


Abstract
Remodelling protein nucleic acid interfaces is an important biological task, which is often carried out by nucleic acid stimulated ATPases of the Swi2/Snf2 superfamily. Here we study the mechano-chemical cycle of such an ATPase, namely the catalytic domain of the Sulfolobus solfataricus Rad54 homologue (SsoRad54cd), by means of fluorescence resonance energy transfer (FRET). The results of the FRET studies show that the enzyme can be found in (at least) two different possible conformations in solution. An open conformation, consistent with a recently reported crystal structure, is converted into a closed conformation after DNA binding. Upon subsequent binding of ATP no further change in conformation can be detected by the FRET measurements. Instead, a FRET detectable conformational change occurs after ATP hydrolysis and prior to ADP release, suggesting a powerstroke that is linked to phosphate release. Based on these data we will present a new model for the mechano-chemical cycle of this enzyme. This scheme in turn provides a working model for understanding the function of other members of the Swi2/Snf2 family.
INTRODUCTION
A common obstacle that is often encountered by enzymes that translocate along DNA—such as polymerases or helicases—are proteins bound to the DNA molecule. These proteins can act as roadblocks for the DNA translocases. DNA-dependent ATPases of the large Swi2/Snf2 superfamily often act to resolve this problem. These ATPases use the free energy liberated by the ATP hydrolysis to remodel the protein nucleic acid interface in order to mobilize the protein (1). The most prominent example of such a process is the mobilization of nucleosomes by chromatin remodelling complexes (2,3). In addition a similar mechanistic problem exists during transcription coupled repair, where the polymerase is stalled at a lesion in the template strand (4). Another member of the Swi2/Snf2 family, Mot1, regulates transcription by displacing TBP from the TATA box (5). Moreover, in homologous recombination Rad54 is (among several other functions) thought to be responsible for clearing proteins bound to DNA during the homology search (6,7). While Swi2/Snf2 DNA-dependent ATPases have been studied in great detail, until now mechanistic details of the mechano-chemical cycle are scarce. Recent single-molecule, biochemical (8) as well as crystallographic studies, however, give first hints as to how these processes occur (9).
The structure of SsoRad54cd alone and in complex with DNA has been solved recently (10) (Figure 1). The structure consists of two domains (domains 1 and 2) each containing a RecA type subdomain (1A and 2A). The classical helicase motifs are shared between the two subdomains. Motifs I, Ia, II and III are located in 1A and motifs IV, V and VI are found in domain 2A. While the structural similarity between this Swi2/Snf2 enzyme and helicases suggests a common mechanism for ATP hydrolysis (11–14), both crystal forms of SsoRad54cd yield the enzyme in an open conformation. Here, the helicase motifs are too far apart from each other to form an ATP binding site that is capable of ATP hydrolysis. Subdomains 1B and 2B in contrast are not structurally related to previously determined helicase domains and thus the role of these domains might be in facilitating DNA translocation. Dürr et al. (10) proposed a model in which the DNA plus ATP bound conformation of the enzyme is closed and repetitive ATP hydrolysis and product release would translocate DNA in an inchworm fashion.
Figure 1.

Labelling sites and expected conformational changes. (A) Open conformation. A model of the SsoRad54cd based on its crystal structure is shown with a bound DNA molecule in the groove generated by the two domains (yellow and blue). Amino acid residues used for mutagenesis are shown in red. The distance between these residues is 75 Å. (B) Closed conformation. Comparison to structures of other related ATPases predicts an active conformation of the enzyme, where one domain underwent an 180° turn (10). The two labelling sites are now in very close proximity of 15 Å.
Fluorescence resonance energy transfer (FRET) has been widely used to study structural aspects of protein–nucleic acid complexes (15,16). Moreover by using FRET on the single-molecule level (single pair FRET, sp-FRET) one is able to follow in real time the motion of enzymes along nucleic acids (17) and investigate the occurring conformational changes (18–20). Furthermore, related single-molecule techniques have been used to study the motion of Rad54 on a DNA template, manifesting Rad54 as a processive DNA translocase (7,21).
