


Abstract
Our previous publication shows that Sulfolobus solfataricus Dpo4 utilizes an ‘induced-fit’ mechanism to select correct incoming nucleotides at 37°C. Here, we provide a comprehensive report elucidating the kinetic mechanism of a DNA polymerase at a reaction temperature higher than 37°C in an attempt to determine the effect of temperature on enzyme fidelity and mechanism. The fidelity of Dpo4 did not change considerably with a 30°C increase in reaction temperature, suggesting that the fidelity of Dpo4 at 80°C is similar to that determined here at 56°C. Amazingly, the incorporation rate for correct nucleotides increased by 18 900-fold from 2°C to 56°C, similar in magnitude to that observed for incorrect nucleotides, thus not perturbing fidelity. Three independent lines of kinetic evidence indicate that a protein conformational change limits correct nucleotide incorporations at 56°C. Furthermore, the activation energy for the incorporation of a correct nucleotide was determined to be 32.9 kcal/mol, a value considerably larger than those values estimated for a rate-limiting chemistry step, providing a fourth line of evidence to further substantiate this conclusion. These results herein provide evidence that Dpo4 utilizes the ‘induced-fit’ mechanism to select a correct nucleotide at all temperatures.
INTRODUCTION
The DNA polymerases of the Y-family are known for their ability to bypass a variety of DNA lesions that, to varying degrees, impede the continuous synthesis of genomic DNA catalyzed by high-fidelity replicative DNA polymerases. In addition to these roles in lesion bypass, recent studies have suggested additional functions of these specialized polymerases in the cell (1,2). These Y-family enzymes are found in all three life domains and are notably devoid of 3′ to 5′ exonuclease activities, which are genuine fidelity checking mechanisms inherent to replicative DNA polymerases. These lesion bypass DNA polymerases are also shown to possess conformationally flexible active sites (3–8), which contributes to both their observed lesion-bypass capabilities and low polymerization fidelities.
It has been shown previously that T7 phage DNA polymerase utilizes an induced-fit mechanism powered by the energy of nucleotide binding, to select a correct nucleotide from a pool of similar nucleotide substrates (9,10). Similar results have been provided as convincingly for DNA polymerase I (Klenow fragment) (11) and HIV-1 reverse transcriptase (12). To our knowledge, pre-steady state kinetic studies have been performed for only three Y-family DNA polymerases with an undamaged DNA substrate. Both Saccharomyces cerevisiae polymerase η (polη) (13) and Sulfolobus solfataricus DNA polymerase IV (Dpo4) (14) have been shown to utilize an induced-fit mechanism for selecting a correct nucleotide while studies of Sulfolobus acidocaldarius DinB homolog (Dbh) (15) very unconvincingly concluded that Dbh uses the chemistry step to choose incoming nucleotides. In the latter study, Cramer and Restle surprisingly argue against an induced-fit mechanism for Dbh solely based on an observed elemental effect of 9.1 for correct nucleotide incorporation. In contrast, the same elemental effect determined by Potapova et al. (16) in an earlier study is 4 and they conclude that Dbh uses the induced-fit mechanism to select a correct nucleotide. These contradictory conclusions are purely based on the measured elemental effect, which is largely considered an unreliable diagnostic for determining the rate-limiting step for DNA polymerases (17). Moreover, Cramer and Restle (15) further report that the koff rate (∼63 s−1) for Dbh•DNA dissociation is at least 10-fold higher than the polymerization rate (kpol), yet are still able to observe a burst in the first turnover. They argue that the unexpected burst is due to a possibly reduced koff rate in the presence of high nucleotide concentrations (15). In order to account for a reduced koff, it is more reasonable to argue that the existence of the induced-fit mechanism leads to the locking of DNA in a tight ternary complex Dbh′•DNA•dNTP. Clearly, more kinetic studies of the Dbh catalytic mechanism need to be performed before a sound conclusion can be reached.
In two previous publications, we used Dpo4 and the methods of pre-steady state kinetics to determine the fidelity (18) and mechanism (14) of single nucleotide incorporation into undamaged DNA at 37°C. These studies demonstrated that similar to reports for other Y-family DNA polymerases, Dpo4 possesses very low incorporation fidelity (10−3 to 10−4), which is several orders of magnitude below that observed for replicative DNA polymerases. However, owing to the fact that the S. solfataricus species propagate in an environment where their physiological temperature varies between 75°C and 85°C (19,20), it becomes important to address what effect temperature may have on the kinetic mechanism of Dpo4 established previously at 37°C (14). Thus, we exploit the thermal stability of Dpo4 in order to probe key mechanistic parameters to determine the effect of temperature on the reaction mechanism. Here, we provide evidence indicating that both the fidelity and induced-fit mechanism remain unchanged with an increase in the reaction temperature although microscopic rate constants are dramatically altered.
MATERIALS AND METHODS
Materials
These chemicals were purchased from the following companies: [α-32P]dTTP and [γ-32P]ATP, GE Healthcare (Picataway, NJ, USA); dNTPs, Gibco-BRL (Rockville, MD, USA); Sp-dTTPαS and Sp-dGTPαS, Biolog – Life Science Institute (Bremen, Germany); ddTTP, Trilink Biotechnologies (San Diego, CA, USA). Full-length Dpo4 fused to a C-terminal His6 tag was overexpressed in Escherichia coli as described in (18). The protein was stored in 20 μl aliquots and stored at −80°C (18). The DNA substrate (D-1) listed in Figure 1 was prepared as described previously (18).
Figure 1.

Concentration dependence on the single turnover rate of dTTP incorporation into the D-1 substrate at 56°C. (A) A preincubated solution of Dpo4 (120 nM) and 5′-[32P]-labeled-D-1 (30 nM) was mixed with various concentrations of dTTP (25 μM, filled circles; 50 μM, open circles; 100 μM, filled squares; 200 μM, open squares; 400 μM, filled triangles; 800 μM, open triangles; 1200 μM, inverted filled triangles) for various time intervals. The solid lines are the best fits to the single exponential equation. (B) The single exponential rates (kobs) were plotted as a function of dTTP concentration. These data were then fit to a hyperbolic equation to yield a kp of 189 ± 13 s−1 and a Kd of 383 ± 65 μM.
Pre-steady state kinetic assays
All experiments using Dpo4, if not specified, were performed in a reaction buffer D containing 50 mM HEPES (pH 7.5 at all temperatures), 5 mM MgCl2, 50 mM NaCl, and 0.1 mM EDTA, 5 mM DTT, 10% glycerol and 0.1 mg/ml BSA. All reactions were carried out at temperatures ranging from 2°C to 56°C using either a rapid chemical quench flow apparatus (KinTek, PA, USA) as described previously (18) or via manual quench. In some cases, we used conditions at subsaturating nucleotide concentration in order to mimic the assays performed previously at 37°C, in addition to the fact that the reaction rates at 56°C were very close to the upper limit that can be measured by instrument, both coupled with those reasons explained previously (18).
