

Abstract
Introduction:
Adenosine A2 agonists improve arterial patency in experimental models of recurrent thrombosis, an effect purportedly triggered by stimulation of platelet A2 receptors and subsequent down-regulation of platelet function. However: (i) there is no direct evidence to substantiate this premise; and (ii) given the recognized differences among species in platelet signaling, it is possible that the mechanisms of A2 receptor stimulation may be model-dependent. Accordingly, we applied an integrated in vivo and in vitro approach, using both canine and human models, to test the hypothesis that the anti-thrombotic effects of A2 agonist treatment are due in part to inhibition of platelet activation.
Methods:
In Protocol 1, recurrent coronary thrombosis was triggered in anesthetized dogs by application of a stenosis at a site of arterial injury. Coronary patency and flow cytometric indices of platelet activation (P-selectin expression; formation of heterotypic aggregates) were compared in dogs pre-treated with the A2 agonist CGS 21680 versus controls. In Protocols 2 and 3, blood samples were obtained from dogs and human volunteers. In vitro aggregation and platelet activation (assessed by impedance aggregometry and flow cytometry, respectively) were quantified in paired aliquots pre-incubated with CGS versus vehicle.
Results:
In the canine models, CGS improved in vivo coronary patency and attenuated in vitro aggregation but, contrary to our hypothesis, did not evoke a down-regulation in platelet activation. In contrast, in human blood samples, CGS attenuated both in vitro aggregation and flow cytometric markers of platelet activation-aggregation.
Conclusion:
The mechanisms contributing to the anti-thrombotic effect of A2 agonist treatment are species-dependent: adenosine A2 receptor stimulation inhibits platelet activation in human, but not canine, models.
Keywords: adenosine, platelets, thrombosis, P-selectin, heterotypic aggregates, fibrinogen
Adenosine and adenosine receptor agonists have been shown to improve arterial patency in experimental models of recurrent thrombosis mimicking unstable angina and augment blood flow in models of coronary hypoperfusion [1-6]. Moreover, release of adenosine from ischemic-reperfused cardiomyocytes has been hypothesized to contribute to the favorable attenuation of recurrent thrombosis evoked by brief antecedent preconditioning ischemia [1-3, 7, 8]. These improvements in patency have been proposed to be a consequence of the well-documented, platelet inhibitory effects of adenosine, initiated via stimulation of adenosine A2 receptors on the platelets' surface [1-6, 8-12]. However, there is at present no direct evidence to substantiate this premise, and the specific site of action of adenosine/adenosine agonists (i.e., stimulation of A2 receptors on platelets versus other blood-borne elements and/or vascular smooth muscle) has not been established. In addition, given the recognized differences among species in platelet responsiveness and signaling [13-17], it is possible that the site and mechanisms of A2 receptor stimulation may be model-dependent and, most notably, may differ in humans. Accordingly, in the current study, we apply an integrated approach, employing a classic in vivo canine model of recurrent coronary thrombosis (the ‘Folts’ model [1, 8, 18, 19]), in vitro analysis of canine blood samples and in vitro analysis of blood samples obtained from healthy human volunteers to test the hypothesis that the anti-thrombotic effects of adenosine A2 receptor agonist treatment involve stimulation of A2 receptors on platelets and a resultant down-regulation in one or more molecular markers of platelet activation-aggregation.
METHODS
All canine studies were approved by the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School, and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (1996). In vitro analysis of human blood samples was approved by the Institutional Review Board of the Medical School.
PROTOCOL 1: In vivo canine model of recurrent thrombosis
In 6 pentobarbital-anesthetized adult mongrel dogs, catheters were positioned in the left jugular vein for administration of fluids and supplemental anesthesia, and in the left carotid artery for measurement of heart rate and arterial pressure and collection of blood samples. A left lateral thoracotomy was performed and two adjacent segments of the left anterior descending coronary artery (LAD) were isolated: the distal segment was instrumented with a Doppler flow probe for continuous measurement of mean coronary blood flow (CBF), while the proximal segment served as the site of later thrombosis (methods described in [8]).
After stabilization, all 6 dogs received a 10 min infusion of the adenosine A2 receptor agonist CGS 21680 (0.5 μg/kg/min), administered via a cannula positioned in the left atrium, followed by a 10 min washout period. CGS has a plasma half-life on the order of 19 min and, at this dose, the estimated maximum plasma concentration is 0.45 μM [20, 21]. The CGS-treated cohort was part of a larger, randomized 3-group study design, with the remaining dogs assigned to receive 10 min of preconditioning ischemia followed by 10 min of reperfusion (n=10), or a matched no-intervention control period (n=12). In vivo data for the control and preconditioned groups have been reported previously [8].
Following the 20 min treatment phase, spontaneous recurrent thrombosis was initiated by squeezing the isolated LAD segment with forceps and applying a stenosis at the site of vessel damage [1, 8, 18, 19, 22]. CBF was monitored for 3 hours without further intervention. At the end of the observation period, the damaged LAD segment was excised and stored in 10% neutral buffered formalin for later histological evaluation [8].
