

Abstract
Cellular expression of ATP-binding cassette (ABC) transport proteins, such as P-glycoprotein (Pgp), multidrug resistance-associated protein (MRP1), or ABCG2, is known to confer a drug-resistant phenotype. Thus, the development of effective transporter inhibitors could be of value to cancer treatment. CBT-1® is a bisbenzylisoquinoline plant alkyloid currently in development as a Pgp inhibitor. We characterized its interactions with the three major ABC transporters associated with drug resistance--Pgp, MRP1 and ABCG2—and compared it to other known inhibitors. CBT-1® completely inhibited rhodamine 123 transport from Pgp-overexpressing cells at a concentration of 1 μM. Additionally, 1 μM completely reversed Pgp-mediated resistance to vinblastine, paclitaxel and depsipeptide in SW620 Ad20 cells. CBT-1® was found to compete [125I]-IAAP labeling of Pgp with an IC50 of 0.14 μM, and low concentrations of CBT-1® (< 1 μM) stimulated Pgp-mediated ATP hydrolysis. In MRP1-overexpressing cells, 10 μM CBT-1® was found completely inhibit MRP1-mediated calcein transport. CBT-1® at 25 μM did not have a significant effect on ABCG2-mediated pheophorbide a transport. Serum levels of CBT-1® in samples obtained from eight patients receiving CBT-1® increased intracellular rhodamine 123 levels in CD56+ cells 2.1- to 5.7-fold in an ex vivo assay. CBT-1® is able to inhibit the ABC transporters Pgp and MRP1, making it an attractive candidate for clinical trials in cancers where Pgp and/or MRP1 might be overexpressed. Further clinical studies with CBT-1® are warranted.
Keywords: P-glycoprotein, MRP1, ABCG2, CBT-1®, drug-resistance, inhibitor
1. Introduction
Cellular mechanisms of drug resistance fall generally into two classes: those that prevent drugs from reaching their target and those that cause genetic changes that affect drug sensitivity [1]. ATP-binding cassette (ABC) transport proteins are known to induce resistance due to their ability to lower intracellular drug concentrations in an energy-dependent manner [1]. P-glycoprotein (Pgp), the product of the MDR1 (ABCB1) gene, has been studied extensively and is known to transport a wide range of chemotherapy drugs such as the anthracyclines, vinca alkaloids, taxanes, etoposide, mitoxantrone, bisantrene and the histone deacetylase inhibitor depsipeptide [1-3]. Subsequent to the discovery of Pgp, the multidrug resistance associated protein, MRP1 (ABCC1), was cloned from lung carcinoma cells selected in doxorubicin [4] and was found to confer resistance to etoposide, vincristine and doxorubicin [5]. The most recently reported ABC transporter associated with drug resistance, ABCG2, is a half-transporter whose substrates include mitoxantrone, topotecan, and flavopiridol [6].
Determining the contribution of Pgp to clinical drug resistance in cancer has not been an easy task, in no small part due to the lack of uniformity in methods used to measure Pgp expression [7]. However, several studies have described increased Pgp expression after initial chemotherapy treatment, especially in leukemia and breast cancer [8]. Pgp expression has also repeatedly been linked to poor outcome in some forms of leukemia [8, 9]. Targeting Pgp has led to the development of Pgp inhibitors that are able to block transport of substrates and increase intracellular accumulation. Many inhibitors have been tested in clinical trials, but definitive proof that inhibition of drug efflux can improve clinical outcome has not been forthcoming. The “first generation” Pgp inhibitors, generally compounds already used to treat other conditions, lacked sufficient potency and early clinical trials were largely unsuccessful [10]. “Second generation” Pgp inhibitors such as valspodar (PSC833) were potent but had deleterious pharmacokinetic interactions leading to some patients receiving inadequate levels of chemotherapy [10]. Some “third generation” compounds, such as elacridar and tariquidar, have been developed and are currently being explored in the clinic [11, 12]; however, the merits of this treatment strategy have been debated and relatively few trials are ongoing.
Large-scale studies linking MRP1 expression to drug resistance in cancer are lacking. MRP1 expression has been found in lung carcinoma samples with incidences of 80% in SCLC to 100% in NSCLC [8, 13, 14]. CNS cancers have also been reported to express MRP1 [15]. MRP1 has been detected in leukemia samples by functional assays [16, 17], and co-expression of MRP1 with Pgp has been found to be a negative prognostic factor in AML [17]. ABCG2, still in its relative infancy as a transporter, has not yet been conclusively linked to clinical drug resistance, although at least one large-scale study linked expression to poor outcome in acute myelogenous leukemia [18].
Recently, a cDNA array analysis of 170 pretreatment acute myeloid leukemia samples classified the samples in 6 separate groups based on unsupervised clustering of the gene expression profiles using the HG_U95Av2 microarray [19]. These groups differed in clinical outcome; impressively, one of the groups with the highest poorest outcome exhibited ABC transporter overexpression [19]. Thus, despite the difficulties experienced to date in the clinical development of ABC transporter inhibitors, results such as these suggest that there is ample reason to continue this effort.
CBT-1® is an orally-administered, bisbenzylisoquinoline plant alkyloid currently being developed as a Pgp inhibitor by CBA Research Inc. Phase I trials with CBT-1® and paclitaxel or doxorubicin have been completed [20, 21] and phase II and III trials are currently in progress. The initial phase I studies demonstrated that CBT-1® did not affect the pharmacokinetics of doxorubicin or paclitaxel and no neurological toxicities were observed [20, 21]. As the clinical development of CBT-1® progressed, it became important to biochemically characterize the interactions between CBT-1® and the ABC transporters shown to transport chemotherapeutic agents, and to compare this agent to known Pgp inhibitors. Thus, we confirmed the ability of CBT-1® to inhibit Pgp-mediated rhodamine transport, prevent Pgp-mediated drug resistance, compete [125I]iodoarylazidoprazosin (IAAP) photolabeling of Pgp, and stimulate ATPase activity. CBT-1® was also found to block MRP1-mediated transport in cell line models, but had no effect on ABCG2-mediated transport.
2. Materials and Methods
2.1. Chemicals
Vinblastine sulfate, rhodamine 123, mitoxantrone and paclitaxel were obtained from Sigma Chemical (St. Lois, MO). Depsipeptide (NSC630176, FR901228, FK228) was provided by the National Cancer Institute Anticancer Drug Screen (Bethesda, MD). Tariquidar was obtained from Xenova Research (Slough, Berkshire, UK). Valspodar was a gift from Novartis Pharmaceuticals (Cambridge, MA). MK-571 was purchased from EMD Biosciences (San Diego, CA). Calcein AM was obtained from Invitrogen Corporation (Carlsbad, CA). Pheophorbide a was purchased from Frontier Scientific (Logan, UT). FTC was isolated by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, National Institutes of Health (Bethesda, MD). [125I]-iodoarylazidoprazosin (IAAP) (2200 Ci/mM) was purchased from Perkin Elmer Life Sciences (Wellesley, MA).
2.2. Cell Lines
SW620 parental and Pgp-overexpressing, doxorubicin-selected SW620 Ad5, Ad20 and Ad300 cells have been previously described and are maintained in RPMI-1640 medium, with the drug selected sublines additionally maintained in 5, 20, or 300 ng/ml adriamycin [22]. Empty-vector transfected human embryonic kidney (HEK293) pcDNA3-10 cells, ABCB1-transfected MDR-19 cells and ABCG2-transfected R-5 cells are maintained in Eagle's medium with 2 mg/ml G418 as previously described [23]. ABCC1-transfected cells are maintained in 5 μM etoposide [24].