Active site titration assay
The equilibrium dissociation constant (Kd) of the Dpo4•DNA binary complex was determined by mixing a preincubated solution of Dpo4 (30 nM) and various concentrations of a 21/41 mer DNA substrate (D-1) with 1.2 mM dTTP and subsequently quenching the reaction after 26 ms (equivalent to seven half-times), in order to achieve the maximal first turnover amplitude. The resulting burst amplitudes were plotted against the concentration of the D-1 substrate and fit via quadratic regression [Equation (5)].
Measurement of the phosphorothioate elemental effect
A preincubated solution of Dpo4 (120 nM) and 5′-labeled D-1 (30 nM) was rapidly mixed with either dTTP (100 µM) or Sp-dTTPαS (100 µM, >95% purity) in buffer D at 56°C. The reactions were quenched with 0.37 M EDTA, analyzed via denaturing gel electrophoresis, and quantitated as described in the Product Analysis section. The data were fit using Equation (1) to yield kobs values. For the incorporation of an incorrect nucleotide, reactions were initiated with either dGTP (100 µM) or Sp-dGTPαS (100 µM, >95% purity). For both correct and incorrect incorporation, the Sp isomer was used as opposed to the Rp isomer due to the stereoselectivity observed previously by Eckstein (21).
Pulse-chase and pulse-quench assays
To provide insight into whether or not a conformational change limited the rate for correct nucleotide incorporation in the first enzyme turnover, two time courses were performed by mixing a preincubated solution of Dpo4 (30 nM) and unlabeled D-1 (30 nM) with a solution containing 40 µM [α-32P]dTTP in buffer D at 56°C. Reactions for both time courses were mixed for times ranging from 20 ms to 2 s. After this variable period, reactions were either immediately quenched with 1 N HCl (pulse-quench) or chased for 15 s (pulse-chase) with a large molar excess of unlabeled dTTP (2.5 mM), before being quenched with 1 N HCl. Dpo4 was subsequently chloroform extracted from each reaction, followed by rapid neutralization with 1 N NaOH/0.1 M Tris. Reactions were quantitated via sequencing gel analysis using a highly crosslinked 20% polyacrylamide matrix as described previously (22) to resolve product from unincorporated [α-32P]dTTP.
Circular dichroism (CD) spectroscopic studies
CD spectra were measured using an AVIV CD spectrometer model 62A DS (Lakewood, NJ, USA). UV spectra were acquired at a fixed wavelength of 222 nm using a 1 mm pathlength cuvette. Samples were dissolved into a degassed buffer (25 mM NaPO4 pH 7.5, 50 mM NaCl, 5 mM MgCl2, 10% glycerol) and filtered with a 0.45 µm membrane to remove residual aggregation. Baseline spectra of buffer alone was obtained and subtracted from sample spectra. The resulting ellipticity (mdeg) was plotted as a function of temperature. To determine the ellipticity of the Dpo4, 30 µM of the protein was added to a solution containing the degassed buffer and was equilibrated at each temperature for 10 min before scanning.
Product analysis
Unless noted, reaction products were analyzed by sequencing gel electrophoresis (17% acrylamide, 8 M urea, 1× TBE running buffer) and quantitated with a Phosphorimager 445 SI (Molecular Dynamics).
Data analysis
Data were fit by nonlinear regression using the program KaleidaGraph (Synergy Software). Data from the single turnover experiments were fit to Equation (1):
| 1 |
Data from the secondary plot for the single turnover experiments were fit to Equation (2):
| 2 |
Data from burst experiments were fit to Equation (3):
| 3 |
Data from the steady-state kinetic experiments were fit to Equation (4):
| 4 |
Data from the active site titration were fit to Equation (5):
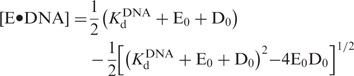 |
5 |
Data from the Arrhenius activation energy treatment were fit to Equation (6):
| 6 |
Here, A represents the enzyme amplitude, k1 is the observed burst rate, k2 is the observed steady-state rate, E0 the active enzyme concentration, kss is the steady-state rate of dNTP incorporation at initial active enzyme concentration of E0, kobs the observed single turnover rate, KdDNA is the equilibrium dissociation constant for E•DNA binary substrate, D0 the DNA concentration, KddNTP is the equilibrium dissociation constant of dNTP from the E•DNA•dNTP complex, Ar is a proportionality constant, Ea is the activation energy, R is the universal gas constant and T is reaction temperature in Kelvin.
RESULTS
Effect of temperature on the nucleotide incorporation fidelity of Dpo4
To determine the effect of temperature on the efficiency and fidelity of nucleotide incorporation catalyzed by Dpo4, single nucleotide incorporation studies were performed under single turnover conditions in order to isolate first-order kinetics without complications from subsequent turnovers. The ground-state binding affinity (1/Kd) for each individual nucleotide was measured through the dNTP concentration dependence of the observed single turnover rate. Dpo4 (120 nM) and 5′-[32P]-D-1 (30 nM) was first preincubated at room temperature, then at 37°C, and finally at 56°C prior to being mixed with various concentrations of a single dNTP to initiate nucleotide incorporation at 56°C. Reactions were quenched at various times and analyzed as described previously (18). For the correct incorporation of dTTP into the D-1 substrate (Figure 1), the observed rates were obtained by fitting the data to a single exponential equation (Materials and Methods section) for each individual time course (Figure 1A). The resulting rates (kobs) were then plotted against the concentration of nucleotide and fit via Equation (2) (Materials and Methods section) (Figure 1B) to yield the maximum nucleotide incorporation rate (kp), which was determined to be 189 ± 13 s−1, and the equilibrium dissociation constant (Kd) of 383 ± 65 μM for the dissociation of dTTP from the Dpo4•D-1•dTTP ternary complex. A similar assay was carried out at 38°C for reasons explained below under the same conditions, where we obtained a kp of 17.7 ± 0.6 s−1 and the Kd of 221 ± 15 μM (Table 1, data not shown), which were both within 2-fold of the corresponding values at 37°C (18). This indicated that with an 18°C increase in reaction temperature, the rate increased 11-fold to a rate that approached the upper limitation for accurate measurement by the rapid chemical quench flow instrument. Yet, the Kd for dTTP binding increased (decreased affinity) by only 1.7-fold. The resulting substrate specificity (kp/Kd) at 56°C was subsequently calculated to be 0.493 µM−1s−1, a value ∼6-fold higher than that calculated at 38°C. In addition, under the assumption that the incoming nucleotide (dNTP) freely diffuses into the Dpo4•D-1 active site (14), the kon was assumed to be equivalent to diffusion control (1 × 108 M−1s−1), and as such, the koff (or k−2 in Scheme 1) of dNTP from the Dpo4•D-1•dNTP ternary complex can be estimated (koff=konKd) to be 38 300 s−1. Notably, we observed full reaction amplitude at all tested temperatures which suggested the integrity of the DNA duplex was not compromised at these higher reaction temperatures. A previous study assessing the overall mutational frequency of Taq DNA polymerase from Thermus aquaticus (23) also suggests that duplex stability had little effect on nucleotide discrimination at 70°C. We believe the stability of the duplex is due to preincubating Dpo4 with DNA at room temperature before exposing the mixture to elevated temperatures. Dpo4 binds DNA tightly (14) and therefore was able to stabilize the duplex DNA in its binding pocket.
Table 1.