Endpoints
Our primary endpoints were coronary patency and flow cytometric markers of platelet function [8]. Vessel patency during the 3 hours of observation was assessed by quantifying the duration of total thrombotic occlusion (CBF = 0); and % flow-time area, defined as the area of the flow-time tracing normalized for each dog to the baseline flow × 180 min. For assessment of platelet activation-aggregation, 5 mL of citrated blood was obtained from all dogs at baseline (before randomization) and at 2 hours after the onset of recurrent thrombosis. Platelet surface P-selectin expression, formation of monocyte-platelet and neutrophil-platelet aggregates, and platelet-fibrinogen binding were quantified by flow cytometry using our previously described methods [8]. All samples were blinded at the time of analysis, and data obtained following recurrent thrombosis were normalized, for each animal, to respective baseline values.
We also, as secondary endpoints, tabulated heart rate and arterial pressure throughout the protocol, and assessed the severity of arterial damage as described previously [8].
Statistical analysis
Results for CGS-treated animals were compared with data obtained from the concurrently randomized control group [8]. As there were 3 groups in the original study design, discrete variables were compared by ANOVA, while variables measured repeatedly throughout the protocol were compared by 2-factor ANOVA with replication. Post-hoc comparisons between CGS-treated animals versus controls were made using the Newman-Keuls test.
PROTOCOL 2: In vitro analysis of canine blood samples
Additional citrated blood samples were obtained at baseline from dogs enrolled in the in vivo protocol and used for in vitro analysis.
Endpoints
Our first endpoint was in vitro aggregation using the standard method of whole blood impedance aggregometry [12, 13, 22]. Two blood aliquots (0.5 mL each) were obtained from all dogs in Protocol 1 (n=28), diluted with 0.5 mL saline, and maintained at 37°C. One aliquot from each pair was incubated for 10 min with exogenous CGS 21680 (final concentration 10 μM), while the second was treated with a matched volume of vehicle (saline). Aggregation was then triggered by the addition of 10 μg collagen and, 10 min later, maximum impedance (in ohms, the index of platelet aggregation) was quantified.
Our second endpoint was the flow cytometric assessment of platelet activation-aggregation in response to in vitro administration of standard stimuli. Pairs of blood aliquots were obtained from 7 dogs and treated with CGS 21680 (10 μM) or saline. Five min later, matched saline- and CGS-treated aliquots were stimulated with ADP (20 μM for 15 min at room temperature) or a thromboxane receptor agonist confirmed to be cross-reactive in dogs (IBOP, 0.25 μM for 15 min at 37°C). Additional saline- and CGS-treated aliquots received control buffer rather that ADP or IBOP and thus served as unstimulated samples. Platelet surface P-selectin expression and formation of monocyte-platelet aggregates were quantified using the same methods employed in Protocol 1.
Statistical analysis
Impedance, P-selectin expression and formation of monocyte-platelet were compared between matched CGS- and saline-treated aliquots by paired t-tests.
PROTOCOL 3: In vitro analysis of human blood samples
Citrated venous blood samples were obtained from 6 healthy adult donors who had not ingested aspirin or other drugs known to alter platelet function for 1 week prior to sampling.
Endpoints
The in vitro treatment and analysis of the human blood samples was analogous to that conducted in Protocol 2 for canine blood. Aliquots were pre-incubated with CGS 21680 (10 μM) or saline, maximum impedance following exposure to collagen (10 μg) was assessed by whole blood impedance aggregometry, and indices of platelet activation-aggregation (P-selectin expression and formation of monoyte-platelet aggregates) were quantified by flow cytometry following addition of ADP (20 μM for 15 min at room temperature), the thromboxane agonist U46619 (20 μM for 15 min at 37°C), or buffer.
Statistical analysis
Data obtained from matched CGS- and saline-treated aliquots were compared using paired t-tests.
RESULTS
Protocol 1
In vivo infusion of CGS 21680 into the left atrium was, as expected, associated with a significant increase in coronary blood flow to 461% of baseline values. This effect was, however, transient, with coronary flow rapidly returning to baseline during the 10 min washout period (i.e., before arterial injury). Administration of CGS into the left atrium did not evoke significant changes in heart rate or cause systemic vasodilation (that is, arterial pressure did not differ versus controls; Table 1).
Table 1.
Hemodynamics ¶
| Pre- Stenosis |
10″ Post- Stenosis |
During CFVs: | ||||
|---|---|---|---|---|---|---|
| Baseline | End-Treat§ | 1 h | 3 h | |||
| Heart rate (beats/min): | ||||||
| Control: | 151±7 | 151±7 | 149±8 | 150±6 | 161±5 | 175±4 † |
| CGS: | 151±11 | 160±15 | 163±16 | 163±16 | 152±12 | 175±6 |
| Mean arterial pressure (mmHg): | ||||||
| Control: | 128±7 | 129±7 | 125±8 | 124±7 | 134±7 | 132±6 |
| CGS: | 134±6 | 116±8 | 121±8 | 119±7 | 135±7 | 121±11 |
| Coronary blood flow (% of Baseline): | ||||||
| Control: | 100% | 104±4% | 103±7% | 34±3% † | na | na |
| CGS: | 100% | 461±46% ‡* | 100±12% | 38±4% † | na | na |
CGS = CGS 21680
Data for control group reported previously in [8].
End-Treat: data obtained at the end of 10 min CGS infusion, or matched time point in controls
na: discrete measurements of CBF not applicable after the onset of CFVs
p<.05 versus Control
p<.05
p<.01 versus Baseline
Arterial damage in CGS-treated dogs was characterized by medial tearing and dissection with minimal adventitial exposure, similar to the vessel injury reported previously for the control cohort [8]. Moreover, all dogs developed spontaneous recurrent thrombosis following arterial injury and displayed persistent cyclic variations in coronary blood flow (CFVs) throughout the 3 hour observation period.