2.3. Flow Cytometry
Cells were trypsinized and incubated for 30 min in complete medium (phenol red-free Richter's medium with 10% FCS and penicillin/streptomycin) with the desired fluorescent substrate (0.5 μg/ml rhodamine 123, 1 μM pheophorbide a (PhA) or 0.2 μM calcein AM) in the presence or absence of the desired inhibitor for 30 min at 37°C in 5% CO2. Subsequently, cells were washed and incubated in substrate-free medium for 1h at 37°C continuing with or without inhibitor. Transport of mitoxantrone (3 μM) was examined in a similar manner [25]. To determine the duration of inhibition by the various Pgp inhibitors, cells were incubated with 0.5 μg/ml rhodamine 123 alone or rhodamine and various concentrations of CBT-1® or other known inhibitors for 30 min at 37°C in 5% CO2. Matched samples were then washed and allowed to incubate in rhodamine-free medium for 30, 60 or 120 minutes, after which intracellular rhodamine fluorescence was determined. To determine significant difference between intracellular fluorescence values, a Student's t-test was performed with p<0.05 being considered significant.
Human serum samples were obtained on day 6 from patients enrolled on a phase II study of oral CBT-1® in a chemotherapy regimen; all patients gave informed consent. Samples were received and analyzed under an Institutional Review Board-approved National Cancer Institute protocol that permits testing of patient samples obtained from outside institutions. Serum samples from the phase I study as well as serum from a normal volunteer were diluted 1:1 with complete medium containing 1 μg/ml rhodamine 123, resulting in a final rhodamine concentration of 0.5 μg/ml, and were added to mononuclear cells from a normal volunteer and were incubated at 37°C for 30 min. Serum from a normal volunteer was run in parallel and diluted 1:1 with medium containing 1 μg/ml rhodamine with 6 μg/ml valspodar, 20 μM, 2 μM or 0.2 μM CBT-1® that was then added to mononuclear cells to serve as positive controls. This resulted in a final concentration of 0.5 μg/ml rhodamine with 3 μg/ml valspodar, 10 μM, 1 μM, or 0.1 μM CBT-1®. Cells were then washed and allowed to incubate for 1 h continuing with control serum or patient serum diluted 1:1 with rhodamine-free medium. Cells incubated with rhodamine and valspodar or CBT-1® were washed and incubated with control serum diluted 1:1 with rhodamine-free medium containing the inhibitors. After one hour at 37°C, the cells were incubated with phycoerythrin-labeled anti-CD56 antibody (BD Bioscience) as previously described, and intracellular rhodamine fluorescence in CD56+ cells was measured [26].
Rhodamine and calcein fluorescence was measured on a FACSort flow cytometer equipped with a 488 nm argon laser and 530 nm bandpass filter; phycoerythrin fluorescence was measured with an 570 nm bandpass filter. Pheophorbide a or mitoxantrone fluorescence was measured with a 635 nm red diode laser and a 561 nm longpass filter. At least 10,000 events were collected for all samples and debris was eliminated by gating on forward versus side scatter; dead cells were excluded based on propidium iodide staining.
2.4. Cytotoxicity Assays
Four-day cytotoxicity assays were performed based on the method of Skehan et al [27]. Briefly, cells were plated (5,000-10,000 cells/well) in 96-well, flat-bottom plates and allowed to attach overnight at 37°C in 5% CO2. Chemotherapeutic agents were subsequently added to the cells at various concentrations and allowed to incubate for 96 hours. Cells were then fixed with 50% (w/v) trichloroacetic acid and then stained with 0.4% sulforhodamine B (w/v) in 1% acetic acid. After washing the plates in 1% acetic acid and allowing them to dry, the dye was then solubilized in 50% Trizma base and plates were read at an absorbance of 540 nm. Each drug concentration was tested in quadruplicate and controls were tested in replicates of eight. Dose modifying factors (DMFs) were calculated for CBT-1® with each drug by dividing the IC50 for each drug without inhibitor by the IC50 for each drug in the presence of inhibitor. To determine significant difference between IC50 values, a Student's t-test was performed with p<0.05 being considered significant.
2.5. Photolabeling of Pgp with [125I]-IAAP
As previously described, crude membranes from HiFive insect cells expressing Pgp were incubated with 10 μM of the desired inhibitor for 10 min after which 3-6 nM [125I]-IAAP (2200 Ci/mole) in 50 mM Tris-Hcl (pH 7.5) was added and the samples were incubated for an additional 5 min in the dark [28]. Samples were then exposed to a UV light source for 10 min at room temperature. Pgp was then immunoprecipitated with the C219 antibody (Signet Laboratories, Dedham, MA). Protein a sepharose beads (100 μL, Amersham Biosciences, Piscataway, NJ) were then added and incubated for 16h at 4°C. The beads were centrifuged at 13,000 rpm for 5 min at 4°C and then washed with RIPA buffer in 1% aprotinin. SDS-PAGE sample buffer (25 μL) was then added and the samples were incubated for 1h at 37°C, followed by the addition of 25 μl of water and an additional incubation at 37°C for 30 min. Samples were separated by PAGE on a 7% Tris-acetate gel, followed by drying, exposure to Bio-Max MR film (Eastman-Kodak, Rochester, NY) and quantitation of the resulting bands. The data were fitted using the program GraphPad Prism 4.0 (GraphPad Software, San Diego, CA).
2.6. Determination of ATPase Activity
The ATPase activity of Pgp was measured as previously described [29]. Briefly, crude membranes isolated from Pgp-expressing HiFive insect cells (100 μg protein/ml) were incubated with varying concentrations of CBT-1® in the presence or absence of 0.2 mM beryllium sulfate and 2.5 mM sodium fluoride (BeFx) in ATPase assay buffer (50 mM Tris-HCl pH 6.8, 100 mM KCl, 10 mM sodium azide, 20 mM EGTA, 20 mM DTT, 20 mM MgCl2) for 5 min at 37°C. The reaction was started by the addition of 5 mM ATP and was stopped by the addition of 0.1 ml of 5% SDS solution. The amount of inorganic phosphate released and the BeFx-sensitive ATPase activity of Pgp was determined [29].
3. Results
3.1. CBT-1® inhibits Pgp-mediated efflux of rhodamine 123
The ability of CBT-1® to inhibit Pgp-mediated efflux of rhodamine 123 was compared to that of the known Pgp inhibitors valspodar, tariquidar, or verapamil. Three concentrations of each inhibitor—0.1, 1 and 10 μM--were tested and are shown in the histograms as a heavy solid line, solid line or dashed line, respectively (Figure 1). As seen in Figure 1, high levels of Pgp expression in the Ad20, Ad300 and MDR-19 cell lines resulted in low intracellular rhodamine concentrations when cells were incubated with rhodamine in the absence of any inhibitor (shaded histogram). CBT-1® was able to partially inhibit rhodamine 123 efflux at a concentration of 0.1 μM (heavy solid line) in the Ad20, Ad300, and MDR-19 cell lines, while complete inhibition was observed at 1 μM (solid line). When cells were incubated with 1 μM CBT in the presence of rhodamine, an increase in fluorescence was consistently observed compared to cells incubated with 10 μM CBT-1® in the presence of rhodamine. This was only statistically significant for the Ad20 subline (p<0.01). Valspodar, shown in the second column, was comparably potent. Tariquidar completely inhibited rhodamine transport at 0.1 μM (heavy solid line) in the Ad20, Ad300, and MDR-19 cell lines, making it the most potent inhibitor examined. Verapamil, shown in the last column, was a relatively poor inhibitor, since 10 μM (dashed line) was required to completely prevent rhodamine transport in all of the Pgp-overexpressing cell lines. Intracellular rhodamine fluorescence values are compiled in Supplemental Table 1.