Kinetic parameters of Dpo4 for an undamaged D-1 substrate with varying temperature
| dNTP | Kd (μM) | kp (s−1) | kp/Kd (μM−1s−1) | Fidelitya | Average Fidelityb |
|---|---|---|---|---|---|
| 26°C | |||||
| dATP | 8.2 × 102 | 0.003 | 4.0 × 10−6 | 1.9 × 10−4 | 2.9 × 10−4 |
| dCTP | 3.8 × 102 | 0.003 | 7.0 × 10−6 | 3.3 × 10−4 | |
| dGTP | 1.2 × 103 | 0.009 | 7.2 × 10−6 | 3.4 × 10−4 | |
| dTTP | 3.0 × 102 | 6.4 | 2.1 × 10−2 | 1 | |
| 38°C | |||||
| dATP | 9.9 × 102 | 0.012 | 1.2 × 10−5 | 1.5 × 10−4 | 4.2 × 10−4 |
| dCTP | 1.6 × 102 | 0.012 | 7.3 × 10−5 | 9.1 × 10−4 | |
| dGTP | 1.7 × 102 | 0.026 | 1.5 × 10−6 | 1.9 × 10−4 | |
| dTTP | 2.2 × 102 | 17.7 | 8.0 × 10−2 | 1 | |
| 44°C | |||||
| dATP | 3.9 × 102 | 0.042 | 1.1 × 10−4 | 9.2 × 10−4 | 5.5 × 10−4 |
| dCTP | 4.0 × 102 | 0.021 | 5.2 × 10−4 | 4.4 × 10−4 | |
| dGTP | 1.3 × 103 | 0.044 | 3.5 × 10−4 | 2.9 × 10−4 | |
| dTTP | 4.8 × 102 | 59.7 | 0.12 | 1 | |
| 50°C | |||||
| dATP | 5.0 × 102 | 0.13 | 2.6 × 10−4 | 5.8 × 10−4 | 4.4 × 10−4 |
| dCTP | 5.2 × 102 | 0.064 | 1.2 × 10−4 | 2.6 × 10−4 | |
| dGTP | 6.8 × 102 | 0.15 | 2.2 × 10−4 | 4.8 × 10−4 | |
| dTTP | 2.6 × 102 | 118 | 0.45 | 1 | |
| 56°C | |||||
| dATP | 1.2 × 103 | 0.42 | 3.6 × 10−4 | 7.4 × 10−4 | 7.9 × 10−4 |
| dCTP | 2.5 × 102 | 0.16 | 6.3 × 10−4 | 1.3 × 10−3 | |
| dGTP | 1.5 × 103 | 0.24 | 1.6 × 10−4 | 3.3 × 10−4 | |
| dTTP | 3.8 × 102 | 189 | 0.49 | 1 |
aCalculated as (kp/Kd)incorrect/[(kp/Kd)correct+(kp/Kd)incorrect].
bCalculated as ∑[(kp/Kd)incorrect/[(kp/Kd)correct+(kp/Kd)incorrect]dATP, dCTP, dGTP]/3.
Scheme 1.

In addition, each of the three incorrect dNTP incorporations into D-1 were performed at both 38 and 56°C as described earlier, to determine the corresponding kinetic parameters (Table 1, data not shown). On average, there was approximately a 19-fold difference (9- to 35-fold) between the kp values for incorrect incorporations at 56°C and those obtained at 38°C, yet only, on average, a 1.2-fold (0.9- to 1.5-fold) increase between the Kd values at these two temperatures. In an attempt to obtain kp data over a larger range of temperatures, the same incorporation assays were performed at 26°C, 44°C and 50°C, in addition to kp estimations performed at 2°C and 14°C, thus varying the temperature by 6°C increments or multiples therein, to cover a 54°C range. Not surprisingly, kp determination at the lower reaction temperatures was severely limited by extremely slow turnover. The kp estimated for correct (dTTP) and incorrect (dGTP) incorporation at 14°C was 0.189 ± 0.008 s−1 and (4.8 ± 0.8)×10−5 s−1, respectively (data not shown). Due to the insignificant product formation for nucleotide incorporation at these lower temperatures, the fidelity could not be determined. However, based on these rates we estimated an increase of 1000-fold and 5000-fold for correct and incorrect nucleotide incorporation, respectively over this 42°C temperature range. Moreover, at 2°C the kp for correct incorporation was estimated to be 0.010 ± 0.001 s−1 (data not shown) while the incorrect incorporation was not observed after three hours of incubation. Thus, upon decreasing the temperature by 54°C, the rate for correct nucleotide incorporation decreased by an amazing 18 900-fold.
In order to determine the change in the fidelity over this range of temperatures, the substrate specificities were calculated for all incorporations between 26°C and 56°C (Table 1). Interestingly, upon calculating the fidelity at each temperature (Table 1) we observed only a 3-fold difference over this 30°C range.
Biphasic kinetics of nucleotide incorporation at 56°C
We previously established the mechanism of single nucleotide incorporation catalyzed by Dpo4 at 37°C (Scheme 1) (14). Yet, the question as to whether or not this mechanism changes with an increase in reaction temperature remains to be determined. In an effort to elucidate the mechanism at a more physiologically relevant temperature for Dpo4, these same studies were performed at 56°C and not at 80°C (see Discussion section). To determine if Dpo4 follows the same biphasic kinetics that were reported at 37°C (14), a pre-steady state kinetic assay at 56°C for the correct incorporation of dTTP into the D-1 substrate (Figure 1) was performed by mixing a preincubated solution of 5′-[32P]-D-1 (120 nM) and Dpo4 (30 nM) with dTTP (100 µM) in buffer D (Materials and Methods section). Reactions were then quenched with 0.37 M EDTA at times ranging from 5 ms to 4.5 s, analyzed via denaturing PAGE, and quantitated using a Phosphorimager. The resulting data were fit to Equation (3) (Materials and Methods section) and demonstrated the same biphasic nature observed at 37°C, characterized by a fast-phase burst rate of 33.6 ± 5.0 s−1 preceding a slow-phase rate of 0.11 ± 0.01 s−1 (Figure 2A). Thus, following the first turnover of the enzyme, the subsequent multi-turnovers occurred at a rate over 300-fold slower due to the dissociation of the DNA product (DNAn+1, Scheme 1) from the binary complex as observed with other enzymes (9,14,24–28). The observed slow phase rate was verified via a subsequent assay performed under steady-state conditions where the DNA substrate was in molar excess (∼200-fold) over the concentration of Dpo4 (Materials and Methods section). Under these conditions, the observed steady-state rate that was determined by fitting the data to Equation 4 (Materials and Methods section) to be 0.23 s−1 (Figure 2B), which is ∼2-fold different from that observed in the burst experiment. However, the rate-limiting step in the first turnover remained to be elucidated (see below).
Figure 2.

