commonly held view that new protein synthesis is needed for the modifications of neuron-neuron functional connectivity. Many of the studies that discuss protein-synthesis dependent memory do so on the basis of findings that protein synthesis inhibitors impair memory, identifying results obtained with the inhibitors as demonstrating a requirement for protein synthesis in memory consolidation. However, other interpretations of these findings are less often considered (Gold, 2006). One is that intact protein synthesis is necessary in a more constitutive manner for replenishment of materials used in memory formation (Routtenberg and Rekart, 2005). Another possibility is that protein synthesis is needed for maintenance of cell health at a level that can sustain the use of cellular resources to fine-tune the connectivity of the nervous system in response to memory (Rudy et al., 2006). Yet another interpretation is that inhibition of protein synthesis results in abnormal neural functions that interfere with memory, a possibility supported by recent evidence from the author’s laboratory (Canal et al., 2007) as described below.
The present paper examines several classes of findings that are not readily incorporated into the view that new protein synthesis is a necessary component of the mechanisms by which new memories are formed. Although this review will mainly focus on contemporary studies, it is important to note extensive evidence from the 1970s and 1980s that led many investigators to conclude that the evidence did not support the idea that protein synthesis was necessary for memory formation, that the effects of protein synthesis inhibitors on memory were very complex, and that the results supported many alternative explanations (e.g., Barraco and Stettner, 1976; Cooper, Bloom and Roth, 1978; Martinez, Jensen and McGaugh, 1981). These points of view were the motivation for the review by Davis and Squire (1984), in which they defended the idea that protein synthesis was necessary for memory. Importantly, whatever conclusion one now draws about this issue, it is not the case that these early studies led to a consensus that protein synthesis was necessary for memory. Nonetheless, statements espousing this view have become standard in the introductions to and rationales for studies of protein synthesis and memory.
2. Is protein synthesis necessary for memory and synaptic plasticity?
2.a. Examples of plasticity resistant to inhibition of protein synthesis
2.a.1. Memory
Although protein synthesis inhibitors often impair memory across species and tasks, it is also the case, as noted by Routtenberg and Rekart (2005), that intact memories are sometimes formed even in the presence of extensive inhibition of protein synthesis. Specific behavioral variables appear to be important. For example, amnesia is attenuated in graded manner by increasing the number of training trials or by increasing the intertrial interval (Quartermain and Botwinick, 1975; Flood et al., 1975), or by increasing the intensity of a training footshock (Flood et al., 1978a). Thus, views regarding the requirement of new protein synthesis for memory formation do not comprehensively incorporate a significant amount of the available data. Results like these, particularly when viewed together with the pharmacological ‘rescue’ studies described later, indicate that protein synthesis is not universally necessary for the formation of memories, specifically including long-lasting memories.
2.a.2. LTP
The multiple variables involved in memory research might confuse the interpretations in favor of or against a role for new protein synthesis in memory. In recent years, many reports have noted that protein synthesis inhibitors have effects on long-term potentiation (LTP) that are analogous to those seen in many memory experiments, specifically intact early LTP with rapid decay in rats and mice treated with protein synthesis inhibitors. However, like memory, LTP is sometimes insensitive to inhibition of protein synthesis and, as in memory experiments, there are specific variables that seem to determine the sensitivity or resistance of LTP to protein synthesis inhibitors. In rat visual cortex, theta burst stimulation resulted in a slowly developing LTP that was blocked by protein synthesis inhibitors and a rapidly established form of LTP that was not blocked by the inhibitors (Kurotani et al., 1996).
Although not directly involving inhibition of protein synthesis, a more recent paper (Steward et al., 2007) provides additional evidence that protein synthesis may not always be necessary for LTP. Perforant path LTP was established in rats using either 250 or 400 Hz stimulation trains. Although the expression of LTP was comparable under these conditions, important molecular markers of LTP induction were very different. LTP induced with 400 Hz tetanizing stimulation trains was accompanied by increased c-Fos and MAP kinase expression. However, LTP induced with 250 Hz trains did not engage these molecular mechanisms. Importantly, the magnitude of LTP was similar under both stimulation conditions, showing that there are at least some forms of synaptic plasticity that do not require new protein synthesis.
A recent report (Fonseca et al., 2006a) provides additional evidence that protein synthesis is not necessary for LTP. The findings of this report suggest that protein synthesis and degradation must be in balance in order for the production of LTP in CA1 (Fonseca et al., 2006a). As reported by any others, late-LTP was impaired in the presence of protein synthesis inhibition. In addition, the report showed that late-LTP was also impaired by pharmacological inhibition of proteasome-mediated protein degradation. Of particular relevance to the present discussion was the finding that simultaneous inhibition of both protein synthesis and degradation did not interfere with the induction and maintenance of LTP. Regardless of the specific interpretation regarding balance of synthesis and degradation, the findings indicate clearly that establishment and maintenance of LTP can occur in the absence of protein synthesis. The ability of pharmacological inhibition of protein degradation to rescue LTP adds to other examples noted below in which drugs can rescue LTP from the effects of protein synthesis inhibition in the absence of reversing that inhibition.
Another set of very interesting results shows that the specific schedule and frequency of test pulses after induction of LTP determine whether early-LTP is or is not impaired by the protein synthesis inhibitor, anisomycin (Fonseca et al., 2006b). The study shows again that anisomycin impairs the establishment of late- but not early-LTP in CA1, with decay of LTP within the hours after tetanus. These effects were seen when the test pulses were administered at the rate of 1/min. However, when test pulses were administered at the rate of 1/10 sec, both early- and late-LTP were impaired; inhibition of protein synthesis also interferes with both early- and late-LTP in the mossy fiber - CA3 system (Calixto et al., 2003). In addition, when anisomycin was applied after establishment of late-LTP, the treatment did not impair LTP unless test pulses (1/10 sec) were administered during the anisomycin exposure. Importantly, the conditions of induction were the same across treatments. The application of test pulses well after induction determined whether LTP was or was not impaired by an inhibitor of protein synthesis. Therefore, it seems reasonable to conclude that the sensitivity to impairment of LTP by anisomycin is not a property of the formation of LTP but is a property of interactions of anisomycin with activity-related alterations of neuronal functions. This view suggests not that neuronal activity determines the protein synthesis dependence of LTP but that the neural activity determines the level of interference with neural functioning – in this case well after LTP induction – to impair the expression of LTP.
2.a.3. Synaptic plasticity in Aplysia
Similar to findings seen with tests of mammalian memory and LTP, protein synthesis is not required for the formation of some forms of synaptic plasticity in the Aplysia. In this preparation, long-term synaptic facilitation of sensory to motor neuron synapses can be produced reliably by repeated tail shocks (Carew et al., 1971). Long-term synaptic facilitation of this system can also be produced by application of 5-HT using either of two procedures, by 5 spaced pulses of 5-HT applied to the ganglia (repeated stimulation) or by a single 25-min application of 5-HT to the sensory neuron soma paired with a single 5-min pulse applied to the soma (asymmetric stimulation) (Sherff and Carew, 1999). With both procedures, the resulting expression of long-term facilitation is evident as a long-lasting increase in motor neuron EPSP amplitude in response to sensory neuron stimulation. Although the expression of the synaptic plasticity appears identical, facilitation induced by asymmetric but not that induced by repeated stimulation is blocked in the presence of a postsynaptic protein synthesis inhibitor (Sherff and Carew, 2004). Thus, the results reveal another instance in which protein synthesis is not required for synaptic plasticity.