Although recurrent thrombosis was seen in all dogs, pre-treatment with CGS was, as anticipated from previous studies [1-4], associated with better maintenance of patency in the damaged and stenotic vessel. Specifically: flow-time area averaged 76±13% and the duration of total thrombotic occlusion was 10±6 min, significant improvements versus the values of 23±5% and 59±14 min observed in the concurrent controls (Figure 1). Surprisingly, however, this enhanced coronary patency seen in CGS-treated animals was not accompanied by a favorable attenuation in molecular markers of platelet activation-aggregation. Formation of heterotypic aggregates, platelet surface P-selectin expression and platelet-fibrinogen binding assessed at 2 hours following the onset of CFVs showed comparable up-regulation versus baseline in both control and CGS-treated groups (Figure 2).
Figure 1.

In vivo patency: Protocol 1. Zero flow duration and % flow-time area in Control and CGS 21680-treated dogs. Data for control group reported previously in [8]. *p<0.05 versus Controls.
Figure 2.
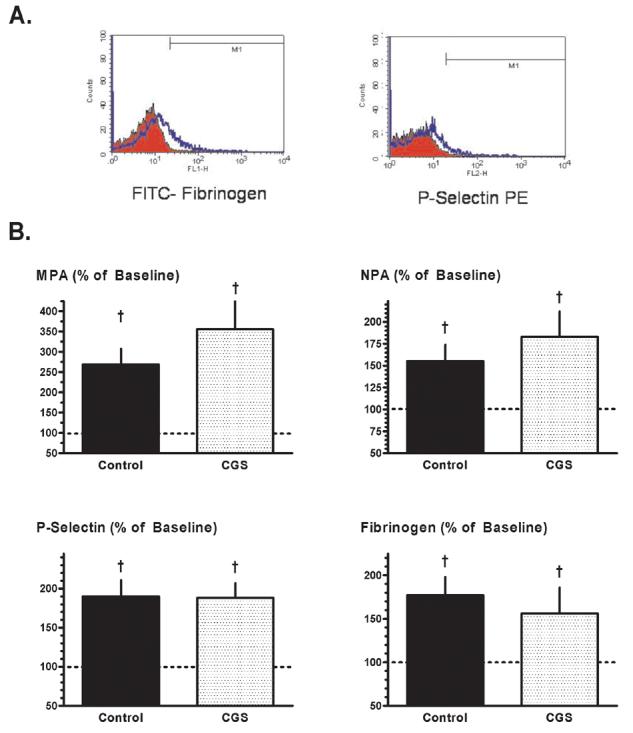
Flow cytometry: Protocol 1. (A) Representative histograms illustrating the increase in platelet-fibrinogen binding (left) and platelet surface P-selectin expression (right) in paired canine blood samples obtained at 2 hours after the onset of recurrent thrombosis (blue profiles) versus baseline (red profiles). (B) Monocyte-platelet aggregates (MPA), neutrophil-platelet aggregates (NPA), platelet surface P-selectin expression and platelet-fibrinogen binding, measured at 2 hours after the onset of recurrent thrombosis and expressed as a % of baseline values, in Control and CGS 21680-treated dogs. Data for control group reported previously in [8]. †p<0.05 versus baseline; no significant differences between groups.
Protocol 2
Maximum in vitro aggregation in canine vehicle-treated aliquots, as determined by whole blood impedance aggregometry, averaged 16 ohms. In contrast, impedance in matched aliquots pretreated with CGS was significantly reduced to a mean of 12 ohms (Figure 3B), a finding consistent with previous in vitro data obtained with adenosine A2 agonists [4, 11, 12, 22] and consistent with the concept that CGS attenuates thrombosis.
Figure 3.
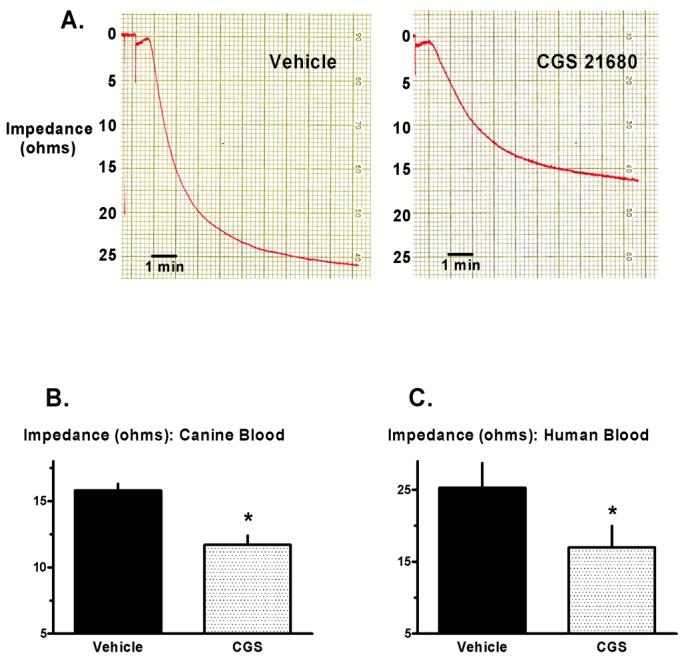
In vitro aggregometry: Protocols 2 and 3. (A) Representative example of in vitro platelet aggregation in a pair of blood aliquots obtained from one human donor. Original recordings show the increase in impedance triggered by collagen in one aliquot pretreated with CGS 21680 versus the matched aliquot pretreated with vehicle. (B) Maximum impedance in matched canine blood aliquots treated with CGS 21680 versus vehicle. (C) Maximum impedance in matched human blood aliquots treated with CGS 21680 versus vehicle. *p<0.05 versus vehicle.