Pre-steady state and steady-state kinetics of dTTP incorporation into D-1 at 56°C. (A) A preincubated solution of Dpo4 (30 nM) and 5′-[32P]-labeled-D-1 (120 nM) was mixed with dTTP (100 μM) for various times followed by quenching with 0.37 M EDTA. The data were fit by nonlinear regression to Equation (3) (Materials and Methods section) with rates of 33.6 ± 5.0 s−1 and 0.11 ± 0.01 s−1 for the exponential and linear phases respectively. (B) dTTP incorporation into D-1 was independently measured under steady-state conditions by preincubating Dpo4 (2.4 nM) and 5′-[32P]-labeled-D-1 (250 nM) and then starting the reactions with the addition of dTTP (0.10 mM). The data were fit to Equation 4 (Materials and Methods section) giving a steady-state rate of 0.23 s−1.
Equilibrium dissociation constant of E•DNA
Before probing the rate-limiting step in the first turnover of Dpo4, we needed to verify that the 19°C increase in temperature did not significantly perturb the ability of Dpo4 to bind DNA as we reported previously (14). For reasons explained previously (14), we can measure the dependence of the burst amplitude on D-1 concentration to ascertain the equilibrium dissociation constant (Kd) of the Dpo4•DNA binary complex at 56°C. As seen in Figure 3, the burst amplitude increased with the addition of the D-1 substrate, reaching maximum amplitude at 400 nM D-1. The solid line (Figure 3) was a fit of the data to Equation (5) (Materials and Methods section) and yielded a Kd value of 39.9 ± 6.3 nM. Using the DNA dissociation rate constant (k−1) of 0.11 s−1 determined above, the apparent second-order association rate constant of the Dpo4•D-1 complex was then calculated to be k1 (or kon) = koff/Kd = 2.8 × 106 M−1s−1, a binding rate constant that was below the diffusion limit and similar to the k1 previously reported for Dpo4 at 37°C as well as for T7 DNA polymerase (9), T4 DNA polymerase (29) and HIV-RT (12). These results indicate that the equilibrium dissociation constant of the Dpo4•D-1 complex at 56°C was <4-fold weaker than what was observed at 37°C. Similar results showing a modestly insignificant change in the equilibrium dissociation constant have been reported for Taq DNA polymerase over a larger range of temperatures (30). Thus, our single turnover conditions as described in Materials and Methods section are valid based on these results.
Figure 3.

Active site titration of Dpo4 at 56°C. Dpo4 (30 nM) was preincubated with various concentrations of 5′-[32P]-labeled-D-1 and subsequently mixed with a solution containing dTTP. The reactions were quenched after 26 ms and the products were analyzed by sequencing gel electrophoresis. The resulting burst amplitudes were plotted as a function of substrate concentration and fit to Equation (5) (Materials and Methods section) which gave a Kd for the Dpo4•D-1 complex of 39.9 ± 6.3 nM.
Determination of the rate-limiting step
We have provided evidence previously that a conformational change limits the correct nucleotide incorporation rate, kp, for Dpo4-catalyzed reactions at 37° C (14). Yet, the identification of the rate-limiting step at a more physiologically relevant temperature remains to be determined. It is possible that as temperature increases, the rate of this proposed rate-limiting conformational change (k3) may approach or overcome the rate of the chemical step (k4) due to increased conformational dynamics. Thus, to determine if the kp at 56°C may be limited by a conformational change, the chemistry step or a combination of both, we measured the value of the α-thio elemental effect. If the α-thio elemental effect at 56°C is similar to what we have observed at 37°C previously (14), this may indicate that the rate-limiting step remains unchanged by the reaction temperature. We performed single turnover studies for the correct incorporation of either dTTP or Sp-dTTPαS (Materials and Methods section). These reactions were quenched at various times and the resulting data were fit with Equation (1) (Materials and Methods section) yielding kobs values of 29.3 ± 2.1 s−1 and 29.1 ± 1.9 s−1 for dTTP and Sp-dTTPαS, respectively (Figure 4A). The α-thio elemental effect (kobsdTTP/kobsdTTPαS) for this correct incorporation was determined to be 1.0 which is close to 1.4 obtained at 37°C previously (14), suggesting that the chemistry step at 56°C as at 37°C (14) was not rate-limiting for the incorporation of correct nucleotides by Dpo4. This conclusion was consistent with additional experimental results described subsequently.
Figure 4.

Elemental effect on the rate for correct and incorrect nucleotide incorporation at 56°C. (A) Dpo4 (120 nM) and 5′-[32P]-labeled-D-1 (30 nM) was mixed with either 100 µM dTTP (filled circles) or Sp-dTTPαS (open circles) in parallel time courses. The data were fit using a single exponential equation [Equation (1)] yielding kobs values of 29.3 ± 2.1 s−1 and 29.1 ± 1.9 s−1 for dTTP and Sp-dTTPαS, respectively giving an elemental effect of 1.0 for correct nucleotide incorporation into D-1. (B) Dpo4 (120 nM) and 5′-[32P]-labeled-D-1 (30 nM) was mixed with either 100 µM dGTP (filled circles) or Sp-dGTPαS (open circles) in parallel time courses. The data were fit by nonlinear regression to the single exponential equation [Equation (1)] yielding kobs values of 0.12 ± 0.01 s−1 and 0.024 ± 0.001 s−1 for dGTP and Sp-dGTPαS, respectively giving an elemental effect of 5.0 for incorrect nucleotide incorporation into D-1.
In addition, we measured the α-thio elemental effect for the incorporation of an incorrect nucleotide, dGTP into the same D-1 substrate (Materials and Methods section). After fitting these data to Equation (1), the observed single turnover rates (kobs) for dGTP and Sp-dGTPαS respectively were 0.12 ± 0.01 s−1 and 0.024 ± 0.001 s−1 (Figure 4B). The corresponding elemental effect of 5.0, which is in the range of 4–11 (31), suggested that the chemistry step likely limited the rate for incorrect nucleotide incorporation (31). This value was almost identical to the value 5.8 derived at 37°C for the same incorrect incorporation of dGTP (14); however, in lieu of the inherent ambiguity surrounding this assay, additional evidence is required to confirm this observation.
Determination of the rates of dissociation from the E′•DNA•dNTP ternary complex
To probe further into whether or not a conformational change is rate-limiting at 56°C, we were able to determine the rate of DNA dissociation from the E′•DNA•dNTP ternary complex under steady-state conditions. In two separate assays, a preincubated solution of Dpo4 (2.4 nM) and 5′-[32P]-D-1 (250 nM) was rapidly mixed with either dideoxyTTP (ddTTP) (1.2 mM) alone or with ddTTP (1.2 mM) and the next correct nucleotide dCTP (1.2 mM). If the incorporation of a correct ddNTP follows the kinetics of the incorporation of a correct dNTP, the latter assay was used to measure the rate of DNA dissociation from a ternary complex that was incapable of performing chemistry due to the incorporation of ddTTP yet could still bind the next correct nucleotide, while the former assay, lacking dCTP, directly measures the DNA dissociation rate from the E•DNA binary complex. In the presence of ddTTP alone, the steady-state rate was measured to be 0.063 ± 0.001 s−1 (Figure 5). To examine whether or not the incorporation of ddTTP into D-1 also followed biphasic kinetics as observed with a correct dNTP in Figure 2A, we performed a similar burst experiment with ddTTP. The product formation with ddTTP (Supplementary Figure 1) does follow the biphasic pattern as observed in Figure 2A. Due to the similarity of the incorporation kinetics observed with ddTTP and dTTP, we concluded that both the slow phase in the burst experiment (Supplemental Figure 1) and the steady-state phase in Figure 5 were limited by DNA dissociation from the E•DNA binary complex. Interestingly, when assayed in the presence of ddTTP and dCTP, a 22-fold slower dissociation rate (0.0028 ± 0.0004 s−1) was observed (Figure 5) and suggested that DNA dissociation was inhibited by the presence of dCTP. This slow dissociation provided strong evidence for a conformational change from the E•DNA•dNTP ternary complex to a tighter binding E′•DNA•dNTP ternary complex, which released DNA with a significantly slower rate constant (k8 in Scheme 1).