Figure 1.

CBT-1® inhibits Pgp mediated rhodamine efflux from Pgp-overexpressing cells. Drug-selected Ad20 and Ad300 cells as well as MDR1-transfected HEK-293 cells (MDR-19) were incubated with 0.5 μg/ml rhodamine 123 in the presence or absence of 0.1, 1 or 10 μM of CBT-1®, verapamil, valspodar or tariquidar for 30 min. Subsequently, cells were washed and allowed to incubate in rhodamine-free medium for 1 h continuing without (shaded histogram) or with 0.1 μM (heavy solid line), 1 μM (solid line), or 10 μM (dashed line) inhibitor. Histograms from one of at least 2 independent experiments are shown.
Similar studies were conducted using CBT-1® and the fluorescent drug mitoxantrone with MDR-19 cells to verify results observed with rhodamine. CBT-1® at 0.1 μM partially inhibited Pgp-mediated mitoxantrone transport, while complete inhibition was afforded by 1 and 10 μM CBT-1® (Supplemental Figure 2A); intracellular fluorescence values for mitoxantrone are given in Supplemental Table 2. Unlike with rhodamine, 10 μM CBT-1® did not decrease intracellular fluorescence of mitoxantrone (Supplemental Figure 2) or BODIPY-prazosin (data not shown) compared to 1 μM.
3.2. Duration of Pgp inhibition
We next sought to determine how long the effects of Pgp inhibition would last once the inhibitor had been removed, and compared CBT-1® to the other known inhibitors in this context. In Ad20 (Figure 2A, B) and Ad300 (Figure 2C, D) cells, in the absence of any inhibitor (filled squares), intracellular rhodamine fluorescence is low at the 0 time point, rapidly decreases over time and is nearly gone after 30 min. Of the inhibitors examined, the effects of verapamil were most rapidly reversed (filled circles). The duration of inhibition by 10 μM CBT-1® (empty circles) and valspodar (empty diamonds) was comparable in the Ad20 cell line (Figure 2B) as both compounds effectively prevented rhodamine efflux over the entire 120 min period after the inhibitor was washed out. At 1 μM, however, in the Ad20 cell line (Figure 2A), the effects of valspodar (empty diamonds) begin to deteriorate after 30 min, whereas the effects of CBT-1® (empty circles) just begin to wear off after 120 min. At the 30, 60 and 120 min time points, rhodamine fluorescence in cells incubated with CBT-1® was significantly higher than in Ad20 cells incubated with valspodar (p<0.02 in all cases). In the Ad300 cell line (Figure 2B, D), the effects of 1 or 10 μM valspodar (empty diamonds) were again significantly more rapidly lost than with a similar concentration of CBT-1® (empty circles) after the 30, 60 and 120 min incubation (p<0.03 in all cases). For both the Ad20 and Ad300 sublines, tariquidar (filled triangles), even at a concentration of 1 μM was able to maintain intracellular rhodamine fluorescence for nearly the entire 2 hours after the inhibitor was washed out of the cells, a result in agreement with previous reports [30].
Figure 2.

Duration of Pgp inhibition by CBT-1®. SW620 Ad20 (A, B) and Ad300 (C, D) cells were incubated in 0.5 μg/ml rhodamine 123 in the absence (C, empty squares) or presence of 10 μM (A, B) or 1 μM (C, D) CBT-1® (CBT, empty circles), valspodar (VAL, filled diamonds), tariquidar (TAR, filled triangles) or verapamil (VER, filled circles) for 30 min and intracellular rhodamine fluorescence was measured to yield time 0 values. Matched samples for each condition were then washed and allowed to incubate in substrate-free medium and intracellular fluorescence was measured at 30, 60 and 120 min after removal of rhodamine 123. Average intracellular rhodamine fluorescence values (channel numbers) from at least 2 experiments are shown; error bars represent standard deviation.
3.3. Modulation of Pgp-mediated drug resistance by CBT-1®
Cytotoxicity assays were subsequently performed to determine if CBT-1® could reverse Pgp-mediated drug resistance in Pgp-overexpressing, drug-selected or transfected cells. Four-day cytotoxicity assays were performed on SW620 parental and drug-selected Ad20 cells with the Pgp substrate drugs vinblastine, paclitaxel, and depsipeptide and the non-Pgp drug 5-FU. Assays on pcDNA3-10 and MDR1-transfected MDR-19 cells were performed using depsipeptide. When cytotoxicity assays were performed with CBT-1® alone, we found that the IC50 values for all cell lines ranged from 4 to 6 μM (data not shown). Assays were thus performed in the presence or absence of 1 μM CBT-1®, which we found to be non-toxic in all cell lines (cell survival > 95%). Dose modifying factors (DMFs) were also calculated by dividing the IC50 for each drug in the absence of inhibitor by the IC50 in the presence of inhibitor. The results are summarized in table 1 and results from an independent cytotoxicity assay performed on SW620 parental cells and Ad20 cells with depsipeptide or vinblastine in the presence or absence of CBT-1® are shown in figure 3.
Table 1.
CBT-1® reverses Pgp-mediated drug-resistance
| Drug | SW620 (mean IC50 ± SD) | Ad20 (mean IC50 ± SD) | RR1 | DMF2 |
|---|---|---|---|---|
| Vinblastine | 0.00094±0.00053 | 0.051±0.022* | 54 | 58 |
| Vinblastine + 1 μM CBT-1® | 0.00038±0.00014 | 0.00088±0.00031 | ||
| Paclitaxel | 0.0060±0.0040 | 0.71±0.025* | 118 | 173 |
| Paclitaxel + 1 μM CBT-1® | 0.0021±0.0012 | 0.0041±0.0016 | ||
| FK228 | 0.00085±0.00022 | 0.025±0.0041* | 29 | 48 |
| FK228 + 1 μM CBT-1® | 0.00040±0.00023 | 0.00052±0.00020* | ||
| 5-FU | 24±16 | 29±23 | 1.2 | 1.3 |
| 5-FU + 1 μM CBT-1® | 34±17 | 45±23 | ||
| Drug | pcDNA3-10 | MDR-19 | ||
| FK228 | 0.0055±0.0048 | 0.87±0.54* | 158 | 48 |
| FK228 + 1 μM CBT-1® | 0.0021±0.0017 | 0.018±0.0067* |
IC50 values (μM) were determined by the sulforhodamine B assay.
IC50 significantly different from parent line (p<0.05)
Relative resistance values were calculated by dividing the IC50 of the resistant line by the IC50 of the parent line.
The dose modifying factor (DMF) was calculated by dividing the IC50 of the resistant cell line (SW620 Ad20 or MDR-19) in the absence of inhibitor by the IC50 in the presence of inhibitor.
Results are from at least 2 independent experiments (mean ± SD).