2.b. Pharmacological rescue of memory impaired by protein synthesis inhibitors
There is extensive evidence, much of it before 1980, showing that many drugs can attenuate amnesia produced by protein synthesis inhibitors. These early studies of attenuation of amnesia by protein synthesis inhibitors and other treatments were reviewed comprehensively by Martinez et al. (1981). A general conclusion from these experiments was that protein synthesis inhibitors impaired memory by depressing arousal levels needed for memory formation. Drugs that reinstated the arousal levels reversed amnesias produced by the inhibitors while drugs that independently depressed arousal levels exaggerated the amnesia.
Early studies of amnesia produced by puromycin found that the memory impairments could be attenuated by a wide range of drugs, including caffeine, ACTH, and vasopressin (Lande et al., 1972; Flexner et al., 1978) administered near the time of training. Also, intracerebral injections of saline administered up to 12 hrs after puromycin treatment reversed the amnesia (Flexner and Flexner, 1967; Rosenbaum et al., 1968), leading to the general conclusion that puromycin impaired memory by mechanisms other than interfering with new protein synthesis necessary for the formation of new memories (cf. Martinez et al., 1981).
In part because this conclusion meant that the question of a role of protein synthesis in memory had not been tested with puromycin, later experiments examined the effects of other protein synthesis inhibitors, primarily anisomycin and cycloheximide. With the experience obtained in puromycin studies, studies of amnesia and pharmacological reversals of amnesia soon appeared. Some examples include findings that anisomycin-, cycloheximide- and / or acetoxycycloheximide-induced amnesias can be attenuated by administration of amphetamine, strychnine, picrotoxin, corticosteroids, dexamethasone, caffeine, nicotine, and phenoxybenzamine (Flood et al., 1977, 1978a,b; Serota et al., 1972; Oliver et al., 1979; Barondes and Cohen, 1968; Nakajima, 1975; Gold and Sternberg, 1978). In one series of experiments (Flood et al., 1977, 1978a), a wide range of stimulants, as above, blocked amnesia. Moreover, general depressants – chloral hydrate and sodium pentobarbital – increased the ability of anisomycin to produce amnesia. An example of the latter effect is as follows. Anisomycin produced amnesia for inhibitory avoidance memory when the training involved a relatively weak footshock but did not produce amnesia if training was accomplished with a more intense footshock. However, anisomycin in combination with post-training injections of either chloral hydrate or pentobarbital did produce amnesia (Flood et al., 1977, 1978a).
Within the experiments that explored attenuation and exaggeration of amnesias induced by protein synthesis inhibitors, some measured the possibility that the drugs altered the level of protein synthesis inhibition (e.g., Flood et al., 1977; Hall et al., 1976; Barondes and Cohen, 1968; Serota et al., 1972; Sershen et al., 1982). Across studies, stimulant drugs that reversed amnesia did not reduce the extent of protein synthesis inhibition; depressant drugs that exaggerated amnesia produced by protein synthesis inhibitors resulted in marginal increases in the extent of protein synthesis inhibition. Thus, these findings support the view that protein synthesis inhibitors interfere with arousal mechanisms and only indirectly interfere with the mechanisms of memory formation.
Davis and Squire (1984) examined the reversal literature and provided an important interpretation of studies that show that stimulants reverse amnesias produced by inhibitors of protein synthesis. Their interpretation was an alternative to the views of others that discounted the relevance of studies that used protein synthesis inhibitors to address questions of the mechanisms of memory. To develop their conclusion, Davis and Squire noted two sets of findings. First, amnesia after administration of a protein synthesis inhibitor, like other classes of treatments, was rarely complete. Second, the stimulants used to reverse amnesia were drugs that themselves enhanced memory. Therefore, Davis and Squire concluded that the stimulants were effective at reversing amnesia because they enhanced the residual weakened memory by mechanisms that do not involve protein synthesis. Presumably, the authors would similarly argue that depressants augment protein synthesis-induced amnesia by themselves impairing memory, as seen at times with anesthetics (e.g., Semba et al., 2005). However, it seems reasonable to believe that drugs that enhance and impair memory do so by altering the processes involved in memory formation (Gold and McGaugh, 1975). Therefore, this view does not offer a neurobiological explanation of how weak memories can be strengthened and strong memories can be weakened in the absence of protein synthesis. In addition, more recent data (Canal et al., 2007, described below) show conditions under which drugs that themselves are not stimulants and that do not enhance memory can nonetheless attenuate anisomycin-induced amnesia, a finding that conflicts directly with the perspective offered by Davis and Squire (1984).
Even recognizing the importance of the perspectives offered by Davis and Squire (1984), it seems curious that the large literature on attenuation of amnesia by protein synthesis inhibitors has been largely ignored or, more precisely, fully overshadowed. Here are a few statistics gleaned from a Science Citations Index search for a few of the key articles above. The Science paper by Flood et al. (1977), showing that anisomycin-induced amnesia could be increased or decreased by stimulants and depressants has been cited 48 times, 7 times since 2000. The review of attenuation of amnesia by Martinez et al. (1981), which concluded that the amnestic effects of protein synthesis inhibitors are nonspecific in nature, has been cited 47 times, 5 since 2000. The review by Davis and Squire (1984), with the conclusion as above that the data make a compelling case that training-related protein synthesis is an essential step in the formation of long-term memory, has been cited 597 times, 320 times since 2000.
A second line of reasoning in the Davis and Squire (1984) review is based on convergent pharmacology showing that multiple protein synthesis inhibitors, each with different side effects, impair memory, thereby supporting the conclusion that protein synthesis is necessary for memory formation. More precisely, however, the convergent pharmacology instead supports the more empirical conclusion that inhibition of protein synthesis impairs memory. One interpretation is that new protein synthesis may be necessary for memory formation. Another interpretation is that there are neural consequences of the massive inhibition of protein synthesis that impair a host of cell functions that might mediate amnesia. This alternative conclusion has support from a recent experiment (Canal et al., 2007) described below.
2.c. Conclusion: New protein synthesis is often not necessary for memory formation
The multiple demonstrations that memory formation can survive extensive inhibition of protein synthesis lead to the conclusion that the requirement is hardly absolute. Findings that behavioral, electrophysiological and pharmacological manipulations can rescue memory and LTP from the insult of protein synthesis inhibition are striking. In many of the contemporary demonstrations showing such results, the authors suggest that they have identified a new memory process that is not dependent on new protein synthesis, even if previous instances were dependent on new protein synthesis. Such arguments keep the dominant views in place even as new data limit the generality of those views. However, the logic seems challenged when treatments administered well after the induction of learning or LTP – e.g. the test pulse patterns administered in Fonseca et al. (2006b) – make the (prior) plasticity dependent or not on protein synthesis.
Thus, there is substantial lack of generality for the findings that protein synthesis inhibitors block memory formation. The lack of generality deserves considerable attention, perhaps with more interest paid to the conditions under which memory is insensitive to protein synthesis inhibitors. In particular, if one believes that new protein synthesis is important for memory formation, it is precisely the conditions in which inhibitors do not impair memory that might be most illuminating, perhaps by revealing the synthesis of the principal proteins – those surviving the inhibition – that are truly necessary for memory formation. Alternatively, if one believes that protein synthesis inhibitors impair memory by inducing cellular or organismic illness, or by blocking new protein synthesis to replenish those proteins used in memory formation, or by simply inserting abnormal neural activity – neural noise (Gold, 2006) – that impairs memory, the issue of why the inhibitors block memory only some of the time is similarly important to these views.