Figure 5.

Measurement of the DNA dissociation rate for the E′•DNA•dNTP complex at 56°C. A preincubated solution of Dpo4 (2.4 nM) and 5′-[32P]-labeled-D-1 (250 nM) was mixed with ddTTP (1.2 mM) in the absence (filled circles) or presence (open circles) of the next correct nucleotide dCTP (1.2 mM). The data were fit to Equation (4) (Materials and Methods section) to give a steady-state rate in the absence and presence of dCTP of 0.063 ± 0.001 s−1 and 0.0028 ± 0.0004 s−1, respectively.
Pulse-chase experiments
To further explore the possibility that the previously suggested conformational change limited nucleotide addition at 56°C, we used a well-established approach to attempt to identify an intermediate along the reaction pathway that would indicate the existence of a conformational change. In this set of experiments, a preincubated solution of Dpo4 and unlabeled D-1 was mixed with [α-32P]-dTTP for various time intervals (Materials and Methods section). Reactions were immediately quenched by the addition of 1 N HCl in the first experiment, while in the second experiment, reactions were instead chased with an excess of unlabeled dTTP before quenching with 1 N HCl. The nucleotide trap in the pulse-chase reactions precludes dissociated [α-32P]-dTTP rebinding to the Dpo4•D-1 complex while allowing any competently bound Dpo4•D-1•[α-32P]-dTTP complexes to partition between dissociation and product formation. As such, additional product formation observed between the pulse-chase relative to the pulse-quench assay has been considered evidence for the existence of at least one distinct ternary complex (E′•DNAn•dNTP) between the ground-state complex (E•DNAn•dNTP) and product resulting from the chemistry step (E•DNAn+1•PPi). Our results were fit to Equation (3) to yield a pulse-chase and pulse-quench amplitudes of 26.9 ± 1.8 nM and 23.3 ± 1.3 nM, respectively (Figure 6). This corresponds to 3.6 nM of intermediate complex that was quenched in the latter assay yet could be chased to product, indicating the existence of an intermediate prior to the chemistry step. As argued previously, this intermediate complex was most likely the E′•DNAn•dTTP (14).
Figure 6.

Pulse-chase and pulse-quench experiment at 56°C. A preincubated solution of Dpo4 (30 nM) and unlabeled D-1 (30 nM) was mixed with [α-32P]-dTTP (40 μM) for time intervals ranging from 0.02 to 2 s. The reactions in these two experiments were either quenched directly with 1 M HCl (pulse-quench) or chased with 2.5 mM unlabeled dTTP (pulse-chase) for 15 s, followed by an HCl quench. Both experiments were fit to the burst equation [Equation (3)] yielding amplitudes of 26.9 ± 1.8 nM and 23.3 ± 1.3 nM, for the pulse-quench (open circles) and pulse-chase (filled circles) experiments, respectively.
Thermal stability
CD spectroscopy was performed to analyze the secondary structure stability of Dpo4 over a range of temperatures measured via the observation of the ellipticity at a fixed wavelength (222 nm). This wavelength lies in a region of the spectrum which detects the protein backbone conformation. Interestingly, the ellipticity slowly increases from 14°C to 86°C, where it then begins to increase sharply suggesting that the protein or specific protein domains begin to thermally unfold (Supplementary Figure 2). This correlates fairly well with the physiological temperature of the organism, which varies between 75°C and 85°C. In addition, we performed a stability assay whereby we incubated solutions containing 55 nM Dpo4 at the following temperatures for 10 min: 2°C, 14°C, 26°C, 32°C, 38°C, 44°C, 50°C, 56°C, 62°C, 70°C, 80°C, 90°C, 95°C and 100°C. Subsequent to the incubation, 10 µl aliquots of the above solutions were then added to 5′-[32P]-D-1 (50 nM) and following a 10 min incubation at 37°C, were rapidly mixed with 1 mM dTTP for 1 min. A plot of relative extension as a function of temperature (Supplemental Figure 3) indicated that Dpo4 was stable over a range of temperatures from 2°C to 90°C, but then abruptly became inactivated at 95°C. These observations also correlate very well with results from our CD studies and strongly suggest that the decrease in activity at 95°C was due to thermal denaturation of the structure of Dpo4. Although one may expect a decrease in stability of Dpo4 at 90°C based on our CD results, we only observed a 5% decrease in relative product formation at this temperature. This could be due to the slight molar excess of Dpo4 over DNA in this assay but is also a function of the less significant conformational uncoupling of Dpo4 secondary structure observed in Supplementary Figure 2 at 90°C compared to 95°C. However, when coupled with our observation of a significant increase in kp with temperature, the increase in ellipticity between 14°C and 86°C suggests an increase in the conformational dynamics of Dpo4 as discussed subsequently.
DISCUSSION
Thermostable enzymes catalyze reactions at much higher rates as the reaction temperature of the medium increases (16,23,30,32–38). These higher enzymatic activities are due to faster conformational dynamics of the enzyme at higher reaction temperatures (35,38) thought to be a function of a balanced relationship between molecular stability and structural flexibility (39). Although the nucleotide incorporation rate has not been quantitatively determined for Dpo4 at 80°C, Dpo4 has been shown to remain active after being incubated for 5 min at a temperature in the range of 37°C to 95°C (40). We naturally expect that the microscopic rate constants in Scheme 1 will increase as the reaction temperature approaches the physiologically relevant temperature of Dpo4 from the work of Arrhenius. Yet, the pre-steady state kinetic parameters of single nucleotide incorporation catalyzed by Dpo4 on an undamaged DNA substrate have only been reported at 37°C. Therefore, the effects of temperature on the fidelity and mechanism of Dpo4 catalysis requires elucidation.