Figure 3.

CBT-1® abrogates Pgp-mediated drug resistance. Four day cytotoxicity assays were performed with depsipeptide or vinblastine on SW620 parental (circles) and Pgp-overexpressing Ad20 cells (squares) in the presence (filled symbols) or absence (empty symbols) of 1 μM CBT-1® as described in the materials and methods section. Representative results are shown and a summary of results is given in Table 1.
Ad20 cells were significantly resistant to all Pgp substrates examined compared to parental SW620 cells (p<0.02 for all drugs). Complete reversal of resistance to vinblastine, paclitaxel, and depsipeptide was observed in the Ad20 cells, as IC50 values for Ad20 cells in the presence of CBT-1® were not statistically significantly different from IC50 values for the parental SW620 line (p>0.05 in all cases), except for depsipeptide, where CBT-1® significantly reversed resistance to levels that were below the parental line (p<0.05). Ad20 cells were only slightly more resistant to 5-FU compared to parental cells, but this did not achieve statistical significance. CBT-1® also nearly completely reversed resistance to depsipeptide in the MDR-19 cells. MDR-19 cells express higher levels of Pgp than the Ad20 cells, which may explain this discrepancy. However, as both Ad20 and MDR-19 cells express Pgp at levels that are higher than what would be observed in patients, it is likely that lower concentrations of CBT-1® would be needed to inhibit physiologic levels of Pgp.
3.4. Effect of CBT-1® on [125I]-IAAP labeling of Pgp and ATP hydrolysis
To further characterize the interaction of CBT-1® with Pgp, we next examined the ability of CBT-1® to prevent photolabeling of Pgp by the prazosin analog [125I]-IAAP in membranes isolated from Pgp-expressing HiFive insect cells. As seen in figure 4A, CBT-1® at a concentration of 10 μM (lane 2) readily competes [125I]-IAAP labeling of Pgp (control shown in lane 1), suggesting it interacts at the drug binding site of [125I]-IAAP. Other well-known Pgp inhibitors valspodar (lane 3), tariquidar (lane 5) and cyclosporine a (lane 6) also prevented photolabeling of Pgp by [125I]-IAAP at a concentration of 10 μM. Verapamil had no effect on [125I]-IAAP labeling of Pgp at 10 μM (lane 4). Comparable protein loading among lanes was verified by colloidal blue stain (Supplemental Figure 2). The IC50 for [125I]-IAAP inhibition by CBT-1® was 0.14 μM (Figure 4B). This compares favorably to previously published values for other known Pgp inhibitors such as biricodar (VX-710), 0.75 μM; cyclosporine a, 3.5 μM and verapamil, >100 μM [31].
Figure 4.
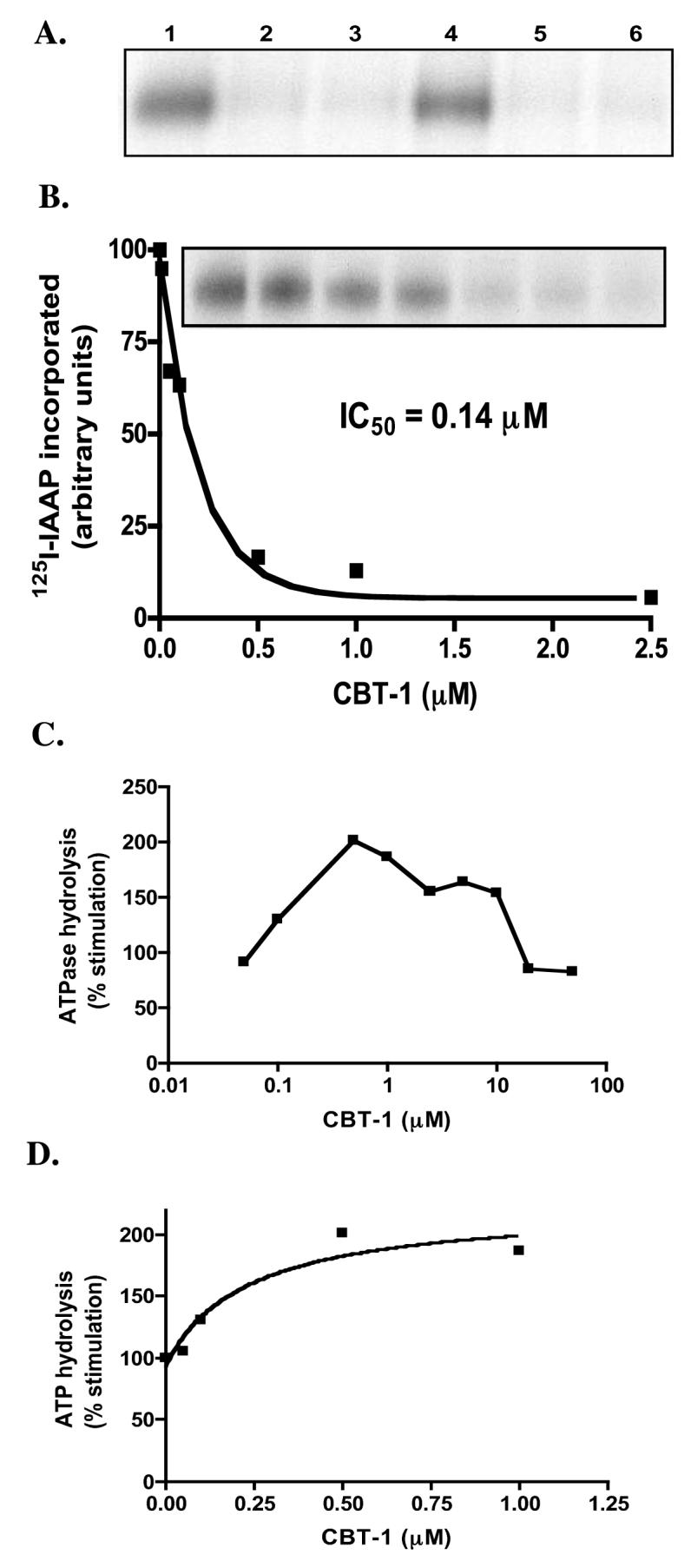
Effect of CBT-1® on [125I]-IAAP labeling and ATPase activity of Pgp. A) Crude membranes isolated from Pgp-overexpressing High-Five insect cells were incubated in the absence (lane 1) or presence of 10 μM CBT-1® (lane 2), valspodar (lane 3), verapamil (lane 4) tariquidar (lane 5) or cyclosporine a (lane 6) with 3-6 nM [125I]-IAAP and crosslinked as described in the materials and methods section. The samples were separated on a 7% Tris-acetate polyacrylamide gel and the amount of [125I]-IAAP crosslinked to Pgp was determined as outlined in the materials and methods section. B) Pgp expressing crude membranes were incubated with 0-25 μM CBT-1® for 5 min and the samples were labeled with [125I]-IAAP as described above. The radioactivity incorporated into the Pgp band was quantitated as described in the materials and methods and the data were fitted using GraphPad Prism 4.0 software. C) The BeFx-sensitive ATPase activity of Pgp was determined in Pgp-expressing High-Five insect cell membranes in the presence of various concentrations of CBT-1® according to the materials and methods section. D) Experiments were performed as in (C) so as to determine the concentration required for 50% of the maximal stimulation of ATPase activity.