3. If protein synthesis is not necessary for memory formation, why do inhibitors impair memory?
The many instances in which memory and synaptic plasticity can occur in the presence of inhibition of protein synthesis indicate clearly that protein synthesis is not necessary for all forms of memory and synaptic plasticity. What of the reports that do show such impairments after administration of protein synthesis inhibitors? If the examples of failures to impair memory with protein synthesis inhibitors mean that new protein synthesis is not necessary in these cases, do reports that inhibitors do impair memory reveal a requirement, at least in these cases, for new protein synthesis? This reverse statement does not necessarily hold true. When, under some conditions, memory and synaptic plasticity are blocked by inhibition of protein synthesis, the results need not imply that protein synthesis is an essential part of the process by which the neural changes are made. Like other conclusions based on loss of function studies, the results indicate what the residual functions are or are not rather than indicating for what the lost function is responsible. More directly, these studies suggest that the absence of protein synthesis is a poor condition under which memories can be made.
The disruption of functional organization of topographic maps in motor cortex is consistent with the view that an inhibitor of protein synthesis, and likely inhibition of protein synthesis itself, disrupts normal cellular processes to impair the functions of those cells. Relating this idea to memory, the mechanisms of amnesia in these cases might reflect cell sickness, abnormal neural electrical activity, or intrusion of neural ‘noise’ masking the primary changes that reflect memory formation (Gold, 2006). Related to these ideas is evidence that protein synthesis inhibitors result in gene superinduction, including genes sometimes associated with memory formation (Edwards and Mahadevan, 1992; Radulovic and Tronson, 2007; Routtenberg and Rekart, 2005; Rudy et al., 2006).
Note that these are potential mechanisms of amnesia. The mechanisms of amnesia need not be the inverse of the mechanisms of memory formation.
An additional example illustrates well the nature and duration of the effects of protein synthesis inhibition on the function of a neural system. Local application of protein synthesis inhibitors results in disruption of topographic maps and increases in the threshold for evoked responses in motor cortex for over 4 days after a single treatment (Kleim et al., 2003); the long-lasting duration of these effects is not evident with other inhibitors of neural function such as application of a local anesthetic. In the same report, anisomycin injections into motor cortex resulted in decreases in synapse number and size, also for more than 4 days after a single injection. This report offers clear evidence that a single injection of anisomycin can have proactive effects on the functional integrity of the brain area receiving the injection.
In recent studies originally intended as a prelude for examinations of the role of protein synthesis in the context of drug abuse research, we obtained evidence suggesting that anisomycin injections into the amygdala result in profound alterations in neurotransmitter release, changes that can readily account for amnesia produced by this treatment (Canal et al., 2007). Rats were trained in a one-trial inhibitory avoidance task and were tested for memory 48 hr later. As shown in Figure 1, pretraining injections of anisomycin into the amygdala impaired memory and impaired training-related increases in expression of c-Fos in the amygdala. Using a combination microdialysis probe and injection cannula, we collected samples of extracellular fluid at the site of injection before, during and after anisomycin injection. The samples were later analyzed for norepinephrine, dopamine and serotonin content. The results of one of these experiments is shown in Figure 2. The first samples collected after anisomycin injections revealed massive increases in release of all three biogenic amines. The peak increases in release were above 1000% for norepinephrine, 11,000% for dopamine and 17,000% for serotonin. It is important to note that the magnitudes of these increases are well beyond those seen after training or stress, where increases of 100–300% are typical (McIntyre et al., 2002; Ma and Morilak, 2005; Shearman et al., 2005; Hassert et al., 2004; Clayton and Williams, 2000; Miyashita and Williams, 2002; Williams et al., 1998).
Figure 1.

Effects of intra-amygdala injections of anisomycin (ANI; 62.5 µg/0.5 µl/side) on memory and on c-Fos immunoreactivity. The anisomycin was injected into the amygdala 20 min prior to inhibitory avoidance training. Left. Anisomycin significantly impaired memory as tested 48 hr after training (p>0.05). Right. Anisomycin blocked training-related increases in c-Fos in sections taken just posterior to the injection site. (From Canal et al., 2007.)
Figure 2.
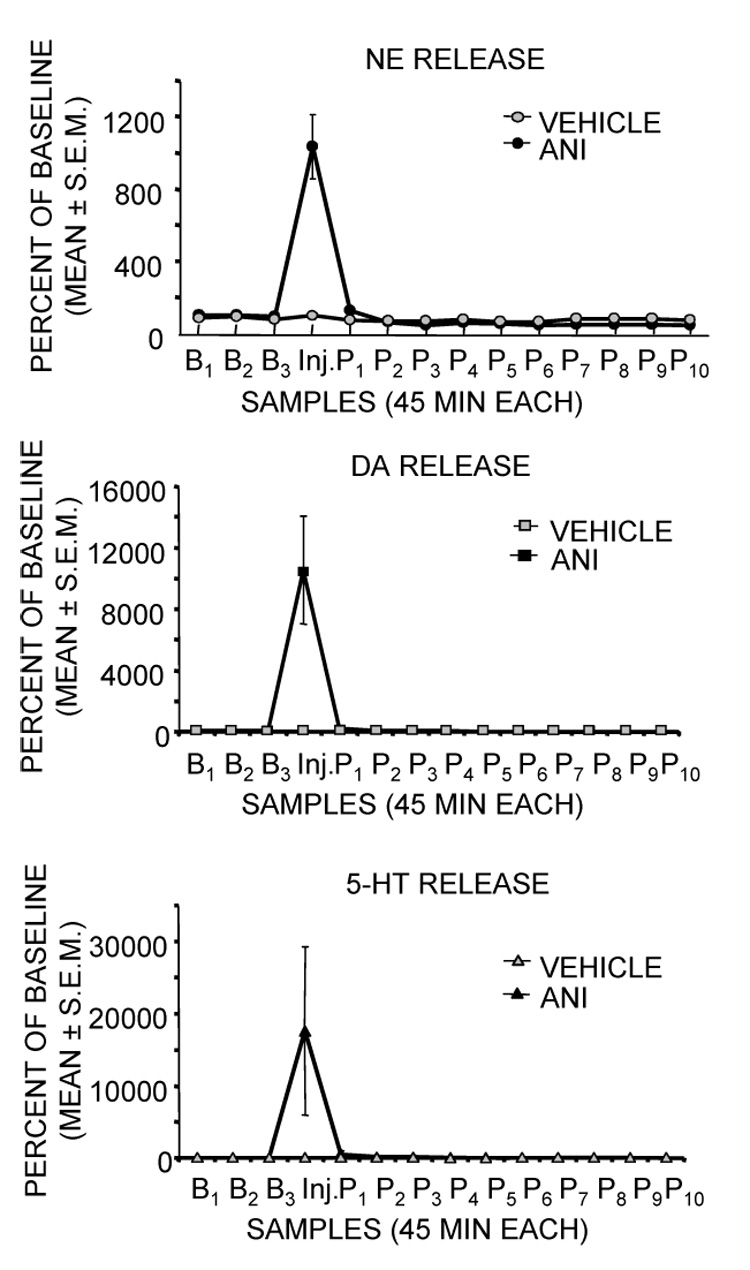
Effects of intra-amygdala injections of anisomycin on release of norepinephrine (NE), dopamine (DA), and serotonin (5-HT) at the site of injection. Note that anisomycin resulted in massive release of each of the biogenic amines during the samples (45 min each) taken immediately after injection. (Data from Canal et al., 2007.)