In this article we report on FRET experiments that allowed us to probe the conformation of a Swi2/Snf2 ATPase in different DNA and nucleotide bound states. Our experiments provide direct evidence that in solution SsoRad54cd switches between an open and a closed conformation as predicted by the crystallographic data. Using double labelled proteins, we observed conformational states in a low FRET regime corresponding to the open conformation as well as significantly higher FRET likely caused by the re-orientation of one domain. Moreover, we were also able to detect a state with an intermediate FRET value. To complement our studies we performed affinity measurements, where we investigated the binding properties and the effect of different nucleotide substrates. Furthermore, we present data from single-pair FRET experiments, which enable us to follow conformational changes of the protein in real time, showing that the enzyme structure is indeed very flexible. Based on our observations we propose a new model for the mechano-chemical cycle of this Swi2/Snf2 DNA translocase.
MATERIALS AND METHODS
Protein expression and purification
SsoRad54cd (residues 430–906) and its mutants were expressed and purified as described previously (10). In brief, proteins were synthesized in Escherichia coli BL21 Rosetta cells (Novagen) using the pET28 expression vector (Novagen) by 4 h shaking in LB containing 0.5 mM IPTG at 37°C and purified to homogeneity by immobilized Ni affinity (utilizing a N-terminal 6xHis-tag), Source-S ion exchange and S-200 gel-filtration chromatography (Pharmacia).
Site-directed mutagenesis
In order to attach dye molecules as probes for conformational changes of the protein, cysteines were introduced using site-directed mutagenesis at positions K629 and K697 (Mutant 2×, see Figure 1). Site directed mutagenesis was performed with the QuickChange kit (Stragagene) according to the manufacturers instructions. All mutant proteins were produced in E. coli and purified according to the wild type protein. The three wildtype cysteines are most likely not accessible for dye labelling (see Supplementary Data). However, additional control experiments were performed, for which in addition to the two engineered cysteines, native cysteine residues (C499 and C670) were replaced with serines to avoid the possibility of labelling at these locations, leaving the only native cysteine residue at position 466 [Mutant 4×, (Supplementary Data)]. Mutagenesis on the last of the three native cysteine residues leads to increased formation of protein aggregates (data not shown).
In order to further dissect the mechano-chemical cycle of the SsoRad54cd protein, a third mutant was also expressed, where in addition to the four mutations of Mutant 4×, the well known DExx -> DQxx box mutation at residue 563 was introduced (Mutant EQ), abolishing the ability of the protein to hydrolyse ATP.
Protein labelling
Labelling was conducted for 1 h at 37°C in PBS (pH 8.0) buffer using six-times molar excess of Cy3-Maleimide (Amersham) as donor dye and ten-times molar excess of Cy5-Maleimide (Amersham) or Alexa647-Maleimide (Molecular Probes) as acceptor dye. The ratio of the different dye molecules was optimized in order to have similar labelling efficiencies for both dye molecules. The two cysteine residues at position 629 and 697 were both very reactive. Therefore there is no preference for the donor or acceptor molecule for any of the two cysteines and thus, the labelling results in a statistical distribution of double (50% Cy3/Cy5) and uniformly labelled enzymes (25% Cy3 and 25% Cy5). We achieved more than 90% labelling efficiency for both dyes while retaining DNA stimulated ATPase activity (data not shown). The protein was separated from the free dye molecules via size exclusion chromatography using two consecutive spin-columns (Amersham, G-50).
The degree of labelling was determined using a Varian Inc. Cary 50 UV-VIS spectrophotometer. The protein concentration was measured at 280 nm using the extinction coefficient ε = 57995 M−1/cm. The absorption of the dye at 280 nm was compensated using correction factors relative to their maximum absorption of 0.08 for Cy3, 0.05 for Cy5 and 0.04 for Alexa 647, respectively. For the donor and acceptor dyes, we used extinction coefficients of 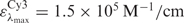 for Cy3,
for Cy3, 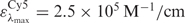 for Cy5 and
for Cy5 and 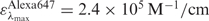 for Alexa 647.
for Alexa 647.