The effects of CBT-1® on ATP hydrolysis were then determined as described in the materials and methods section. Figure 4C shows that CBT-1® initially stimulates Pgp-hydrolysis at low concentrations (< 1 μM), then loses this stimulatory effect with increasing concentrations of CBT-1®. This has also been reported for other Pgp inhibitors such as MS-209 [31]. The concentration of CBT-1® required for 50% stimulation of the maximum ATP hydrolysis value was found to be 0.28 μM (Figure 4D).
3.5. CBT-1® inhibits MRP1-mediated transport
To determine whether CBT-1® interacts with MRP1, calcein transport was examined in ABCC1-transfected HEK293 cells in the presence of varying concentrations of CBT-1®. As seen in figure 5A, CBT-1® at a concentration of 10 μM (solid line) completely inhibited MRP1-mediated calcein efflux (shaded histogram) in MRP1-transfected HEK293 cells and was found to be as effective as 50 μM of the known MRP1 inhibitor MK-571 (dashed line). No inhibition of MRP1-mediated transport was observed with 1 μM CBT-1®. Results with calcein are summarized in Supplemental Table 3. Similar results were obtained when mitoxantrone was used as a substrate (Supplemental Figure 2B), and results are compiled in Supplemental Table 2. Since CBT-1® was found to be toxic at 10 μM, combination studies could not be performed. Our results show that CBT-1® is able to prevent transport mediated by either Pgp or MRP1. Inhibition of MRP1, however, occurs at a higher concentration than Pgp, suggesting CBT-1® has a greater affinity for Pgp than MRP1.
Figure 5.
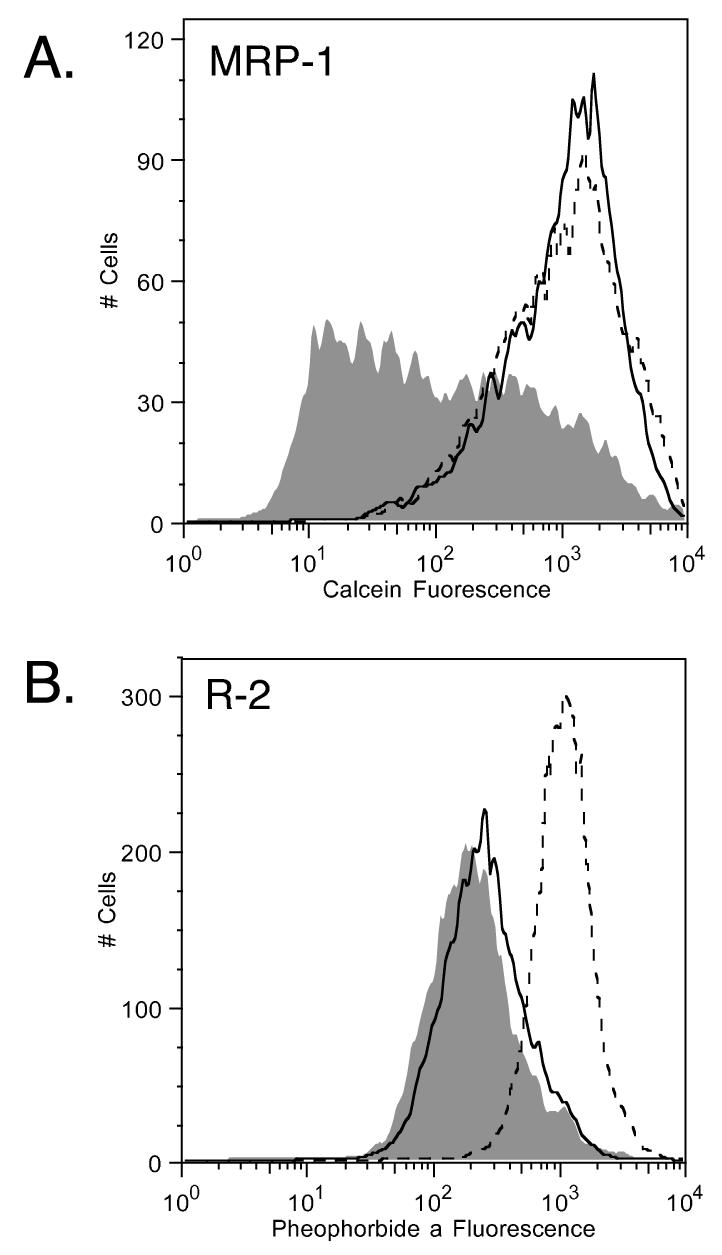
CBT-1® inhibits MRP1-mediated calcein transport but not ABCG2-mediated pheophorbide a (PhA) transport. A) ABCC1-transfected cells were incubated with 0.1 μM calcein AM in the absence (shaded histogram) or presence of 10 μM CBT-1® (solid line) or 25 μM of the known MRP1 inhibitor MK-571 (dashed line). B) Wild-type ABCG2-transfected cells were incubated with 1 μM PhA in the absence (shaded histogram) or presence of 10 μM CBT-1® (solid line) or 25 μM of the known ABCG2 inhibitor FTC (dashed line). Histograms from 1 of 2 independent experiments are shown.
3.6. CBT-1® does not modulate ABCG2-mediated pheophorbide a or mitoxantrone transport
We have previously reported that some Pgp inhibitors, such as tariquidar and elacridar, can additionally inhibit ABCG2-mediated transport [32, 33]. To determine if this was the case for CBT-1®, ABCG2-transfected cells were incubated with 1 μM PhA in the presence of 25μM CBT-1® or 10 μM FTC. As shown in figure 5B, 10 μM FTC (dashed line) readily prevented ABCG2-mediated PhA transport (shaded histogram) from ABCG2-transfected cells while CBT-1® was ineffective even at concentrations of 25 μM (solid line). Similar results were obtained when mitoxantrone was used as the ABCG2 substrate (Supplemental Figure 2C). Results with pheophorbide a are presented in Supplemental Table 4, while results with mitoxantrone are summarized in Supplemental Table 2.
3.7. CBT-1® present in serum samples obtained from patients receiving CBT-1® inhibits Pgp activity of CD56+ cells in an ex vivo assay
To determine whether CBT-1® levels in patients were high enough to inhibit Pgp, we obtained serum samples from eight patients receiving CBT-1® on a phase I study. Samples were obtained from 3 patients who had only been exposed to CBT-1®, two patients who had received continuous CBT-1 and one dose of paclitaxel, two patients who had received CBT-1® and 2 cycles of paclitaxel, and one patient who received CBT-1® and 3 cycles of paclitaxel. Due to protocol limitations, no samples were obtained from any of the patients prior to CBT-1® administration. Post-treatment samples were compared to serum obtained from a normal volunteer. Results from six of the eight patients are shown in figure 6. CD56+ cells incubated in rhodamine with serum obtained from patients receiving CBT-1® had a 2.1- to 5.7-fold increase in intracellular rhodamine fluorescence compared to cells incubated with rhodamine in control serum. CD56+ cells incubated with control serum in the presence of rhodamine do not have high intracellular rhodamine fluorescence due to the high levels of Pgp expressed by these cells (solid line). However, cells that were incubated in serum obtained from the six patients receiving CBT-1® displayed higher intracellular fluorescence (dashed line) due to the CBT-1® present in the serum. Cells were also incubated with normal serum containing 0.1 μM, 1 μM, or 10 μM CBT-1® or 3 μg/ml valspodar; results with valspodar and 0.1 μM CBT-1® are shown in Figure 6. The exogenously added inhibitors caused a 2.2-, 3-, 3.4-, or 3.2-fold increase in rhodamine in CD56+ cells, respectively, compared to cells incubated with rhodamine in control serum. CBT-1® levels in serum samples ranged from 123 to 573 ng/ml (386 to 917 nM). Due to limited amounts of patient material, the experiments were performed twice, with results from one experiment shown in Figure 6.