When release was measured at long times after injection, the dramatic early increases in release were followed by later substantial decreases in release of both dopamine and norepinephrine, perhaps reflecting either a failure of neurotransmitter synthesis to maintain sufficient supplies for release or a shutdown of release by presynaptic autoreceptors. Norepinephrine release approached zero and dopamine release approached 40% of control values at about 90 min after injection (Figure 3). The decreases in release of norepinephrine and dopamine were sustained for over 8 hrs, returning to baseline at 48 hrs (data not shown).
Figure 3.
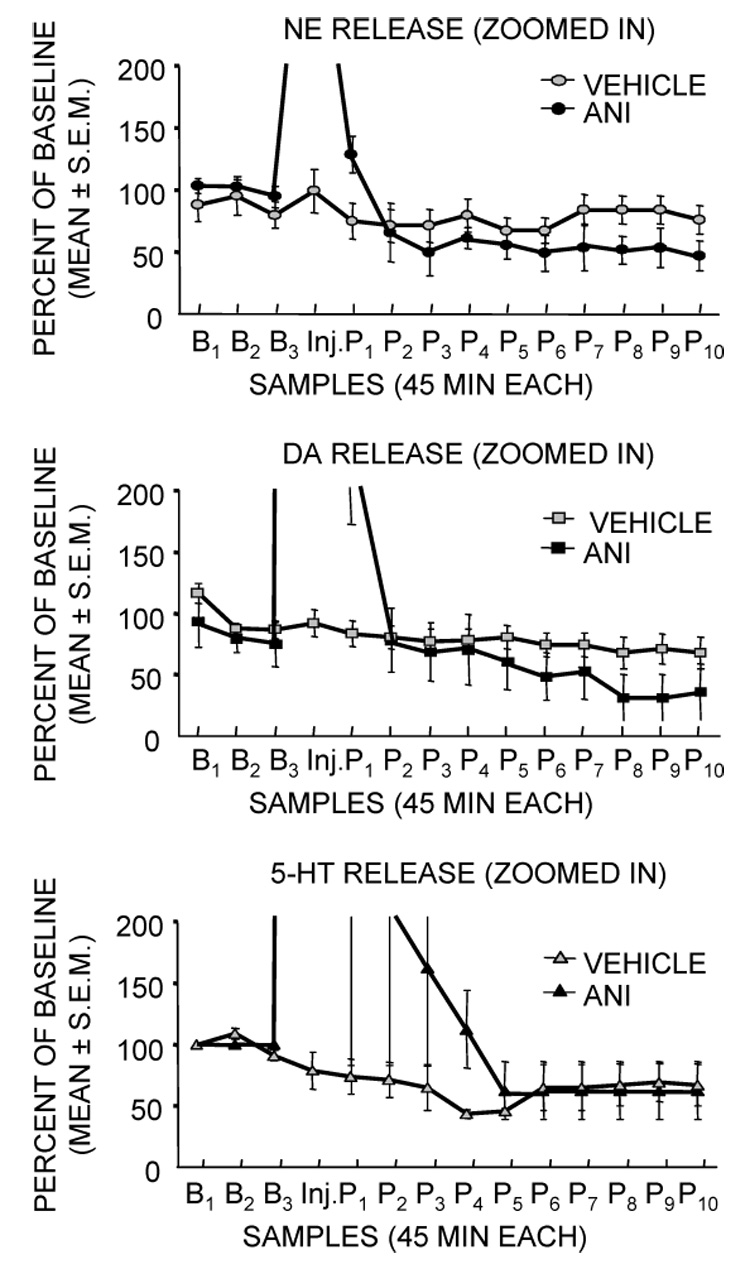
Effects of intra-amygdala injections of anisomycin on release of norepinephrine, dopamine and serotonin in samples collected during the 8 hrs after injection. These figures are based on the data in Figure 2, except that the y-axis is greatly expanded to reveal the extensive decreases in release of norepinephrine and dopamine that were evident even 8 hr after injection. These levels returned to baseline at 48 hrs (not shown). (Data from Canal et al., 2007.)
These findings raise the possibility that anisomycin produces amnesia by initiating these increases and decreases in neurotransmitter release. To begin to test this idea, we examined the particular role of norepinephrine in mediating the amnesia. Specifically, we attempted to determine whether it was the increase or the decrease in release that induced amnesia. To address this issue, we administered adrenergic receptor antagonists timed to block the consequences of the peak release or, in other groups, we administered adrenergic receptor agonists timed to compensate for the decrease in release. The results, and timelines to explain the experimental conditions, are shown in Figure 4. Anisomycin was injected into the amygdala 2 hrs before training and produced substantial amnesia on memory tests 48 hr after training. When the β-adrenergic receptor antagonist, propranolol, which itself can impair memory with other timing relative to training (Debiec and LeDoux, 2006; Lennartz et al., 1996; McIntyre et al., 2005; Miranda et al., 2003), was administered 10 min prior to injection of the protein synthesis inhibitor, the anisomycin-induced amnesia was attenuated. Conversely, a high dose of norepinephrine was administered 20 min prior to training, i.e. imitating the high levels of release produced by anisomycin, was as effective in inducing amnesia as was anisomycin. The ability of norepinephrine to enhance memory at low doses and to impair memory at high doses is seen with many drug treatments (Koob, 1991; Gold, 1995, 2006), and may reflect the basis for the inverted-U relationship between arousal and memory proposed a century ago by Yerkes and Dodson (1908).
Figure 4.
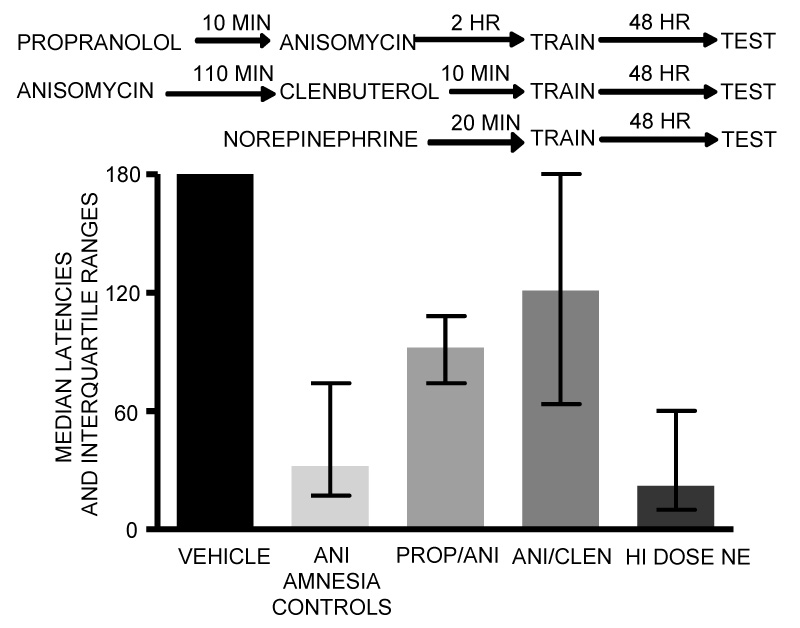
Attenuation and induction of amnesia with noradrenergic drugs. The top of the figure shows the timelines for these experiments. The β-adrenergic receptor antagonist, propranolol, administered 10 min prior to anisomycin, attenuated the amnesia, apparently blunting the impact of the large increase in release of NE in response to anisomycin injection. Similarly, the β-adrenergic receptor agonist, clenbuterol, administered 110 min after anisomycin and 10 min prior to training, attenuated the amnesia, apparently blunting the impact of the substantial decrease in release of NE evident 2 hr after training. These results suggest that both the increase and decrease in NE release contribute to anisomycin-induced amnesia. Finally, injections 20 min before training of a high dose of NE alone, i.e. without anisomycin, was sufficient to impair later memory. (Data from Canal et al., 2007.)