Fidelity
We have incrementally determined the fidelity of nucleotide incorporation catalyzed by Dpo4 at several temperatures ranging from 26°C to 56°C using the same single turnover experiments described previously (18). We were able to extract the maximum rate of nucleotide incorporation (kp) and ground-state binding affinity (Kd) for each nucleotide incorporation event into the undamaged DNA substrate, D-1 (Figure 1). These parameters along with the substrate specificities and overall fidelity were calculated and listed in Table 1. The resulting kpcorrect values and average kpincorrect values were then plotted as a function of temperature including those kp values estimated at 2°C and 14°C (Supplemental Figure 4). In addition, the average overall fidelity over this same temperature range was plotted (Figure 7). We found that with an increase in the reaction temperature of 30°C (26°C to 56°C), the kp for correct and incorrect nucleotides increase by 30-fold and 27- to 143-fold, respectively (Table 1). Similar results were observed for another thermophilic Y-family member Dbh, a homolog from S. acidocaldarius, which demonstrated a 40-fold higher rate at 65°C compared to 22°C (16). Conversely, the differences in ground-state binding affinities (Kd) for both correct and incorrect nucleotides differ by only 2- to 3-fold over this temperature range, therefore not significantly altering the ΔΔG value nor the conclusion we reported previously for Dpo4 in regard to its inability to discriminate between correct and incorrect nucleotides in the ground-state (18). Due to the lack of a significant change in the Kd with temperature coupled with the observation of a similar magnitude change of kp for both correct and incorrect incorporations, the overall fidelity (Table 1) from 26°C to 56°C decreased by a very modest 2.7-fold (Figure 7). Interestingly, the error frequency of Taq DNA polymerase, a thermostable enzyme from Thermus aquaticus, has been shown to increase only 2-fold over a temperature range of 15°C, from 55°C to 70°C using a base substitution reversion assay (23). These differences could be a function of the experimental error inherent to these assays or due to the relative insensitivity of this assay, since a forward mutation assay for Taq polymerase showed no observable difference in the error frequency with the same increase in temperature (23). A similar magnitude (∼2-fold) difference in error frequency was also observed for Thermococcus litoralis DNA polymerase with a temperature difference of 17°C (41). Unfortunately, our quest to perform analogous fidelity studies at temperatures exceeding 56°C employing the same single turnover experiments are limited by the following factors: (i) the instrument is not suitable for usage at temperatures above 70°C (18); (ii) based on our kinetic results above for correct dNTP incorporation at 56°C, the expected rate at 80°C (>9900 s−1) will far exceed the capabilities of the instrument since the reaction will complete within the instrument's dead time of mixing (∼1 ms); (iii) at 80°C the 21/41 mer D-1 substrate will be largely melted requiring the use of a considerably longer primer-template that would preclude comparison to our previous results (14,18) not to mention the difficulty in separating the product from substrate via gel electrophoresis for this longer substrate. Thus we hypothesize that the fidelity of Dpo4 at 80°C will not be significantly different from its fidelity determined at 56°C in this report. Incidentally, a combination of steady-state kinetics and studies using a forward mutation assay reveal that the substitution error rate of Dpo4 with undamaged DNA (6.5 × 10−3) at 70°C (42) does not differ significantly from the misinsertion fidelity (10−3 to 10−4) estimated at 37°C (40).
Figure 7.

Temperature dependence of nucleotide incorporation fidelity.
Conformational flexibility and mechanism
As the temperature of a reaction is increased, there is an ensuing exponential increase in the rate for catalysis until the enzyme structure succumbs to thermal denaturation which is thought to be brought about by local or global unfolding of the catalytically competent conformation (39). We have observed a similar exponential-like increase in the rates of Dpo4 for both correct and incorrect nucleotide incorporation when temperature is increased (Supplementary Figure 4). In both cases this exponential increase is preceded by a small angle linear increase in the rate. In this linear phase, which occurs at the lower reaction temperatures, the rates remain similar in magnitude and are relatively slow presumably due to the lack of sufficient energy for a relatively large portion of Dpo4 molecules to overcome the activation energy (Ea) to catalyze the reaction at these temperatures (39). This decrease in the ability of an enzyme performing the steps and conformational changes requisite for catalysis has been correlated with increased rigidity at these low nonphysiological temperatures (43,44). The data in Supplementary Figure 4 indicate that the rates for both correct and incorrect incorporations slowly transition to an exponential phase at roughly 40°C. However, the highest temperature that we have assayed for enzyme activity is 56°C, which is roughly 20°C to 30°C below its physiological temperature. This trend suggests that the rate for correct incorporation at its physiological temperature is over 9000 s−1. Moreover, we observe a dramatic decrease of 18 900-fold in the rate for correct incorporation when the temperature decreases from 56°C to 2°C. Likewise, a 42°C decrease in temperature decreases the rate for incorrect incorporation 5000-fold and eliminates the observation of these events completely at 2°C under our assay conditions. These dramatic decreases are not due to protein denaturation based on the results from our CD spectroscopic analysis and stability assay (Supplementary Figures 2 and 3) but instead are a function of the rigid structure of Dpo4 at low and nonphysiological temperatures.
However, the increased conformational dynamics and decreased rigidity of Dpo4 at 56°C did not change the minimal mechanism for single nucleotide incorporation (Scheme 1) which is analogous to that reported at 37°C and indicates Dpo4 still utilizes an induced-fit mechanism for the selection of correct nucleotides. Free Dpo4 and DNA associate with a second order rate constant of 2.8×106 M−1s−1 to form the Dpo4•DNA binary complex. Once competently bound, free dNTP diffuses into the Dpo4 active site at an association rate constant assumed to be equivalent to diffusion control. Observation of the biphasic nature for correct incorporation from the burst experiment indicates that the slowest step in the overall reaction scheme occurs subsequent to the chemistry step and is due to DNA dissociation from the enzyme since the slow linear phase is quantitatively equivalent to our steady-state kinetic results (Figure 2). Further kinetic interrogation of the burst phase revealed several lines of evidence suggesting that the incorporation of a correct nucleotide was limited by a protein conformational change that preceded the chemistry step. First, we failed to observe an elemental effect for the incorporation of a correct dNTP analog containing a sulfur substitution at the α-phosphate position. This concept was originally proposed by Herschlag, Piccirilli, and Cech (31) after observing that a rate-limiting chemical step involving the making or breaking of a phosphate bond in the hydrolysis of phosphate diesters shows a phosphorothioate elemental effect in the range of 4- to 11-fold. Although the elemental effect has been described as an unreliable diagnostic due to the possible steric clashes of the proposed pentacoordinated sulfur-containing intermediate with the enzyme, we believe that these studies may be relevant in the context of Dpo4, due to its significantly less restrictive active site (3) making it less likely to succumb to the steric effects that limit the reliability of this assay, or at least applicable to be used indirectly to probe whether or not the rate-limiting step is altered by reaction temperature. Secondly, using steady-state kinetics, we determined a significantly smaller DNA dissociation rate constant from a ternary complex formed in the presence of both ddTTP and dCTP (0.0028 ± 0.0004 s−1) than form a ternary complex only in the presence of ddTTP (0.063 ± 0.001 s−1). This observation provides evidence for the existence of two distinct ternary complexes with different affinities for DNA and suggests that the ternary complex in the presence of the next correct nucleotide (dCTP) undergoes certain changes in conformation to decrease its DNA dissociation rate constant while unable to perform the chemistry step. Finally, analysis of the reaction amplitudes of the pulse-quench/pulse-chase experiments have indicated that there exists a population of ternary complexes (E′•DNAn•dNTP) that can proceed to product under chase conditions yet are not observed when quenched in the former assay. This provides direct evidence for the slow formation of an enzyme-bound ternary complex preceding the chemistry step as has been observed with T7 DNA polymerase (9), the Klenow fragment (11), HIV-RT (12) and polη (13). The said conformational change was observed to occur at a rate of 189 s−1 at 56°C and was 30-fold faster than the proposed rate-limiting conformational change measured at 37°C. The argument for the nature of this rate-limiting conformational change was described previously (14) based on structural arguments and will not be discussed in detail here. In comparison to the corresponding rate constants gleaned at 37°C, all the rate constants determined at 56°C are faster (Table 2).