Figure 6.
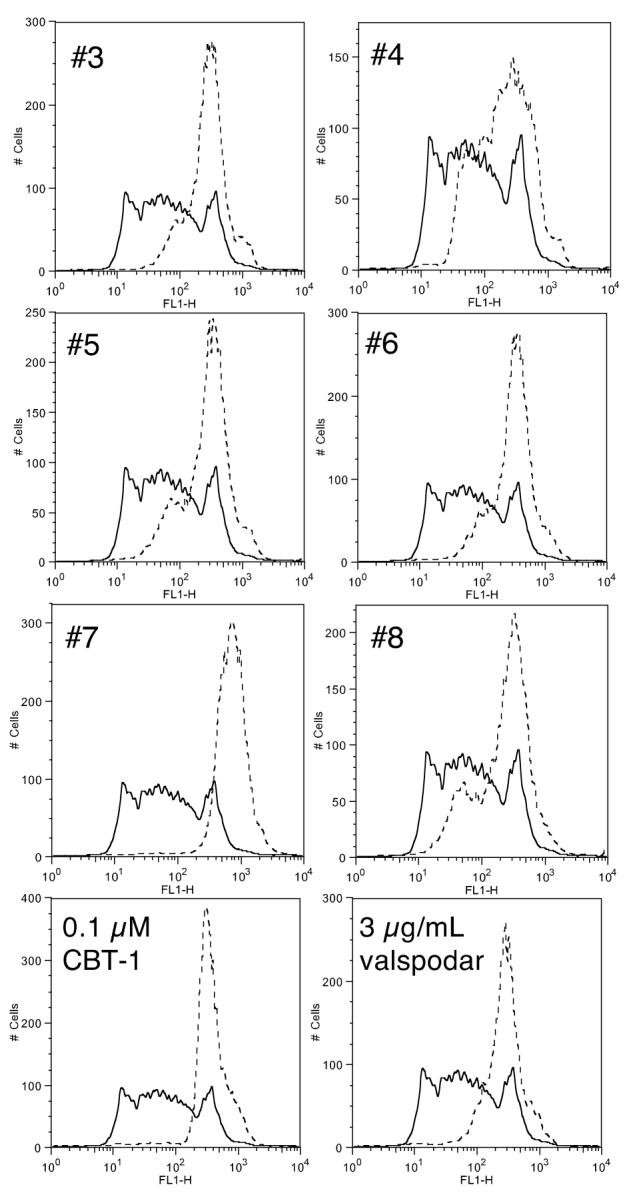
Serum levels of CBT-1® prevent Pgp-mediated rhodamine 123 from CD56+ cells. Mononuclear cells were incubated for 30 min in serum obtained on day 6 from 6 patients receiving CBT-1® diluted 1:1 with medium containing 1 mg/ml rhodamine 123. As a control, mononuclear cells were incubated for 30 min in control serum containing 6 μg/ml valspodar, 20 μM, 2 μM or 0.2 μM CBT-1® that was also diluted 1:1 with complete medium. Cells were then washed and incubated for 1 h with control serum or patient serum diluted 1:1 with rhodamine-free complete medium. Similarly, cells incubated in the presence of inhibitors were incubated in control serum containing the desired inhibitor diluted 1:1 with complete medium. Intracellular rhodamine fluorescence was subsequently examined in CD56+ cells. The histograms compare rhodamine fluorescence in CD56+ cells incubated with normal serum (solid line) to that in cells incubated with patient serum or normal serum containing exogenously added CBT-1® (0.1 μM) or valspodar (3 μg/ml) (dashed lines). Results with serum samples of patients #3-#8 from 1 of 2 experiments are shown.
4. Discussion
In the present study, we characterized the interactions of the Pgp inhibitor CBT-1® with Pgp, MRP1 and ABCG2—ABC transporters associated with drug resistance. We found that 1 μM CBT-1® completely inhibited Pgp-mediated rhodamine efflux from Pgp-overexpressing, drug selected SW620 Ad5, Ad20 and Ad300 cell lines as well as MDR1-transfected HEK293 cells. The potency of CBT-1® was greater than verapamil, approximately equivalent to valspodar, but less than tariquidar, one of the most potent Pgp inhibitors we have studied to date. The duration of Pgp inhibition by CBT-1® was less than that of tariquidar, but greater than that observed with valspodar or verapamil. In combination studies with CBT-1®, 1 μM effectively abrogated resistance to vinblastine, paclitaxel and depsipeptide in SW620 Ad20 cells as demonstrated by 4-day cytotoxicity assays. Interaction at the drug binding site of Pgp was confirmed in experiments showing competition of [125I]-IAAP labeling of Pgp by CBT-1®. In addition to acting as a Pgp inhibitor, 10 μM CBT-1® was able to completely inhibit MRP1-mediated transport of calcein in ABCC1-transfected HEK293 cells. CBT-1® was not found to prevent ABCG2-mediated transport of PhA in ABCG2-transfected HEK293 cells. CBT-1® serum levels in patients receiving CBT-1® as part of a chemotherapy regimen were high enough to prevent Pgp-mediated rhodamine 123 efflux from CD56+ cells in an ex vivo assay. Our results thus show CBT-1® is able to effectively inhibit Pgp- and MRP1-mediated transport.
We noted that, at the 10 μM concentration of CBT-1®, intracellular rhodamine fluorescence values were decreased compared to the 1 μM concentration. This appeared to be specific to rhodamine, as we did not observe this phenomenon when using calcein AM, BODIPY-prazosin or mitoxantrone as the fluorescent substrate. Although we are not sure of the exact mechanism that causes the decrease in fluorescence, CBT-1® may interact with rhodamine, quenching its fluorescence at high concentrations.
Initial phase I studies of CBT-1® in combination with adriamycin or paclitaxel have shown that no dose reduction of the chemotherapeutic agent was necessary, suggesting that CBT-1® has no pharmacokinetic interactions with adriamycin or paclitaxel. When the “second generation” Pgp inhibitor valspodar was used in combination with chemotherapy, it was necessary to reduce the dose of the chemotherapy drug to prevent toxicity [34]. The need for dose reduction was later found to be due to interactions with the cytochrome P450 pathway by valspodar, thus inhibiting drug metabolism and decreasing drug clearance [8]. Biricodar has also been shown reduce clearance of paclitaxel, suggesting a potential pharmacokinetic interaction as well [35]. We have argued this pharmacokinetic interaction represented an insurmountable problem for valspodar. Such a considerable variability in the pharmacokinetic interaction was observed that, despite reduction of the anticancer drug dose, about one-third of patients still had toxicity related to the anticancer drug [10]. Furthermore, approximately one-third of patients experienced little drug toxicity and were effectively undertreated [10]. A Pgp inhibitor without major drug interactions would be valuable. CBT-1® may thus be an improvement over current inhibitors of ABC transporters.