In parallel with these pharmacological experiments, we also assessed a possible role for the substantial decreases in release of norepinephrine seen 2–8+ hr after injection of anisomycin. In this experiment, a β-adrenergic receptor agonist was administered after anisomycin injection and prior to training to compensate for the decreased release. As shown in Figure 4, this treatment also substantially attenuated amnesia induced by anisomycin injections into the amygdala.
These findings suggest that the peak increases in release of norepinephrine are necessary and sufficient to mediate amnesia produced by intra-amygdala injections of anisomycin. In this respect, the experiments support the view that there are neural consequences of the massive inhibition of protein synthesis that can mediate amnesia, an interpretation that stands in contrast to the standard view that amnesias produced by protein synthesis inhibitors reveal a requirement of de novo protein synthesis for generating the neural bases of new memory formation.
Note that an interpretation explaining the effects of protein synthesis inhibitors on memory in terms of abnormal release of neurotransmitters is entirely consistent with the rapid forgetting, most often taken as decay of short-term memory. In particular, the long-duration decrease in neurotransmitter release might contribute to the inability of memory systems to maintain the neural changes responsible for memory formation.
There are many questions regarding the generality of these findings that remain to be tested. These include assessments of other neurotransmitter changes. As Martinez et al. (1981) note, the finding that a single noradrenergic drug reverses amnesia produced by many treatments (e.g. phenoxybenzamine as in Gold and Sternberg, 1978) leaves open the possibility that there are multiple points of entry into the mechanisms of amnesia beyond norepinephrine and the biogenic amines. In addition, it is important to determine whether these neurochemical effects mediate amnesias produced by other protein synthesis inhibitors and whether these effects are specific to the amygdala. The outcomes of such experiments will of course inform and therefore frame further discussions about the mechanisms of amnesia. However, in a positive sense, the focus of those discussions may shift away from single consideration of the mechanisms of memory to additional considerations of the mechanisms of amnesia.
Together with several other neurotransmitters, there is considerable evidence that norepinephrine – particularly in the amygdala - modulates memory formation. At moderate levels of release or with moderate injection doses, norepinephrine sets a condition in which the probability that a strong memory will be formed is increased, rather than creating that memory per se. At high levels of release or high doses near the time of training, norepinephrine impairs later memory. Modulation of memory was initially intended to explain the roles of endogenous hormones and neurotransmitters in regulating memory formation (Gold and McGaugh, 1975). While the application of the term, modulation, in the context of protein synthesis inhibitors seems somewhat at odds with the original meaning of memory modulation, these recent findings suggest that anisomycin may indeed modulate memory through the neurochemical processes responsible for modulation of memory formation rather than for the substrate bases of memory formation. This provides an interesting point of convergence between studies of modulation and consolidation of memory.
4. Proteins and protein synthesis as both modulators and substrates of memory
With considerable evidence against a general requirement for new protein synthesis in order to form new memories, additional data point to an important role for proteins, and for protein synthesis, in memory. The evidence includes training-related changes in immediate early gene expression, changes in gene microarray profiles and changes in specific proteins. One alternative to a need for new protein synthesis is that post-translational modifications of existing protein may be sufficient to represent memory (Routtenberg and Rekart, 2005). These modifications would necessarily be long-lasting and would participate as a basis for memory for a relatively long time after training. Is there evidence for such changes and is there evidence that the long-lasting changes are important to maintenance of memory?
Most tests of gene and protein expression are performed during the few hours after training. This aspect of experimental design appears to be based on the idea that these molecular changes are engaged by training to mediate later brain changes, particularly structural changes, responsible for long-lasting memory (e.g., Kandel, 2001). Although the temporal features of these experiments presume a time course of several hours to form long-lasting memory, the evidence indicates that there is a wide range of times for decay of short-term memory and neural plasticity. As reviewed elsewhere (Gold, 2006), the time-courses for decay of memory in the presence of inhibition of protein synthesis vary from 1 to at least 24 hrs (cf. Gold, 2006). In LTP experiments, the time for decay in the presence of protein synthesis inhibitors is at least from 10 min to 4 hrs. Given the variability in these time courses, the rate of decay of memory and synaptic plasticity seems likely to reflect a large set of molecular participants. A recent interesting review of the time courses for many factors, including cAMP, PKA, pCREB, and c-Fos among others, notes that each has a different time course when measured in the hippocampus during the several hours after training (Izquierdo et al., 2006). In addition, for many measures, there were multiple peaks of activation during this time. Izquierdo et al. (2006) conclude that memory formation includes a network of serial and parallel molecular events, using multiple time courses, that produce memory. While their analyses included only times proximal to training, of particular interest here are a growing number of demonstrations that many signaling and transcription factors remain expressed or activated at times well beyond training.
The phosphorylation of CREB offers an example of a molecular change that seems to last beyond times often suggested for the formation of memory. Given the proposed role of pCREB in initiating synthesis of proteins needed for memory formation during the first few hours after an experience (cf. Yin and Tully, 1996; Silva et al., 1998; Abel and Kandel, 1998), the long lasting increase in phosphorylation of CREB is surprising. Nonetheless, there are several examples in which pCREB is activated after training and in which the activation lasts well beyond the end of training. In one report (Stanciu et al., 2001), pCREB increased in the hippocampus after fear conditioning at 30 min and then decayed, in concert with many ideas about short-term memory. However, pCREB then increased again at 6 hrs after training. Longer durations after training were not measured in this experiment, but the 6 hr time-point is arguably at the end of or beyond putative short-term memory processes. Longer times have been measured in other experiments. In one such study, Taubenfeld et al. (2001) showed that pCREB was activated in the hippocampus even 20 hr after inhibitory avoidance training, a time well beyond traditional definition of short-term memory. In an experiment still in progress in this author’s laboratory, preliminary evidence suggests that pCREB expression is increased for up to 48 hr after inhibitory avoidance training (unpublished results). In a report that did not explicitly examine memory (Bilang-Bleuel et al., 2002), rats were exposed to forced swimming, presumably engaging memory mechanisms as in various versions of swim tasks. pCREB increased after that experience in the dentate gyrus and neocortex for beyond 48 hr, the longest training-measurement time tested. pCREB is similarly elevated in the dentate gyrus for at least 24 hr after induction of perforant path LTP (Schulz et al., 1999).
pCREB is not unique in exhibiting molecular changes that appear to outlast times typically offered for memory formation. A recent article in this journal (Trifilieff et al., 2007) shows biphasic increases in phosphorylated ERK (pERK) in both the hippocampus and amygdala after fear conditioning. The first peak in ERK activation was evident at 15 min after training, returning toward baseline at 3 hr. Important to the present discussion, the second peak was evident in CA3 of the hippocampus even 24 hr after training, the longest time tested, in mice that had received either unpaired or paired conditioning trials. The time course was different in the basolateral and lateral amygdala where increases in pERK were seen at 15 min, returned toward baseline at 3 hr, peaked again at 9 hr, and returned to baseline at 24 hr. Thus, the time courses vary with brain area but, in both cases, there were increases evident long after training. In the hippocampus, the duration of the effect is unknown because times later than 24 hr were not assessed. The long time courses of molecular changes appear to have generality not only across proteins but also across species. In tests of facilitation of the gill withdrawal reflex in the Aplysia, PKA activity increases soon after administration of serotonin, then decreases to baseline at 3 hrs, before increasing again at 20 hrs after application of serotonin (Sutton et al., 2001).