Table 2.
Estimated kinetic constants of Dpo4
| Parameters | Constants 37°Ca | Constants 56°C |
|---|---|---|
| k1 | 1.9 μM−1s−1 | 2.8 μM−1s−1 |
| k−1 | 0.02 s−1 | 0.10 s−1 |
| Kd, DNA | 10.6 nM | 39.9 nM |
| k2 | 100 μM−1s−1 | 100 μM−1s−1 |
| k−2 | 23 000 s−1 | 38 300 s−1 |
| Kd, dNTP | 230 μM | 383 μM |
| k3 | 9.4 s−1 | 189 s−1 |
| k8 | 0.004 s−1 | 0.0028 s−1 |
Activation energy
We would like to present additional evidence for the existence of a rate-limiting protein conformational change for Dpo4 using results from our dNTP concentration dependence studies described above. The kp values extracted for the incorporation of a correct nucleotide at these varying temperatures were treated and fit via the Arrhenius equation [Equation (6), Materials and Methods section] as shown in Figure 8 to yield an activation energy (Ea) of 32.9 kcal/mol. This Ea for the incorporation of a correct nucleotide theoretically represents the energy barrier for either k3, k4 or a combination of both in Scheme 1 since this assignment is dependent upon which step in the first turnover is rate-limiting. Now it is well known that numerous, if not all DNA and RNA polymerases, use a two-metal ion mechanism to catalyze the addition of an incoming nucleotide to a DNA or RNA substrate (45). Although the nucleophilic attack of the pentacovalent transition state can produce a completely associative, completely dissociative, or an intermediate semi-associative transition state complex, Herschlag et al. (31) and references therein in addition to recent crystal structure analyses of β-phosphoglucomutase (46) and a group I intron (47) have indicated the existence of a partially associative (semi-associative) transition state. Although modeled on an associative mechanism and under the assumption of a rate-limiting chemistry step, Florián et al. (48) used computer simulation to conclude that for T7 DNA polymerase, a chemistry step involving the transfer of a proton to activate the 3′-hydroxyl nucleophile, accounts for an activation energy of 12.3 kcal/mol (48). In addition, Radhakrishnan and Schlick (49) used quantum mechanics/molecular mechanics dynamics simulations and quasi-harmonic free energy calculations to show that the rate-limiting chemistry step for correct incorporation catalyzed by DNA polymerase β occurred with a free energy of activation of 17 kcal/mol. Moreover, for uncatalyzed phosphodiester bond formation in solution, the Ea for its rate-limiting chemistry step is estimated to be 21.1 kcal/mol (48). This value should be significantly lower when this rate-limiting chemical reaction occurs in an enzyme active site based on Pauling's transition state theory (50) due to the likely stabilization of the ionic transition state from favorable interactions conferred from the enzyme. Our calculated Ea of 32.9 kcal/mol for the correct incorporation of dTTP into D-1 is considerably larger than that derived via these various simulation methods which each measured the Ea of the chemistry step and as such, suggests that k3 (protein conformational change) rather than k4 (chemistry step) is limiting the correct nucleotide incorporation catalyzed by Dpo4. Although the precise nature of the proposed rate-limiting conformational change is unknown, this conclusion seems reasonable because even local structural rearrangements within an enzyme active site will involve movements of many chemical bonds and should have a relatively large activation energy. In addition, the large Ea reported here suggests that the structure of Dpo4 is rigid at low temperature and becomes more dynamic at higher temperature (35,38,51), leading to increasingly higher kp values at more physiological temperatures (see above discussion). Moreover, our results above gleaned an elemental effect of 5.0 for the incorrect incorporation of dGTP into D-1 which lead us to suggest that the chemistry step limited incorrect nucleotide incorporation. Interestingly, we calculated an Ea of 24.2 kcal/mol for this incorrect incorporation which was close to the range of 12.3 to 21.1 kcal/mol estimated for a rate-limiting chemistry step (see above). Thus we propose that determination of Ea is a new mechanistic method of determining the rate-limiting step in a kinetic mechanism of DNA polymerase-catalyzed polymerization.
Figure 8.

Activation energy for correct nucleotide incorporation. The extracted kp values were plotted as a function of reaction temperature according to Equation (6) (Materials and Methods section) to yield an activation energy (Ea) of 32.9 kcal/mol.
In summary, we have probed the effects of reaction temperature on the fidelity and elementary steps of nucleotide incorporation into undamaged DNA catalyzed by a Y-family DNA polymerase. Our pre-steady state kinetic results reveal that Dpo4 uses an induced-fit mechanism to select correct nucleotides at 56°C. We also have shown that the overall fidelity remains unchanged over a range of 30°C, suggesting that the fidelity at 80°C will also be in the range of 10−3 to 10−4. The direct implication of these results is that the conformational flexibility of Dpo4 does not impinge upon the faithfulness of Dpo4 to incorporate nucleotides into undamaged DNA at higher temperatures.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation Career Award to Z.S. (Grant MCB-0447899). K.A.F. was the American Heart Association Predoctoral Fellow (Grant 0415129B) and The Presidential Fellow from the Ohio State University. J.A.B was a Fellow of the National Institutes of Health Chemistry and Biology Interface Program at the Ohio State University (Grant 5 T32 GM008512-11). Funding to pay the Open Access publication charges for this article was provided by the National Science Foundation Career Award (Grant MCB-0447899).
Conflict of interest statement. None declared.