Activity measurements
ATP-hydrolysis was measured with a malachite-green based phosphate detection reagent (Biomol Green, Biomol). About 2.5 × 10−9 M protein was incubated with 5 × 10−8 M poly dA-poly dT-DNA (Amersham) and 1 mM ATP in ATPase buffer (20 mM MES pH 5.5, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA) at 60°C for 30 min in a volume of 50 μl. The reaction was stopped by addition of 500 μl of Biomol Green. After vortexing, the mixture was incubated for 25 min. at room temperature. The amount of released phosphate was determined by comparison to a pre-recorded calibration curve using the maximum absorption value Amax.
FRET measurements
All measurements were conducted at 60°C using a steady state fluorescence spectrometer (Edinburgh Instruments F900) with polarizer at magic-angle conditions. Excitation wavelengths were 530 nm for the donor and 630 nm for the acceptor. Spectral resolution was 3 nm for the excitation and 7 nm for the emission. Freshly labelled protein was used at a concentration of 2.5 × 10−7 M in ATPase buffer together with 1.2 × 10−7 M poly dA-poly dT-DNA (Amersham), corresponding to roughly 120 basepairs per protein molecule. For comparison we tested different types and lengths of DNA but all gave similar results (data not shown). Nucleotides were used at 0.67 mM (ATP and ADP). For experiments with the transition state analogue ADP · AlFx, 0.67 mM ADP, 1.33 mM AlCl3 and 6.66 mM NaF were used.
Calculations of FRET efficiency
FRET efficiencies were calculated by measuring the enhancement of the fluorescence emission of the acceptor in the presence of the donor (as compared with the acceptor only sample) when exciting the donor (15). One advantage of this method is that the fluorescence quantum yields do not have to be determined and that only the labelling efficiency of the donor has to be known, as that of the acceptor does not enter the analysis. As an example the FRET efficiency E for the FRET wavelength pair at 530 nm/670 nm (donor excitation/acceptor fluorescence emission) is calculated as follows (15):
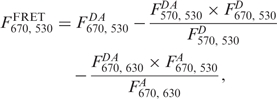 |
1 |
| 2 |
where F is the normalized fluorescence emission intensity and d is the percentage of donor labelling of SsoRad54cd. The upper indices DA (donor-acceptor experiment), D (donor-only control) and A (acceptor-only control) describe the sample composition. Emission and excitation wavelengths are given by the first and second of the lower indices. In Equation (1) the second term stands for the spectral crosstalk of the donor into the spectral region of the acceptor fluorescence and the third term is the direct excitation of the acceptor. The experiments were performed on a statistical distribution of labelled complexes (see above) and thus a factor of two has to be included in Equation (2) in comparison with the equation derived earlier by Clegg (15) in order to compute the correct FRET efficiency. Both donor and acceptor extinction coefficients at the desired wavelengths were calculated from the normalized excitation spectra. FRET efficiencies were calculated in the range of 540–640 nm for donor emission and 640–700 nm for acceptor emission (Supplementary data). Within the spectral range of 565–575 nm for donor emission and 665–685 nm for acceptor emission the observed FRET values varied by only a few percent, and thus the values computed over this wavelength range were averaged. In order to compare FRET efficiencies from different protein preparations, we normalized the observed FRET efficiencies to the value for DNA and protein. This procedure gives reliable information, even if the percentage of active protein varies from one protein preparation to the next.
While FRET efficiencies can be converted into distances, if one knows or determines a characteristic parameter called Förster distance (22), in the present work we are only interested in detecting large conformational changes and not in determining the distance between donor and acceptor molecule at a particular state.