These examples of long lasting changes were evident as changes in the activation levels of constitutively expressed proteins. However, downstream targets of gene transcription are likely to follow activation of PKA, ERK and pCREB. For instance, c-Fos and zif-268 both exhibit increases in levels of expression as late as 24 hr, after inhibitory avoidance training (Bekinschtein et al., 2007).
These long-lasting molecular changes do not fit easily into a rubric of a molecular cascade important for memory formation because, in most views, the memory formation is complete well before the molecular events have subsided. One possibility is that the multiple waves of molecular activation reveal multiple waves of synaptic plasticity involved in memory formation. Of course, this is a view of memory formation that is quite different from most hypotheses that led from memory consolidation to molecular mechanisms of memory formation; it is also not a view for which there is direct evidence. It is also important to note that the structural changes often believed to underlie memory formation can occur rapidly, within 0 – 30 min in many circumstances. Among many examples (cf. Segal, 2001; Halpain, 2000), rapid structural changes have been seen in studies of LTP (Kadota and Kadota, 2002; Toni et al., 2001; Engert and Bonhoeffer, 1999; Maletic-Savatic, et al.., 1999), application of glutamate (Matsuzaki et al., 2004; Halpain et al., 1998), estradiol (MacLusky et al., 2005) or leptin (O’Malley et al., 2007). Thus, the onset of the product – restructuring of dendritic spines – appears to precede the onset and termination of many molecular events thought to generate the product. Particularly when viewed in the context of the rapidity (seconds to minutes) with which brain structural changes can occur, the ability for transcription, translation and transport to cause these changes seems unlikely.
One alternative view, much as proposed by Routtenberg and Rekart (2005), involves the use of proteins already in place at synapses and ready to engage structural changes. Consistent with this view, a recent report (Holahan and Routtenberg, 2007) demonstrates that a broad serine/threonine kinase inhibitor, H-7, blocked memory for fear conditioning even if injected into the cingulate cortex 3 weeks after training. To test the possibility that the effect was on expression or retrieval of the learned response, rather than a component of that learned response, a similar experiment was performed that showed that the treatment similarly blocked extinction of fear, i.e. the inverse behavioral response. These findings thereby showed that the kinase inhibitor did not only interfere with the expression of memory. Instead, as interpreted by the investigators, post-translational modifications including kinase activation patterns might be a component of the basis for long-lasting memory. This novel approach needs additional exploration across neural systems and across molecular candidates.
These results, together with the several examples offered earlier of long-lasting molecular changes after learning or LTP, suggest an additional, related possibility. Most theories of the neurobiology of memory focus on changes in neural circuitry, e.g. changes in neuron-neuron relationships. Alternatively, perhaps long lasting changes in activation of some existing proteins, as pCREB above and as in the posttranslational modifications involved in the Holahan and Routtenberg (2007) experiment, establish intracellular memories. Restated, some long-lasting molecular responses to an experience might themselves be a neurobiological memory, a long-lived residual of experience. Instead of storing details of an experience, these changes may represent the relatively non-specific information that something significant happened, resetting the likelihood that a population of neurons will participate in future episodes of plasticity. Thus, in contrast to memories that might contain specific information about an experience, these intracellular changes may be the basis for altered readiness to engage in future plasticity. This idea is very similar to one expressed several years ago in an intriguing article, “The genomic action potential” (Clayton, 2000). The genomic action potential prepares a neuron to be ready for a new experience, modulating the ability of that neuron to change under a new condition requiring plasticity. I suggest here updating this view to include long-lasting molecular changes, changes that may represent a new state for the neuron in which that neuron is ready – for a substantial time – to change again. It is at this point that the molecular biologies of memory consolidation and memory modulation converge to become cellular memories. In this sense, the long-lasting molecular change is a cellular memory of significant change, modulating the ability of the cell to change again.
Footnotes
Research from the author’s laboratory and preparation of this manuscript supported by NIA (AG007648) and NIDA (DA016951 and DA024129).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abel T, Kandel ER. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Research - Brain Research Reviews. 1998;26:360–378. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- Barondes SH, Cohen HD. Arousal and the conversion of "short-term" to "long-term" memory. Proceedings of the National Academy of Sciences USA. 1968;61:923–929. doi: 10.1073/pnas.61.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraco RA, Stettner LJ. Antibiotics and memory. Psychological Bulletin. 1976;83:242–302. [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LRM, Izquierdo I, Medina JH. Persistence of long-term memory storage requires a late protein synthesis- and BDNF-dependent phase in the hippocampus. Neuron. 2007;53:261–277. doi: 10.1016/j.neuron.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Bilang-Bleuel A, Rech J, De Carli S, Holsboer F, Reul JM. Forced swimming evokes a biphasic response in CREB phosphorylation in extrahypothalamic limbic and neocortical brain structures in the rat. European Journal of Neuroscience. 2002;15:1048–1060. doi: 10.1046/j.1460-9568.2002.01934.x. [DOI] [PubMed] [Google Scholar]
- Calixto E, Thiels E, Klann E, Barrionuevo G. Early maintenance of hippocampal mossy fiber long-term potentiation depends on protein and RNA synthesis and presynaptic granule cell integrity. Journal of Neuroscience. 2003;23:4842–4849. doi: 10.1523/JNEUROSCI.23-12-04842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Amnesia produced by altered release of neurotransmitters after intraamygdala injections of a protein synthesis inhibitor. Proceedings of the National Academy of Science. 2007;104:12500–12505. doi: 10.1073/pnas.0705195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew TJ, Castellucci VF, Kandel ER. An analysis of dishabituation and sensitization of the gill-withdrawal reflex in Aplysia. International Journal of Neuroscience. 1971;2:79–98. doi: 10.3109/00207457109146995. [DOI] [PubMed] [Google Scholar]
- Clayton EC, Williams CL. Adrenergic activation of the nucleus tractus solitarius potentiates amygdala norepinephrine release and enhances retention performance in emotionally arousing and spatial memory tasks. Behavioural Brain Research. 2000;112:151–158. doi: 10.1016/s0166-4328(00)00178-9. [DOI] [PubMed] [Google Scholar]
- Clayton DF. The genomic action potential. Neurobiology of Learning & Memory. 2000;74:185–216. doi: 10.1006/nlme.2000.3967. [DOI] [PubMed] [Google Scholar]