REFERENCES
- 1.Ogi T, Lehmann AR. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006;8:640–642. doi: 10.1038/ncb1417. [DOI] [PubMed] [Google Scholar]
- 2.Liu G, Chen X. DNA polymerase eta, the product of the xeroderma pigmentosum variant gene and a target of p53, modulates the DNA damage checkpoint and p53 activation. Mol. Cell Biol. 2006;26:1398–13413. doi: 10.1128/MCB.26.4.1398-1413.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 4.Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Mol. Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 5.Silvian LF, Toth EA, Pham P, Goodman MF, Ellenberger T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat. Struct. Biol. 2001;8:984–989. doi: 10.1038/nsb1101-984. [DOI] [PubMed] [Google Scholar]
- 6.Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- 7.Uljon SN, Johnson RE, Edwards TA, Prakash S, Prakash L, Aggarwal AK. Crystal structure of the catalytic core of human DNA polymerase kappa. Structure. 2004;12:1395–1404. doi: 10.1016/j.str.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- 9.Patel SS, Wong I, Johnson KA. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 10.Tsai YC, Johnson KA. A new paradigm for DNA polymerase specificity. Biochemistry. 2006;45:9675–9687. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahlberg ME, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry. 1991;30:4835–4843. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh JC, Zinnen S, Modrich P. Kinetic mechanism of the DNA-dependent DNA polymerase activity of human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 1993;268:24607–24613. [PubMed] [Google Scholar]
- 13.Washington MT, Prakash L, Prakash S. Yeast DNA polymerase eta utilizes an induced-fit mechanism of nucleotide incorporation. Cell. 2001;107:917–927. doi: 10.1016/s0092-8674(01)00613-4. [DOI] [PubMed] [Google Scholar]
- 14.Fiala KA, Suo Z. Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2116–2125. doi: 10.1021/bi035746z. [DOI] [PubMed] [Google Scholar]
- 15.Cramer J, Restle T. Pre-steady-state kinetic characterization of the DinB homologue DNA polymerase of Sulfolobus solfataricus. J. Biol. Chem. 2005;280:40552–40558. doi: 10.1074/jbc.M504481200. [DOI] [PubMed] [Google Scholar]
- 16.Potapova O, Grindley ND, Joyce CM. The mutational specificity of the Dbh lesion bypass polymerase and its implications. J. Biol. Chem. 2002;277:28157–28166. doi: 10.1074/jbc.M202607200. [DOI] [PubMed] [Google Scholar]
- 17.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 18.Fiala KA, Suo Z. Pre-steady-state kinetic studies of the fidelity of sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2106–2115. doi: 10.1021/bi0357457. [DOI] [PubMed] [Google Scholar]
- 19.Zillig W, Stetter KO, Wunderl S, Schultz W, Preiss H, Scholz I. The Sulfolobus-‘caldariella’ group: taxonomy on the basis of the structure of DNA-dependent RNA polymerases. Arch. Microbiol. 1980;125:259–269. [Google Scholar]
- 20.Zillig W, Arnold HP, Holz I, Prangishvili D, Schweier A, Stedman K, She Q, Phan H, Garrett R, et al. Genetic elements in the extremely thermophilic archaeon Sulfolobus. Extremophiles. 1998;2:131–140. doi: 10.1007/s007920050052. [DOI] [PubMed] [Google Scholar]
- 21.Eckstein F. Nucleoside phosphorothioates. Annu. Rev. Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- 22.Arnold JJ, Ghosh SK, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Divalent cation modulation of primer, template, and nucleotide selection. J. Biol. Chem. 1999;274:37060–37069. doi: 10.1074/jbc.274.52.37060. [DOI] [PubMed] [Google Scholar]
- 23.Tindall KR, Kunkel TA. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry. 1988;27:6008–60013. doi: 10.1021/bi00416a027. [DOI] [PubMed] [Google Scholar]
- 24.Wong I, Patel SS, Johnson KA. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 25.Donlin MJ, Patel SS, Johnson KA. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry. 1991;30:538–546. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- 26.Kati WM, Johnson KA, Jerva LF, Anderson KS. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992;267:25988–25997. [PubMed] [Google Scholar]
- 27.Johnson AA, Johnson KA. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 2001;276:38097–38107. doi: 10.1074/jbc.M106046200. [DOI] [PubMed] [Google Scholar]
- 28.Ahn J, Werneburg BG, Tsai MD. DNA polymerase beta: structure-fidelity relationship from pre-steady-state kinetic analyses of all possible correct and incorrect base pairs for wild type and R283A mutant. Biochemistry. 1997;36:1100–1107. doi: 10.1021/bi961653o. [DOI] [PubMed] [Google Scholar]
- 29.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 30.Datta K, LiCata VJ. Thermodynamics of the binding of Thermus aquaticus DNA polymerase to primed-template DNA. Nucleic Acids Res. 2003;31:5590–5597. doi: 10.1093/nar/gkg774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herschlag D, Piccirilli JA, Cech TR. Ribozyme-catalyzed and nonenzymatic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry. 1991;30:4844–48454. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]
- 32.Bryant FO, Adams MW. Characterization of hydrogenase from the hyperthermophilic archaebacterium, Pyrococcus furiosus. J. Biol. Chem. 1989;264:5070–5079. [PubMed] [Google Scholar]
- 33.Cheng TC, Ramakrishnan V, Chan SI. Purification and characterization of a cobalt-activated carboxypeptidase from the hyperthermophilic archaeon Pyrococcus furiosus. Protein Sci. 1999;8:2474–2486. doi: 10.1110/ps.8.11.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh M, Grunden AM, Dunn DM, Weiss R, Adams MW. Characterization of native and recombinant forms of an unusual cobalt-dependent proline dipeptidase (prolidase) from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 1998;180:4781–4789. doi: 10.1128/jb.180.18.4781-4789.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandel AM, Akke M, Palmer A.G., III. Dynamics of ribonuclease H: temperature dependence of motions on multiple time scales. Biochemistry. 1996;35:16009–16023. doi: 10.1021/bi962089k. [DOI] [PubMed] [Google Scholar]
- 36.Harwood VJ, Denson JD, Robinson-Bidle KA, Schreier HJ. Overexpression and characterization of a prolyl endopeptidase from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 1997;179:3613–3618. doi: 10.1128/jb.179.11.3613-3618.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laderman KA, Davis BR, Krutzsch HC, Lewis MS, Griko YV, Privalov PL, Anfinsen CB. The purification and characterization of an extremely thermostable alpha-amylase from the hyperthermophilic archaebacterium Pyrococcus furiosus. J. Biol. Chem. 1993;268:24394–243401. [PubMed] [Google Scholar]
- 38.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 39.Fields PA. Review: protein function at thermal extremes: balancing stability and flexibility. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001;129:417–431. doi: 10.1016/s1095-6433(00)00359-7. [DOI] [PubMed] [Google Scholar]
- 40.Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattila P, Korpela J, Tenkanen T, Pitkanen K. Fidelity of DNA synthesis by the Thermococcus litoralis DNA polymerase–an extremely heat stable enzyme with proofreading activity. Nucleic Acids Res. 1991;19:4967–4973. doi: 10.1093/nar/19.18.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kokoska RJ, Bebenek K, Boudsocq F, Woodgate R, Kunkel TA. Low fidelity DNA synthesis by a y family DNA polymerase due to misalignment in the active site. J. Biol. Chem. 2002;277:19633–19638. doi: 10.1074/jbc.M202021200. [DOI] [PubMed] [Google Scholar]
- 43.Jaenicke R. Protein stability and molecular adaptation to extreme conditions. Eur. J. Biochem. 1991;202:715–728. doi: 10.1111/j.1432-1033.1991.tb16426.x. [DOI] [PubMed] [Google Scholar]
- 44.Jaenicke R, Bohm G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 45.Steitz TA. A mechanism for all polymerases. Nature. 1998;391:231–232. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- 46.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science. 2003;299:2067–2071. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 47.Stahley MR, Strobel SA. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science. 2005;309:1587–1590. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- 48.Florian J, Goodman MF, Warshel A. Computer simulation of the chemical catalysis of DNA polymerases: discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 2003;125:8163–8177. doi: 10.1021/ja028997o. [DOI] [PubMed] [Google Scholar]
- 49.Radhakrishnan R, Schlick T. Correct and incorrect nucleotide incorporation pathways in DNA polymerase beta. Biochem. Biophys. Res. Commun. 2006;350:521–529. doi: 10.1016/j.bbrc.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pauling L. Molecular architecture and biological reactions. Chem. Eng. News. 1946;24:1375–1377. [Google Scholar]
- 51.Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist WI, Kern D. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl Acad. Sci. USA. 2002;99:5247–5252. doi: 10.1073/pnas.082100499. [DOI] [PMC free article] [PubMed] [Google Scholar]