To date, no MRP1-specific inhibitors have been evaluated in the clinic. The only other inhibitors shown to inhibit Pgp and MRP1 and subsequently used in clinical trials are dofequidar (MS-209) [36-38] and biricodar (VX-710) [39, 40]. Dofequidar in combination with cyclophosphamide, doxorubicin and fluorouracil (CAF) was found to increase overall response rate compared to CAF alone [36]. Early in vitro studies showed biricodar to be a potent inhibitor of both Pgp and MRP1 [31, 41], while subsequent studies also demonstrated it to be an inhibitor of ABCG2 [25]. While the value of inhibitors that are capable of inhibiting multiple transporters has yet to be determined clinically, some groups have reported expression of multiple transporters in clinical samples, particularly in leukemia [19, 42]. Daunorubicin and mitoxantrone, both substrates of Pgp and MRP1, are often used in chemotherapy regimens used to treat leukemia [18]. Inclusion of CBT-1® into treatment strategies for diseases where MRP1 or both MRP1 and Pgp are expressed may therefore increase response. However, as relatively high concentrations of CBT-1® were necessary to inhibit MRP1 in transfected cells where levels are relatively high, we will need to evaluate whether levels of CBT-1® achieved in patients would be able to inhibit physiologic levels of MRP1 that would tend to be lower than those observed in transfected cells.
Few Pgp inhibitors remain in development. To our knowledge, only elacridar, tariquidar and dofequidar are in trials in addition to CBT-1®. The in vitro studies presented here demonstrate that tariquidar is more potent than CBT-1®. However, it is not certain that the ideal modulator will be the most potent available. Indeed, the only patients with renal cell carcinoma remaining free of disease following treatment with valspodar and vinblastine on our Phase I trial received the oral formulation; no other responses were observed with the more potent IV agent [43]. It may be that an agent with somewhat lesser potency will have fewer drug interactions; or that there will be less effect on bone marrow stem cells. Given the importance of ABC transporters in protecting bone marrow stem cells, a very potent Pgp inhibitor may require reduction of the anticancer agent dose to prevent prolonged neutropenia. If CBT-1® can avoid both of these pitfalls and distribute well into tumors, it may become the simplest strategy for improving the efficacy of Pgp substrate drugs.
As cancer therapy has become increasingly outpatient-based, there has been a drive to develop oral agents. Thus, an oral Pgp inhibitor could be combined with an oral anticancer agent for a more prolonged schedule of administration. We know from the phase I and II trials already conducted that seven days of CBT-1® is well tolerated. This could serve as the basis of a trial combining CBT-1® with substrates such as etoposide or imatinib, both orally administered agents. Obviously, the tolerability of continuous dosing for CBT-1® would have to be demonstrated for such a strategy to succeed.
CBT-1® has been in clinical trials for over a decade, in small studies that have consistently confirmed its safety. The past few years have seen numerous trials with other modulators open and close, in many cases due to toxicity; in others, increasingly due to poor trial design; and still others with a clearly negative result. If CBT-1® were to succeed as a Pgp inhibitor, it would be proof that effective treatments need not come from large pharmaceutical companies.
Supplementary Material
Acknowledgements
We appreciate the help of Dr Krishnamachary Nandigama in providing crude membranes from the Pgp-expressing HiFive cells for Pgp ATP hydrolysis experiments. We thank Dr. Orsolya Polgar for critical review of the manuscript. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and the Commissioned Corps of the Public Health Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Robert Oldham is a consultant to CBA Research. Daryl Barnett is an employee of CBA International, Inc., and an affiliate of CBA Research, Inc. who provided the investigational agent, without charge, to the National Cancer Institute. Daryl Barnett is also a minority stock-holder in CBA Pharma, Inc which is the parent of CBA Research, Inc. Neither CBA Research, Inc. nor its affiliates provided any funding for this research.
References
- 1.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 2.Zhang XP, Ritke MK, Yalowich JC, Slovak ML, Ho JP, Collins KI, et al. P-glycoprotein mediates profound resistance to bisantrene. Oncol Res. 1994;6:291–301. [PubMed] [Google Scholar]
- 3.Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH, et al. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood. 2004;103:4636–43. doi: 10.1182/blood-2003-09-3068. [DOI] [PubMed] [Google Scholar]
- 4.Cole SPC, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–4. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 5.Kruh GD, Zeng H, Rea PA, Liu G, Chen ZS, Lee K, et al. MRP subfamily transporters and resistance to anticancer agents. J Bioenerg Biomembr. 2001;33:493–501. doi: 10.1023/a:1012827221844. [DOI] [PubMed] [Google Scholar]
- 6.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 7.Beck WT, Grogan TM, Willman CL, Cordon-Cardo C, Parham DM, Kuttesch JF, et al. Methods to detect P-glycoprotein-associated multidrug resistance in patients' tumors: Consensus recommendations. Cancer Res. 1996;56:3010–20. [PubMed] [Google Scholar]
- 8.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–24. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 9.Marie JP, Zhou DC, Gurbuxani S, Legrand O, Zittoun R. MDR1/P-glycoprotein in haematological neoplasms. Eur J Cancer. 1996;32A:1034–8. doi: 10.1016/0959-8049(96)00055-x. [DOI] [PubMed] [Google Scholar]
- 10.Leonard GD, Polgar O, Bates SE. ABC transporters and inhibitors: new targets, new agents. Curr Opin Investig Drugs. 2002;3:1652–9. [PubMed] [Google Scholar]
- 11.Planting AS, Sonneveld P, van der Gaast A, Sparreboom A, van der Burg ME, Luyten GP, et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2005;55:91–9. doi: 10.1007/s00280-004-0854-6. [DOI] [PubMed] [Google Scholar]
- 12.Abraham J, Edgerley M, Wilson R, Chen C, Medina W, Hermonstine L, et al. A phase I study of the novel p-glycoprotein (Pgp) antagonist, XR9576 in combination with vinorelbine. Proc Am Soc Clin Oncol. 2001;37 [Google Scholar]
- 13.Wright SR, Boag AH, Valdimarsson G, Hipfner DR, Campling BG, Cole SP, et al. Immunohistochemical detection of multidrug resistance protein in human lung cancer and normal lung. Clin Cancer Res. 1998;4:2279–89. [PubMed] [Google Scholar]
- 14.Nooter K, Bosman FT, Burger H, van Wingerden KE, Flens MJ, Scheper RJ, et al. Expression of the multidrug resistance-associated protein (MRP) gene in primary non-small-cell lung cancer. Ann Oncol. 1996;7:75–81. doi: 10.1093/oxfordjournals.annonc.a010484. [DOI] [PubMed] [Google Scholar]
- 15.Mohri M, Nitta H, Yamashita J. Expression of multidrug resistance-associated protein (MRP) in human gliomas. J Neurooncol. 2000;49:105–15. doi: 10.1023/a:1026528926482. [DOI] [PubMed] [Google Scholar]