- Cooper JR, Bloom FE, Roth RH. The Biochemical Basis of Neuropharmacology. 3rd edition. New York: Oxford University Press; 1978. [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychological Bulletin. 1984;96:518–559. [PubMed] [Google Scholar]
- Debiec J, LeDoux JE. Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Annals of the New York Academy of Sciences. 2006;1071:521–524. doi: 10.1196/annals.1364.056. [DOI] [PubMed] [Google Scholar]
- Dudai Y. Molecular bases of long-term memories: a question of persistence. Current Opinion in Neurobiology. 2002;12:211–216. doi: 10.1016/s0959-4388(02)00305-7. [DOI] [PubMed] [Google Scholar]
- Edwards DR, Mahadevan LC. Protein synthesis inhibitors differentially superinduce c-fos and c-jun by three distinct mechanisms: lack of evidence for labile repressors. EMBO Journal. 1992;11:2415–2424. doi: 10.1002/j.1460-2075.1992.tb05306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Flexner JB, Flexner LB. Restoration of expression of memory lost after treatment with puromycin. Proceedings of the National Academy of Sciences USA. 1967;57:1143–1144. doi: 10.1073/pnas.57.6.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner JB, Flexner LB, Walter R, Hoffman PL. ADH and related peptides: Effect of pre- or posttraining treatment on puromycin amnesia. Pharmacology Biochemistry and Behavior. 1978;8:93–95. doi: 10.1016/0091-3057(78)90129-6. [DOI] [PubMed] [Google Scholar]
- Flood JF, Bennett EL, Orme AE, Rosenzweig MR. Effects of protein synthesis inhibition on memory for active avoidance training. Physiology & Behavior. 1975;14:177–184. doi: 10.1016/0031-9384(75)90163-8. [DOI] [PubMed] [Google Scholar]
- Flood JF, Jarvik ME, Bennett EL, Orme AE, Rosenzweig MR. The effect of stimulants, depressants, and protein synthesis inhibition on retention. Behavioral Biology. 1977;20:168–183. doi: 10.1016/s0091-6773(77)90734-9. [DOI] [PubMed] [Google Scholar]
- Flood JF, Bennett EL, Orme AE, Rosenzweig MR, Jarvik ME. Memory: modification of anisomycin-induced amnesia by stimulants and depressants. Science. 1978a;199:324–326. doi: 10.1126/science.619461. [DOI] [PubMed] [Google Scholar]
- Flood JF, Vidal D, Bennett EL, Orme AE, Vasquez S, Jarvik ME. Memory facilitating and anti-amnesic effects of corticosteroids. Pharmacology, Biochemistry & Behavior. 1978b;8:81–87. doi: 10.1016/0091-3057(78)90127-2. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nagerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006a;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Nagerl UV, Bonhoeffer T. Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nature Neuroscience. 2006b;9:478–480. doi: 10.1038/nn1667. [DOI] [PubMed] [Google Scholar]
- Glassman E. The biochemistry of learning: an evaluation of the role of RNA and protein. Annual Review of Biochemistry. 1969;38:605–646. doi: 10.1146/annurev.bi.38.070169.003133. [DOI] [PubMed] [Google Scholar]
- Gold PE. Modulation of emotional and non-emotional memories: Same pharmacological systems, different neuroanatomical systems. In: McGaugh JL, Weinberger NM, Lynch GS, editors. Brain and Memory: Modulation and Mediation of Neural Plasticity. NY: Oxford Press; 1995. pp. 41–74. [Google Scholar]
- Gold PE. The many faces of amnesia. Learning & Memory. 2006;13:506–514. doi: 10.1101/lm.277406. [DOI] [PubMed] [Google Scholar]
- Gold PE, McGaugh JL. A single trace, two process view of memory storage processes. In: Deutsch D, Deutsch JA, editors. In Short term memory. New York: Academic Press; 1975. pp. 355–390. [Google Scholar]
- Gold PE, Sternberg DB. Retrograde amnesia produced by several treatments: Evidence for a common neurobiological mechanism. Science. 1978;201:367–369. doi: 10.1126/science.208153. [DOI] [PubMed] [Google Scholar]
- Hall ME, Schlesinger K, Stamm E. Prevention of memory loss following puromycin treatment. Pharmacology Biochemistry & Behavior. 1976;5:525–528. doi: 10.1016/0091-3057(76)90256-2. [DOI] [PubMed] [Google Scholar]
- Halpain S. Actin and the agile spine: how and why do dendritic spines dance? Trends in Neuroscience. 2000;23:141–146. doi: 10.1016/s0166-2236(00)01576-9. [DOI] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. Journal of Neuroscience. 1998;18:9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassert DL, Miyashita T, Williams CL. The effects of peripheral vagal nerve stimulation at a memory-modulating intensity on norepinephrine output in the basolateral amygdala. Behavioral Neuroscience. 2004;118:79–88. doi: 10.1037/0735-7044.118.1.79. [DOI] [PubMed] [Google Scholar]
- Holahan MR, Routtenberg A. Post-translational synaptic protein modification as substrate for long-lasting, remote memory An initial test. Hippocampus. 2007;17:93–97. doi: 10.1002/hipo.20245. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Bevilaqua LRM, Rossato JI, Bonini JS, Medina JH, Cammarota M. Different molecular cascades in different sites of the brain control memory consolidation. Trends in Neuroscience. 2006;29:496–505. doi: 10.1016/j.tins.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: A dialog between genes and synapses. Bioscience Reports. 2001;21:565–611. doi: 10.1023/a:1014775008533. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH. Molecular biology of learning: Modulation of transmitter release. Science. 1982;218:433–443. doi: 10.1126/science.6289442. [DOI] [PubMed] [Google Scholar]
- Kadota T, Kadota K. Rapid structural remodeling of shaft synapses associated with long-term potentiation in the cat superior cervical ganglion in situ. Neuroscience Research. 2002;43:135–146. doi: 10.1016/s0168-0102(02)00028-7. [DOI] [PubMed] [Google Scholar]
- Kleim JA, Bruneau R, Calder K, Polock DI, VandenBerg PM, MacDonald E, Monfils HH, Sutherland RJ, Nader K. Functional organization of adult motor cortex is dependent upon continued protein synthesis. Neuron. 2003;40:167–176. doi: 10.1016/s0896-6273(03)00592-0. [DOI] [PubMed] [Google Scholar]
- Koob GF. Arousal, stress and inverted-U shaped curves: Implications for cognitive function. In: Lister RG, Weingartner HJ, editors. Perspectives on cognitive neuroscience. London: Oxford University Press; 1991. pp. 300–313. [Google Scholar]
- Kurotani T, Higashi S, Inokawa H, Toyama K. Protein and RNA synthesis-dependent and -independent LTPs in developing rat visual cortex. NeuroReport. 1996;8:35–39. doi: 10.1097/00001756-199612200-00008. [DOI] [PubMed] [Google Scholar]
- Lande S, Flexner JB, Flexner LB. Effect of corticotropin and desglycinamide-lysine vasopressin on suppression of memory by puromycin. Proceeding of the National Academy of Sciences USA. 1972;69:558–560. doi: 10.1073/pnas.69.3.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennartz RC, Hellems KL, Mook ER, Gold PE. Inhibitory avoidance impairments induced by intra-amygdala propranolol are reversed by glutamate but not glucose. Behavioral Neuroscience. 1996;110:1033–1039. doi: 10.1037//0735-7044.110.5.1033. [DOI] [PubMed] [Google Scholar]
- Ma S, Morilak DA. Chronic intermittent cold stress sensitises the hypothalamic-pituitary-adrenal response to a novel acute stress by enhancing noradrenergic influence in the rat paraventricular nucleus. Journal of Neuroendocrinology. 2005;17:761–769. doi: 10.1111/j.1365-2826.2005.01372.x. [DOI] [PubMed] [Google Scholar]