Vehicle-treated aliquots stimulated with IBOP or ADP showed the anticipated, robust increases in flow cytometric markers of platelet activation-aggregation when compared with quiescent, unstimulated samples. However, as in Protocol 1, pretreatment with CGS had no inhibitory effect on P-selectin expression or formation of monocyte-platelet aggregates in canine blood (Figure 4A).
Figure 4.
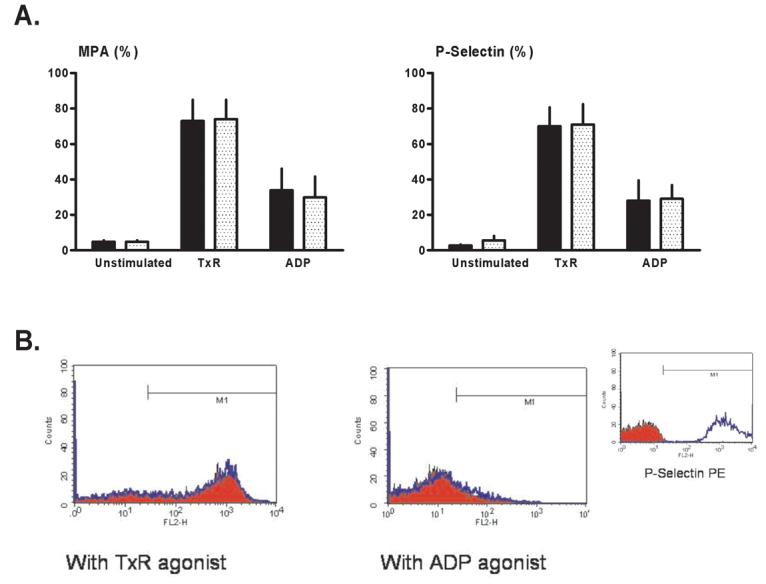
Flow cytometry: Protocol 2 (canine blood samples). Monocyte-platelet aggregates (MPA) and platelet surface P-selectin expression were quantified in unstimulated (no agonist, quiescent) blood aliquots, aliquots stimulated with a stable thromboxane receptor agonist (TxR), and aliquots stimulated with ADP. (A) Mean values for samples pre-treated with CGS 21680 (stippled bars) versus paired vehicle-controls (solid bars). (B) Representative histograms showing platelet surface P-selectin expression in CGS-treated blood aliquots (red profiles) versus vehicle-controls (blue profiles). (Insert) Positive and negative controls: histograms for isotype-negative (red) and PMA-positive (phorbol myristate acetate: blue) controls for P-selectin in canine blood.
Protocol 3
Whole blood aggregometry conducted on human blood samples revealed that, as in the canine model, impedance was significantly reduced in CGS-prereated aliquots when compared with vehicle-controls (Figures 3A and 3C). However, in contrast to data obtained in canine samples, platelet activation-aggregation in response to exogenous stimulation with ADP or U46619 was significantly attenuated in human samples pre-incubated with CGS when compared with saline-controls (Figure 4B).
DISCUSSION
We report that brief pretreatment with the adenosine A2 receptor agonist CGS 21680 significantly improves coronary patency in the in vivo canine model of recurrent thrombosis. This favorable effect of A2 receptor stimulation was not, however, accompanied by a down-regulation in flow cytometric indices of platelet activation-aggregation: i.e., results of our in vivo and in vitro analyses revealed that, contrary to our hypothesis, CGS does not appear to attenuate platelet reactivity in canine blood. Our results further demonstrate that platelet responsiveness to adenosine A2 receptor stimulation is species-dependent, with CGS 21680 evoking a marked inhibition of agonist-induced activation in human blood samples.
Adenosine A2 receptor stimulation, coronary patency and platelet function in the canine model
Previous studies conducted in anesthetized dogs have shown that adenosine and adenosine agonists attenuate recurrent thrombosis in damaged and stenotic coronary arteries [1-4], and inhibit the formation of microemboli in the setting of stable coronary hypoperfusion [5, 6]. Moreover, although the mechanisms by which these agents improve coronary patency are poorly understood, stimulation of platelet adenosine A2 receptors and subsequent sustained suppression of platelet activation-aggregation has been proposed to play a role [1-6]. The outcome of Protocol 1 corroborates the concept that A2 agonists augment coronary patency (Figure 1). However, the improved patency seen in CGS-treated dogs was not accompanied by a favorable down-regulation in flow cytometric markers of platelet activation-aggregation (Figure 2). A similar unanticipated incongruity was observed in Protocol 2. CGS had the expected inhibitory effect on in vitro aggregation in canine samples as measured by whole blood impedance aggregometry (Figure 3B), but did not attenuate platelet surface P-selectin expression or the formation of monocyte-platelet aggregates as detected by flow cytometry (Figure 4A).