- 16.Legrand O, Simonin G, Perrot JY, Zittoun R, Marie JP. Pgp and MRP activities using calcein-AM are prognostic factors in adult acute myeloid leukemia patients. Blood. 1998;91:4480–8. [PubMed] [Google Scholar]
- 17.Legrand O, Simonin G, Beauchamp-Nicoud A, Zittoun R, Marie JP. Simultaneous activity of MRP1 and Pgp is correlated with in vitro resistance to daunorubicin and with in vivo resistance in adult acute myeloid leukemia. Blood. 1999;94:1046–56. [PubMed] [Google Scholar]
- 18.Benderra Z, Faussat AM, Sayada L, Perrot JY, Chaoui D, Marie JP, et al. Breast cancer resistance protein and P-glycoprotein in 149 adult acute myeloid leukemias. Clin Cancer Res. 2004;10:7896–902. doi: 10.1158/1078-0432.CCR-04-0795. [DOI] [PubMed] [Google Scholar]
- 19.Wilson CS, Davidson GS, Martin SB, Andries E, Potter J, Harvey R, et al. Gene expression profiling of adult acute myeloid leukemia identifies novel biologic clusters for risk classification and outcome prediction. Blood. 2006;108:685–96. doi: 10.1182/blood-2004-12-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oldham RK, Reid WK, Preisler HD, Barnett D. A phase I and pharmacokinetic study of CBT-1 as a multidrug resistance modulator in the treatment of patients with advanced cancer. Cancer Biother Radiopharm. 1998;13:71–80. doi: 10.1089/cbr.1998.13.71. [DOI] [PubMed] [Google Scholar]
- 21.Oldham RK, Reid WK, Barnett D. Phase I study of CBT-1 and Taxol in patients with Taxol resistant cancers. Cancer Biother Radiopharm. 2000;15:153–9. doi: 10.1089/cbr.2000.15.153. [DOI] [PubMed] [Google Scholar]
- 22.Lai G-M, Chen Y-N, Mickley LA, Fojo AT, Bates SE. P-glycoprotein expression and schedule dependence of Adriamycin cytotoxicity in human colon carcinoma cell lines. Int J Cancer. 1991;49:696–703. doi: 10.1002/ijc.2910490512. [DOI] [PubMed] [Google Scholar]
- 23.Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, et al. Mutations at amino acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–8. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller M, Yong M, Peng XH, Petre B, Arora S, Ambudkar SV. Evidence for the role of glycosylation in accessibility of the extracellular domains of human MRP1 (ABCC1) Biochemistry. 2002;41:10123–32. doi: 10.1021/bi026075s. [DOI] [PubMed] [Google Scholar]
- 25.Minderman H, O'Loughlin KL, Pendyala L, Baer MR. VX-710 (biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res. 2004;10:1826–34. doi: 10.1158/1078-0432.ccr-0914-3. [DOI] [PubMed] [Google Scholar]
- 26.Robey R, Bakke S, Stein W, Meadows B, Litman T, Patil S, et al. Efflux of rhodamine from CD56+ cells as a surrogate marker for reversal of P-glycoprotein-mediated drug efflux by PSC 833. Blood. 1999;93:306–14. [PubMed] [Google Scholar]
- 27.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 28.Shukla S, Robey RW, Bates SE, Ambudkar SV. The calcium channel blockers, 1,4-dihydropyridines, are substrates of the multidrug resistance-linked ABC drug transporter, ABCG2. Biochemistry. 2006;45:8940–51. doi: 10.1021/bi060552f. [DOI] [PubMed] [Google Scholar]
- 29.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–14. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 30.Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001;61:749–58. [PubMed] [Google Scholar]
- 31.Germann UA, Shlyakhter D, Mason VS, Zelle RE, Duffy JP, Galullo V, et al. Cellular and biochemical characterization of VX-710 as a chemosensitizer: reversal of P-glycoprotein-mediated multidrug resistance in vitro. Anticancer Drugs. 1997;8:125–40. doi: 10.1097/00001813-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 32.de Bruin M, Miyake K, Litman T, Robey R, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999;146:117–26. doi: 10.1016/s0304-3835(99)00182-2. [DOI] [PubMed] [Google Scholar]
- 33.Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, et al. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–6. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- 34.Bates SE. Solving the problems of multidrug resistance: ABC transporters in clinical oncology. In: Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. ABC Proteins: From Bacteria to Man. Elsevier Science; London: 2002. pp. 359–91. [Google Scholar]
- 35.Rowinsky EK, Smith L, Wang YM, Chaturvedi P, Villalona M, Campbell E, et al. Phase I and pharmacokinetic study of paclitaxel in combination with biricodar, a novel agent that reverses multidrug resistance conferred by overexpression of both MDR1 and MRP. J Clin Oncol. 1998;16:2964–76. doi: 10.1200/JCO.1998.16.9.2964. [DOI] [PubMed] [Google Scholar]
- 36.Saeki T, Nomizu T, Toi M, Ito Y, Noguchi S, Kobayashi T, et al. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J Clin Oncol. 2007;25:411–7. doi: 10.1200/JCO.2006.08.1646. [DOI] [PubMed] [Google Scholar]
- 37.Kimura Y, Aoki J, Kohno M, Ooka H, Tsuruo T, Nakanishi O. P-glycoprotein inhibition by the multidrug resistance-reversing agent MS-209 enhances bioavailability and antitumor efficacy of orally administered paclitaxel. Cancer Chemother Pharmacol. 2002;49:322–8. doi: 10.1007/s00280-001-0419-x. [DOI] [PubMed] [Google Scholar]
- 38.Shrivastava P, Hanibuchi M, Yano S, Parajuli P, Tsuruo T, Sone S. Circumvention of multidrug resistance by a quinoline derivative, MS-209, in multidrug-resistant human small-cell lung cancer cells and its synergistic interaction with cyclosporin A or verapamil. Cancer Chemother Pharmacol. 1998;42:483–90. doi: 10.1007/s002800050849. [DOI] [PubMed] [Google Scholar]
- 39.Peck RA, Hewett J, Harding MW, Wang YM, Chaturvedi PR, Bhatnagar A, et al. Phase I and pharmacokinetic study of the novel MDR1 and MRP1 inhibitor biricodar administered alone and in combination with doxorubicin. J Clin Oncol. 2001;19:3130–41. doi: 10.1200/JCO.2001.19.12.3130. [DOI] [PubMed] [Google Scholar]
- 40.Seiden MV, Swenerton KD, Matulonis U, Campos S, Rose P, Batist G, et al. A phase II study of the MDR inhibitor biricodar (INCEL, VX-710) and paclitaxel in women with advanced ovarian cancer refractory to paclitaxel therapy. Gynecol Oncol. 2002;86:302–10. doi: 10.1006/gyno.2002.6762. [DOI] [PubMed] [Google Scholar]
- 41.Germann UA, Ford PJ, Shlyakhter D, Mason VS, Harding MW. Chemosensitization and drug accumulation effects of VX-710, cyclosporin A, MS-209 and GF120918 in multidrug resistant HL60/ADR cells expressing the multidrug resistance-associated protein MRP. Anticancer Drugs. 1997;8:141–55. doi: 10.1097/00001813-199702000-00005. [DOI] [PubMed] [Google Scholar]
- 42.Plasschaert SL, Van Der Kolk DM, De Bont ES, Vellenga E, Kamps WA, De Vries EG. Breast cancer resistance protein (BCRP) in acute leukemia. Leuk Lymphoma. 2004;45:649–54. doi: 10.1080/10428190310001597928. [DOI] [PubMed] [Google Scholar]
- 43.Bates S, Kang M, Meadows B, Bakke S, Choyke P, Merino M, et al. A Phase I study of infusional vinblastine in combination with the P-glycoprotein antagonist PSC 833 (valspodar) Cancer. 2001;92:1577–90. doi: 10.1002/1097-0142(20010915)92:6<1577::aid-cncr1484>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.