- MacLusky NJ, Luine VN, Hajszan T, Leranth C. The 17α and 17β isomers of estradiol both induce rapid spine synapse formation in the CA1 hippocampal subfield of ovariectomized female rats. Endocrinology. 2007;146:287–293. doi: 10.1210/en.2004-0730. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Martinez JL, Jr, Jensen RA, McGaugh JL. Attenuation of experimentally-induced amnesia. Progress in Neurobiology. 1981;16:155–186. doi: 10.1016/0301-0082(81)90011-3. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CK, Hatfield T, McGaugh JL. Amygdala norepinephrine levels after training predict inhibitory avoidance retention performance in rats. European Journal of Neuroscience. 2002;16:1223–1226. doi: 10.1046/j.1460-9568.2002.02188.x. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Miyashita T, Setlow B, Marjon KD, Steward O, Guzowski JF, McGaugh JL. Memory-influencing intra-basolateral amygdala drug infusions modulate expression of Arc protein in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10718–10723. doi: 10.1073/pnas.0504436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda MI, LaLumiere RT, Buen TV, Bermudez-Rattoni F, McGaugh JL. Blockade of noradrenergic receptors in the basolateral amygdala impairs taste memory. European Journal of Neuroscience. 2003;18:2605–2610. doi: 10.1046/j.1460-9568.2003.03008.x. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Williams CL. Glutamatergic transmission in the nucleus of the solitary tract modulates memory through influences on amygdala noradrenergic systems. Behavioral Neuroscience. 2002;116:13–21. doi: 10.1037//0735-7044.116.1.13. [DOI] [PubMed] [Google Scholar]
- Nakajima S. Amnesic effect of cycloheximide in the mouse mediated by adrenocortical hormones. Journal of Comparative and Physiological Psychology. 1975;88:378–385. doi: 10.1037/h0076207. [DOI] [PubMed] [Google Scholar]
- Oliver GWO, Rose GP, Brimblecombe RW, Livett BH. Analysis of Y-maze learning in mice using cycloheximide. General Pharmacology. 1979;10:489–497. doi: 10.1016/0306-3623(79)90014-4. [DOI] [PubMed] [Google Scholar]
- O’Malley D, MacDonald N, Mizielinski S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Molecular and Cellular Neuroscience. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartermain D, Botwinick CY. Role of the biogenic amines in the reversal of cycloheximide-induced amnesia. Journal of Comparative and Physiological Psychology. 1975;88:386–401. doi: 10.1037/h0076208. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Tronson NC. Protein synthesis inhibitors, gene superinduction and memory: too little or too much protein? Neurobiology of Learning and Memory. 2007 doi: 10.1016/j.nlm.2007.08.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum M, Cohen HD, Barondes SH. Effect of intracerebral saline on amnesia produced by inhibitors of cerebral protein synthesis. Communications in Behavioral Biology. 1968;2:47–50. [Google Scholar]
- Routtenberg A, Rekart JL. Post-translational modification as the substrate for long-lasting memory. Trends in Neuroscience. 2005;28:12–19. doi: 10.1016/j.tins.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, Moineau J, Bolding K. Anisomycin and the reconsolidation hypothesis. Learning " Memory. 2006;13:1–3. doi: 10.1101/lm.157806. [DOI] [PubMed] [Google Scholar]
- Schulz S, Siemer H, Krug M, Höllt V. Direct evidence for biphasic cAMP responsive element-binding protein phosphorylation during long-term potentiation in the rat dentate gyrus in vivo. Journal of Neuroscience. 1999;19:5683–5692. doi: 10.1523/JNEUROSCI.19-13-05683.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Rapid plasticity of dendritic spine: hints to possible functions? Progress in Neurobiology. 2001;63:61–70. doi: 10.1016/s0301-0082(00)00021-6. [DOI] [PubMed] [Google Scholar]
- Semba K, Adachi N, Arai T. Facilitation of serotonergic activity and amnesia in rats caused by intravenous anesthetics. Anesthesiology. 2005;102:616–623. doi: 10.1097/00000542-200503000-00021. [DOI] [PubMed] [Google Scholar]
- Sershen H, Reith ME, Lajtha A. On the interaction between nicotine and cycloheximide. Brain Research. 1982;251:183–185. doi: 10.1016/0006-8993(82)91290-2. [DOI] [PubMed] [Google Scholar]
- Serota RG, Roberts RB, Flexner LB. Acetoxycycloheximide-induced transient amnesia: Protective effects of adrenergic stimulations. Proceedings of the National Academy of Sciences USA. 1972;69:340–342. doi: 10.1073/pnas.69.2.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman E, Rossi S, Sershen H, Hashim A, Lajtha A. Locally administered low nicotine-induced neurotransmitter changes in areas of cognitive function. Neurochemical Research. 2005;30:1055–1066. doi: 10.1007/s11064-005-7132-9. [DOI] [PubMed] [Google Scholar]
- Sherff CM, Carew TJ. Coincident induction of long-term facilitation in Aplysia: cooperativity between cell bodies and remote synapses. Science. 1999;285:1911–1914. doi: 10.1126/science.285.5435.1911. [DOI] [PubMed] [Google Scholar]
- Sherff CM, Carew TJ. Parallel somatic and synaptic processing in the induction of intermediate-term and long-term synaptic facilitation in Aplysia. Proceedings of the National Academy of Sciences USA. 2004;101:7463–7468. doi: 10.1073/pnas.0402163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annual Review of Neuroscience. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory and Brain. New York: Oxford University Press; 1987. [Google Scholar]
- Stanciu M, Radulovic J, Spiess J. Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Molecular Brain Research. 2001;94:15–24. doi: 10.1016/s0169-328x(01)00174-7. [DOI] [PubMed] [Google Scholar]
- Steward O, Huang F, Guzowski JF. A form of perforant path LTP can occur without ERK1/2 phosphorylation or immediate early gene induction. Learning " Memory. 2007;14:433–447. doi: 10.1101/lm.554607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Masters SE, Bagnall MW, Carew TJ. Molecular mechanisms underlying a unique intermediate phase of memory in Aplysia. Neuron. 2001;31:143–154. doi: 10.1016/s0896-6273(01)00342-7. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Wiig KA, Monti B, Dolan B, Pollonini G, Alberini CM. Fornix-dependent induction of hippocampal CCAAT enhancer-binding protein β and δ co-localizes with phosphorylated cAMP response element-binding protein and accompanies long-term memory consolidation. Journal of Neuroscience. 2001;21:84–91. doi: 10.1523/JNEUROSCI.21-01-00084.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, Muller D. Remodeling of synaptic membranes after induction of long-term potentiation. Journal of Neuroscience. 2001;21:6245–6251. doi: 10.1523/JNEUROSCI.21-16-06245.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Calandreau L, Herry C, Mons N, Micheau J. Biphasic ERK1/2 activation in both the hippocampus and amygdala may reveal a system consolidation of contextual fear memory. Neurobiology of Learning and Memory. 2007 doi: 10.1016/j.nlm.2007.05.004. in press. [DOI] [PubMed] [Google Scholar]
- Williams CL, Men D, Clayton EC, Gold PE. Norepinephrine release in the amygdala after systemic injection of epinephrine or escapable footshock: contribution of the nucleus of the solitary tract. Behavioral Neuroscience. 1998;112:1414–1422. doi: 10.1037//0735-7044.112.6.1414. [DOI] [PubMed] [Google Scholar]
- Yerkes RM, Dodson JD. The relation of strength of stimulus to rapidity of habit-formation. Journal of Comparative and Neurological Psychology. 1908;18:459–482. [Google Scholar]
- Yin JC, Tully T. CREB and the formation of long-term memory. Current Opinions in Neurobiology. 1996;6:264–268. doi: 10.1016/s0959-4388(96)80082-1. [DOI] [PubMed] [Google Scholar]