This unexpected dissociation between indices of thrombosis and platelet activation-aggregation may in theory be explained by limitations in study design. For example, in Protocol 1, it could be argued that the enhanced patency seen with CGS was transient and flow cytometric assessment of platelet activation-aggregation was made at an inappropriate time. Temporal analysis of the patency data does not support this concept. Rather, flow-time area remained stable in both cohorts throughout the protocol, averaging 68%, 81% and 79% in CGS-treated dogs versus 21%, 23% and 24% in controls during the first, second and third hours of observation, respectively. Second, while brief infusion of CGS initiated a sustained improvement in patency, the blood sample used for flow cytometric analysis was obtained well beyond the 19 min plasma half-life of the A2 agonist. However, the outcome of Protocol 2 demonstrated that direct addition of 10 μM CGS to blood aliquots (a dose 20-fold higher than the estimated maximum in vivo concentration of CGS in Protocol 1), similarly failed to inhibit up-regulation of P-selectin and monocyte-platelet aggregates in canine blood (Figure 4A). With regard to Protocol 2, it could be argued that pretreatment with CGS failed to attenuate flow cytometric indices of platelet function because supra-maximal concentrations ADP and IBOP were used. This was also not the case, as we confirmed in preliminary dose-optimization studies that the stimuli were sub-maximal. Finally, our observations that CGS attenuated in vitro aggregation in the absence of a concomitant, favorable effect on the formation of monocyte-platelet aggregates may be considered especially puzzling. However, given the fact that monocyte-platelet aggregates consist of a single monocyte with one or more platelets attached [23], and that in normal dogs there is a 500-fold difference in the number of circulating platelets versus monocytes (on the order of 250,000 platelets versus 500 monocytes per μL, respectively), monocyte-platelet aggregates would not be expected to make a significant numerical contribution to in vitro thrombosis assessed by impedance aggregometry.
An alternative explanation for the unexpected outcomes of Protocols 1 and 2 is that, in the canine model, the attenuated thrombosis seen with CGS 21680 may be a consequence of adenosine A2 receptor stimulation at sites other than platelets. Specifically, we propose that the improved in vivo patency and attenuated in vitro aggregation seen with CGS were initiated by adenosine A2 receptor stimulation on neutrophils (rather than platelets), and that A2-mediated alterations in adhesion (rather than platelet activation-aggregation) play a pivotal role [24-29]. Despite well-documented evidence that adenosine attenuates neutrophil adhesion via an A2-mediated down-regulation of CD11b/CD18 and L- selectin [24-27], further prospective studies will be required to substantiate this hypothesis.
Mechanistic implications for the improved patency seen with preconditioning
Our group has shown that a brief, 10 min episode of preconditioning ischemia improves subsequent vessel patency in damaged and stenotic canine coronary arteries [1, 3]. Furthermore, this augmented patency is associated with a down-regulation in multiple flow cytometric markers of platelet activation-aggregation [8]. Our working hypothesis has been that the favorable, anti-platelet effect of preconditioning is initiated by release of adenosine from cardiomyocytes during brief ischemia-reperfusion and stimulation of A2 receptors on the platelets' surface [1-3]. However, the outcome of Protocols 1 and 2, showing that direct administration of an A2 agonist does not evoke a down-regulation in platelet function, fails to support this concept. Additional evidence arguing against the adenosine A2-platelet hypothesis was provided by recent in vivo experiments in which control and preconditioned dogs were pre-treated with the selective A2 receptor antagonist ZM 241385. ZM abrogated the preconditioning-induced improvement in coronary patency, without blocking the benefits of preconditioning on platelet activation-aggregation [30]. Taken together, these data strongly suggest that the better maintenance of coronary patency seen with preconditioning is not triggered by an adenosine A2-mediated attenuation of platelet reactivity.
Adenosine A2 receptor stimulation and platelet function in human blood samples
A small number of studies have provided in vitro evidence for an anti-thrombotic effect of adenosine A2 receptor agonists in human blood [31-33]. Our results in Protocol 3, showing that in vitro aggregation in response to collagen was significantly attenuated in human blood aliquots pre-incubated with CGS 21680 (Figures 3A and 3C) is consistent with these earlier reports, and similar to our results obtained in canine samples (Figure 3B). However, to our knowledge no previous studies have used flow cytometry to investigate whether CGS evokes a favorable down-regulation in molecular indices of platelet activation-aggregation. In contrast to our findings in the canine model, pre-treatment with CGS inhibited the increase in platelet surface P-selectin expression and formation of monocyte-platelet aggregates triggered by in vitro stimulation with ADP and U46619 (Figure 4B), thereby implying that, in human blood, the site(s) of action of CGS include the platelet.
The only technical difference between our in vitro analyses of platelet function in canine versus human samples was the use of IBOP versus U46619. We found in pilot experiments that U46619 failed to trigger platelet activation-aggregation in dog blood. This observation is in agreement with some previous reports [34, 35], and has been attributed to a genetic defect in G protein function that purportedly renders most dogs refractory to thromboxane A2 receptor stimulation [36, 37]. In contrast, others have successfully used U46619 in canine models [38], while in our hands IBOP effectively stimulated platelet activation-aggregation. Although resolution of this issue is beyond the scope of the current study, Protocols 2 and 3 underscore the general concept of differences among species in platelet signaling [13-17] and, most notably, differences in adenosine A2 receptor responsiveness.