DNA affinity—fluorescence anisotropy measurements
Binding of large proteins to short pieces of DNA drastically changes the rotational diffusion time of the DNA molecule, since the rotational diffusion time increases with molecular weight. The rotational diffusion time can be monitored by measuring the fluorescence anisotropy of an attached dye molecule (23). By titrating protein into a constant DNA concentration one can follow the increase of fluorescence anisotropy upon binding. All anisotropy measurements were conducted both at room temperature as well as 40°C. Higher temperatures were not compatible with the short DNA oligomer due to increased thermal fluctuations of the DNA ends. The instrumental G-factor was determined individually for each measurement. About 6.25 × 10−9 M of a 25 bp duplex oligonucleotide (Biomers) labelled with 6-Tetramethylrhodamine (TAMRA) was mixed with varying amounts of SsoRad54cd protein (5 × 10−8 M–1.2 × 10−6 M) in 60 μl ATPase buffer. A ds DNA oligomer was used, consisting of TAMRA-TAT TTA TGA AAC TTT TCC GGT TTA A as well as the complementary strand without a dye molecule. Nucleotides were used at 1 mM concentration.
For the analysis the mean value over all anisotropy values was taken in the range of 570–610 nm. In the experiment one measures the ensemble anisotropy, which is defined as the average anisotropy of the two species, bound and unbound DNA. Anisotropy measurements were done in triplicates and the mean anisotropy values were then fitted globally using a modified Hill equation in order to take into account the background anisotropy of the unbound DNA:
| 3 |
where r corresponds to the measured anisotropy values of the samples under investigation, the oligonucleotide sample without protein rRef, the samples containing protein rSample and the extrapolated anisotropy value at saturating conditions r100%. P is the total amount of protein used and n is the Hill coefficient.
While the above procedure worked well in the absence of nucleotides (Figure 2), when we performed these experiments in presence of nucleotides we found a reproducible tendency of the enzyme to form aggregates before a saturating level could be reached. Interestingly this behaviour could also be seen in the absence of nucleotides when the saturation level was reached and the protein concentration was increased even further (>1.5 μM). In order to extract dissociation constants in the presence of nucleotides, we used a different approach. The protein and DNA concentration was kept constant at two different ratios, namely at Kd or 0.5 Kd with respect to the binding in the absence of nucleotides, while nucleotides were added at saturating conditions. This approach gives us both, high sensitivity with respect to changes of the dissociation constant as well as a high dynamic range in case of formation of aggregates. The obtained anisotropy values could then be computed maintaining all parameters but Kd, by re-writing Equation (3), yielding:
| 4 |
The total protein concentration is known, so one is able to compute the desired Kd values (Table 1).
Figure 2.

DNA affinity—Fluorescence anisotropy measurements. The recorded fluorescence anisotropy values show characteristic saturation behaviour (blue), which can be fitted with a modified Hill equation [Equation (3)]. An increase of temperature (40°C) leads to a decrease of the dissociation constant (red).
Table 1..
Comparison of DNA binding constants
| Kd (nM)—Wildtype (25°C) | Kd (nM)—Wildtype (40°C) | |
|---|---|---|
| w/o | 3.2 × 102 ± 0.3 × 102 | 1.7 × 102 ± 0.03 × 102 |
| ATP | 2.1 × 102 ± 0.2 × 102 | 0.6 × 102 ± 0.06 × 102 |
| ADP | 1.6 × 102 ± 0.2 × 102 | 0.7 × 102 ± 0.06 × 102 |
Preparation of sample chamber for sp-FRET measurements
Micro-fluidic chambers coated with a mixture of biotinylated and non-biotinylated PEG molecules were prepared as described previously (24). A 100 bp DNA duplex was attached to the biotin-PEG surface of the micro-fluidic chamber via Neutravidin/Biotin attachment. To this end the chamber was incubated with 0.5 mg/ml Neutravidin (Molecular Probes) in PBS (pH 7.4). After exchanging the buffer with ATPase buffer, the protein was loaded followed by extensive washing to remove unbound protein.