Summary and limitations
The adenosine A2 receptor agonist CGS 21680 attenuates recurrent thrombosis in the in vivo canine model of unstable angina, and attenuates in vitro aggregation in both canine and human blood samples. However, our results further reveal that the site(s) and mechanisms of action of CGS are complex and species-dependent, with the improvement in patency seen in the canine model achieved in the absence of a concomitant down-regulation in platelet function.
It must be acknowledged that, in Protocol 1, our conclusions are based on measurement of platelet activation-aggregation at one time point after the onset of recurrent thrombosis. The temporal requirements of the flow cytometric methods, together with our emphasis on initiating all analyses within 15 min of sample collection, precluded the feasibility of collecting sequential data. Time constraints also explain the fact that, in the in vitro protocols, we focused on 2 (rather than 4) indices of platelet activation-aggregation, and did not assess the full battery of agonists on all endpoints. Rather, for each endpoint, we focused on the agonist(s) that yielded the most reproducible and quantitatively largest response (collagen for aggregometry, thromboxane receptor agonists and ADP for flow cytometry). Nonetheless, in vitro analysis of human blood samples revealed that CGS 21680 evoked a significant down-regulation in platelet activation-aggregation, thereby raising the possibility that adenosine A2 receptor stimulation may provide a potential therapeutic target for inhibition of platelet function in the clinical setting.
Figure 5.
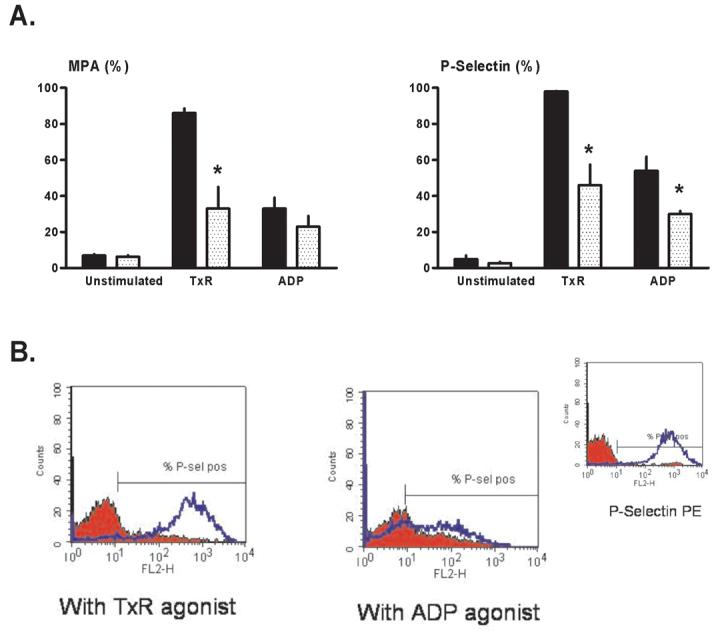
Flow cytometry: Protocol 3 (human blood samples). Monocyte-platelet aggregates (MPA) and platelet surface P-selectin expression were quantified in unstimulated (no agonist, quiescent) blood aliquots, aliquots stimulated with a stable thromboxane receptor agonist (TxR), and aliquots stimulated with ADP. (A) Mean values for samples pre-treated with CGS 21680 (stippled bars) versus paired vehicle-controls (solid bars). *p<0.05 versus matched vehicle-treated aliquots. (B) Representative histograms showing platelet surface P-selectin expression in CGS-treated blood aliquots (red profiles) versus vehicle-controls (blue profiles). (Insert) Positive and negative controls: histograms for isotype-negative (red) and PMA-positive (phorbol myristate acetate: blue) controls for P-selectin in human blood.
Acknowledgments
Supported by R01-HL72684 from the National Institutes of Health (to KP)
Abbreviations
- ADP
adenosine diphosphate
- CBF
coronary blood flow
- CFVs
cyclic variations in coronary blood flow
- IBOP
[15-(1a,2b(5Z),3a-(1E,3S),4a)]-7-[3-hydroxy-4-(p-iodophenoxy)-1-butenyl]-7-oxabicycloheptenoic acid
- LAD
left anterior descending coronary artery
Footnotes
Presented in part at the 7th Annual American Heart Association Conference on Arteriosclerosis, Thrombosis and Vascular Biology, Denver CO, April 27-29, 2006.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hata K, Whittaker P, Kloner RA, Przyklenk K. Brief antecedent ischemia attenuates platelet-mediated thrombosis in damaged and stenotic canine coronary arteries: role of adenosine. Circulation. 1998;97:692–702. doi: 10.1161/01.cir.97.7.692. [DOI] [PubMed] [Google Scholar]
- 2.Hata K, Whittaker P, Kloner RA, Przyklenk K. Brief myocardial ischemia attenuates platelet thrombosis in remote, damaged, and stenotic carotid arteries. Circulation. 1999;100:843–8. doi: 10.1161/01.cir.100.8.843. [DOI] [PubMed] [Google Scholar]
- 3.Przyklenk K, Whittaker P. Brief antecedent ischemia enhances recombinant tissue plasminogen activator-induced coronary thrombolysis by adenosine-mediated mechanism. Circulation. 2000;102:88–95. doi: 10.1161/01.cir.102.1.88. [DOI] [PubMed] [Google Scholar]
- 4.Bullough DA, Zhang C, Montag A, Mullane KM, Young MA. Adenosine-mediated inhibition of platelet aggregation by acadesine. A novel antithrombotic mechanism in vitro and in vivo. J Clin Invest. 1994;94:1524–32. doi: 10.1172/JCI117493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitakaze M, Hori M, Sato H, Takashima S, Inoue M, Kitabatake A, Kamada T. Endogenous adenosine inhibits platelet aggregation during myocardial ischemia in dogs. Circ Res. 1991;69:1402–8. doi: 10.1161/01.res.69.5.1402. [DOI] [PubMed] [Google Scholar]
- 6.Minamino T, Kitakaze M, Asanuma H, Tomiyama Y, Shiraga M, Sato H, Ueda Y, Funaya H, Kuzuya T, Matsuzawa Y, Hori M. Endogenous adenosine inhibits P-selectin-dependent formation of coronary thromboemboli during hypoperfusion in dogs. J Clin Invest. 1998;101:1643–53. doi: 10.1172/JCI635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreotti F, Pasceri V. Ischaemic preconditioning. Lancet. 1996;348:204. doi: 10.1016/s0140-6736(05)66158-2. [DOI] [PubMed] [Google Scholar]
- 8.Linden MD, Whittaker P, Frelinger AL, Barnard M, Michelson AD, Przyklenk K. Preconditioning ischemia attenuates molecular indices of platelet activation-aggregation. J Thromb Haemost. 2006;4:2670–7. doi: 10.1111/j.1538-7836.2006.02228.x. [DOI] [PubMed] [Google Scholar]
- 9.Dionisotti S, Conti A, Sandoli D, Zocchi C, Gatta F, Ongini E. Effects of the new A2 adenosine receptor antagonist 8FB-PTP, an 8 substituted pyrazolo-triazolo-pyrimidine, on in vitro functional models. Br J Pharmacol. 1994;112:659–65. doi: 10.1111/j.1476-5381.1994.tb13126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hourani SM, Cusack NJ. Pharmacological receptors on blood platelets. Pharmacol Rev. 1991;43:243–98. [PubMed] [Google Scholar]
- 11.Sandoli D, Chiu PJ, Chintala M, Dionisotti S, Ongini E. In vivo and ex vivo effects of adenosine A1 and A2 receptor agonists on platelet aggregation in the rabbit. Eur J Pharmacol. 1994;259:43–9. doi: 10.1016/0014-2999(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 12.Przyklenk K, Whittaker P. In vitro platelet responsiveness to adenosine-mediated ‘preconditioning’ is age-dependent. J Thromb Thrombolysis. 2005;19:5–10. doi: 10.1007/s11239-005-0849-0. [DOI] [PubMed] [Google Scholar]
- 13.Galvez A, Badimon L, Badimon JJ, Fuster V. Electrical aggregometry in whole blood from human, pig and rabbit. Thromb Haemost. 1986;56:128–32. [PubMed] [Google Scholar]
- 14.Andre P, Bal dit Sollier C, Bonneau M, Pignaud G, Hainaud P, Azzam K, Drouet L. Which experimental model to choose to study arterial thrombosis and evaluate potentially useful therapeutics? Haemostasis. 1996;26(Suppl 4):55–69. doi: 10.1159/000217286. [DOI] [PubMed] [Google Scholar]
- 15.Soloviev MV, Okazaki Y, Harasaki H. Whole blood platelet aggregation in humans and animals: a comparative study. J Surg Res. 1999;82:180–7. doi: 10.1006/jsre.1998.5543. [DOI] [PubMed] [Google Scholar]
- 16.Nieman MT, Warnock M, Hasan AA, Mahdi F, Lucchesi BR, Brown NJ, Murphey LJ, Schmaier AH. The preparation and characterization of novel peptide antagonists to thrombin and factor VIIa and activation of protease-activated receptor 1. J Pharmacol Exp Ther. 2004;311:492–501. doi: 10.1124/jpet.104.069229. [DOI] [PubMed] [Google Scholar]
- 17.Nylander S, Mattsson C, Lindahl TL. Characterisation of species differences in the platelet ADP and thrombin response. Thromb Res. 2006;117:543–9. doi: 10.1016/j.thromres.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 18.Folts JD, Crowell EB, Jr., Rowe GG. Platelet aggregation in partially obstructed vessels and its elimination with aspirin. Circulation. 1976;54:365–70. doi: 10.1161/01.cir.54.3.365. [DOI] [PubMed] [Google Scholar]
- 19.Folts J. An in vivo model of experimental arterial stenosis, intimal damage, and periodic thrombosis. Circulation. 1991;83:IV3–14. [PubMed] [Google Scholar]
- 20.Jordan JE, Zhao ZQ, Sato H, Taft S, Vinten-Johansen J. Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280:301–9. [PubMed] [Google Scholar]
- 21.Balwierczak JL, Sharif R, Krulan CM, Field FP, Weiss GB, Miller MJ. Comparative effects of a selective adenosine A2 receptor agonist, CGS 21680, and nitroprusside in vascular smooth muscle. Eur J Pharmacol. 1991;196:117–23. doi: 10.1016/0014-2999(91)90416-n. [DOI] [PubMed] [Google Scholar]
- 22.Przyklenk K, Kloner RA. Sildenafil citrate (Viagra) does not exacerbate myocardial ischemia in canine models of coronary artery stenosis. J Am Coll Cardiol. 2001;37:286–92. doi: 10.1016/s0735-1097(00)01049-4. [DOI] [PubMed] [Google Scholar]
- 23.Neumann FJ, Marx N, Gawaz M, Brand K, Ott I, Rokitta C, Sticherling C, Meinl C, May A, Schomig A. Induction of cytokine expression in leukocytes by binding of thrombin-stimulated platelets. Circulation. 1997;95:2387–94. doi: 10.1161/01.cir.95.10.2387. [DOI] [PubMed] [Google Scholar]
- 24.Wollner A, Wollner S, Smith JB. Acting via A2 receptors, adenosine inhibits the upregulation of Mac-1 (Cd11b/CD18) expression on FMLP-stimulated neutrophils. Am J Respir Cell Mol Biol. 1993;9:179–85. doi: 10.1165/ajrcmb/9.2.179. [DOI] [PubMed] [Google Scholar]
- 25.Thiel M, Chambers JD, Chouker A, Fischer S, Zourelidis C, Bardenheuer HJ, Arfors KE, Peter K. Effect of adenosine on the expression of beta(2) integrins and L-selectin of human polymorphonuclear leukocytes in vitro. J Leukoc Biol. 1996;59:671–82. doi: 10.1002/jlb.59.5.671. [DOI] [PubMed] [Google Scholar]
- 26.Vinten-Johansen J, Thourani VH, Ronson RS, Jordan JE, Zhao ZQ, Nakamura M, Velez D, Guyton RA. Broad-spectrum cardioprotection with adenosine. Ann Thorac Surg. 1999;68:1942–8. doi: 10.1016/s0003-4975(99)01018-8. [DOI] [PubMed] [Google Scholar]
- 27.Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. 1999;43:860–78. doi: 10.1016/s0008-6363(99)00187-x. [DOI] [PubMed] [Google Scholar]
- 28.Eguchi H, Ikeda H, Murohara T, Yasukawa H, Haramaki N, Sakisaka S, Imaizumi T. Endothelial injuries of coronary arteries distal to thrombotic sites: role of adhesive interaction between endothelial P-selectin and leukocyte sialyl LewisX. Circ Res. 1999;84:525–35. doi: 10.1161/01.res.84.5.525. [DOI] [PubMed] [Google Scholar]
- 29.Russell-Smith NC, Flower RJ, Cardinal DC. Measuring platelet and leucocyte aggregation/adhesion responses in very small volumes of whole blood. J Pharmacol Methods. 1981;6:315–33. doi: 10.1016/0160-5402(81)90071-1. [DOI] [PubMed] [Google Scholar]
- 30.Linden MD, Michelson AD, Barnard MR, Frelinger AL, III, Furman MI, Przyklenk K. Does adenosine A2 receptor stimulation trigger the favorable ‘anti-platelet’ effect of preconditioning ischemia? Arterioscler Thromb Vasc Biol. 2006;25:e-46. abstract. [Google Scholar]
- 31.Dionisotti S, Zocchi C, Varani K, Borea PA, Ongini E. Effects of adenosine derivatives on human and rabbit platelet aggregation. Correlation of adenosine receptor affinities and antiaggregatory activity. Naunyn Schmiedebergs Arch Pharmacol. 1992;346:673–6. doi: 10.1007/BF00168741. [DOI] [PubMed] [Google Scholar]
- 32.Cristalli G, Vittori S, Thompson RD, Padgett WL, Shi D, Daly JW, Olsson RA. Inhibition of platelet aggregation by adenosine receptor agonists. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:644–50. doi: 10.1007/pl00004904. [DOI] [PubMed] [Google Scholar]
- 33.Varani K, Borea PA, Guerra L, Dionisotti S, Zocchi C, Ongini E. Binding characteristics of the adenosine A2 receptor ligand [3H]CGS 21680 to human platelet membranes. Biochem Pharmacol. 1994;48:1658–61. doi: 10.1016/0006-2952(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 34.Bush LR, Smith SG. Antagonism of U46619-induced aggregation of human and canine platelets by four TXA2 receptor antagonists. Thromb Res. 1986;44:377–89. doi: 10.1016/0049-3848(86)90012-5. [DOI] [PubMed] [Google Scholar]
- 35.Yao SK, McNatt J, Cui K, Anderson HV, Maffrand JP, Buja LM, Willerson JT. Combined ADP and thromboxane A2 antagonism prevents cyclic flow variations in stenosed and endothelium-injured arteries in nonhuman primates. Circulation. 1993;88:2888–93. doi: 10.1161/01.cir.88.6.2888. [DOI] [PubMed] [Google Scholar]
- 36.Johnson GJ, Leis LA, King RA. Thromboxane responsiveness of dog platelets is inherited as an autosomal recessive trait. Thromb Haemost. 1991;65:578–80. [PubMed] [Google Scholar]
- 37.Johnson GJ, Leis LA, Dunlop PC. Thromboxane-insensitive dog platelets have impaired activation of phospholipase C due to receptor-linked G protein dysfunction. J Clin Invest. 1993;92:2469–79. doi: 10.1172/JCI116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong TT, Huang J, Driscoll E, Lucchesi BR. Preclinical evaluation of S18886 in an experimental model of coronary arterial thrombosis. J Cardiovasc Pharmacol. 2006;48:239–48. doi: 10.1097/01.fjc.0000248234.08277.7e. [DOI] [PubMed] [Google Scholar]