For the experiments without DNA, the enzyme itself was biotinylated. A five times excess of Biotin-XX-sulfosuccinimidyl ester (Molecular Probes, USA) was added parallel to the labelling reaction with fluorescent dyes. The modified protein was then loaded into the sample chamber after Neutravidin had been applied. Unbound protein molecules were removed by rinsing extensively with buffer.
Experimental setup for sp-FRET
Single-pair FRET experiments were performed on a homebuilt prism-based total internal reflection fluorescence microscope (TIRFM). Details about the experimental setup have been published previously (24). Alternating between FRET (532 nm) and direct acceptor excitation (633 nm) allows for the discrimination of dynamics in FRET efficiencies due to conformational changes against changes in FRET efficiencies caused by fluctuations in acceptor brightness (22). All measurements were recorded with an exposure time of 100 ms per frame for 25 s in duration.
Single-molecule data analysis
The acquired data were analysed using custom software written in MATLAB. We used a fully automated routine to find FRET pairs and to calculate and subtract the local background. For the calculation of FRET efficiency of the individual FRET pairs, we used the following formula (24,25):
| 5 |
where 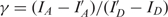 and
and 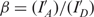 . IA and ID are background corrected intensities from the acceptor and donor channels and IA, D and I′A, D are the intensities before and after acceptor photobleaching, respectively. β and γ are experimental correction factors: β accounts for the leakage of the donor emission into the acceptor channel, while γ is a factor that includes the quantum yields of the fluorophores and the detection efficiencies of the two channels. We determined the correction factors for all FRET pairs individually by time averaging the intensities I and I′. FRET pairs where no acceptor bleaching was observed were discarded from the analysis, since for these FRET pairs γ could not be determined. Direct excitation of the acceptor was so low (<5%) that it did not lead to detectable changes in the histograms (ΔE < 1%) and was therefore disregarded in the analysis. FRET efficiencies were calculated for every time point without filtering the original data sets. The histograms of these FRET values represent transfer efficiency distributions.
. IA and ID are background corrected intensities from the acceptor and donor channels and IA, D and I′A, D are the intensities before and after acceptor photobleaching, respectively. β and γ are experimental correction factors: β accounts for the leakage of the donor emission into the acceptor channel, while γ is a factor that includes the quantum yields of the fluorophores and the detection efficiencies of the two channels. We determined the correction factors for all FRET pairs individually by time averaging the intensities I and I′. FRET pairs where no acceptor bleaching was observed were discarded from the analysis, since for these FRET pairs γ could not be determined. Direct excitation of the acceptor was so low (<5%) that it did not lead to detectable changes in the histograms (ΔE < 1%) and was therefore disregarded in the analysis. FRET efficiencies were calculated for every time point without filtering the original data sets. The histograms of these FRET values represent transfer efficiency distributions.
The total intensity is computed by Itotal = ID × γ + IA.
RESULTS
Dye labels as sensitive reporters for the conformation of a Swi2/Snf2 ATPase
Structural studies of SsoRad54cd predicted large conformational changes during the enzymatic cycle of this enzyme (10). In the structure of the protein alone as well as in complex with DNA, the protein is found in an open conformation (Figure 1A), in which domain 2 is flipped by ∼180° in comparison to the orientation of the equivalent domain in ATP bound DExx box helicases (26). Therefore a conformational change is necessary, presumably a rotation of domain 2 by ∼180° (Figure 1B) to bring the protein into an active conformation capable of hydrolysing ATP (10). In order to probe the conformational changes of SsoRad54cd during its mechano-chemical cycle we performed site-directed mutagenesis introducing new cysteines at two positions (K629 and K697, Mutant 2×) and attached Cy3 and Cy5 molecules as a donor acceptor pair (see Materials and Methods section). The relative positions of these residues were expected to change significantly (Figure 1) upon the re-orientation of domain 2. As a consequence, the fluorescence energy transfer between the fluorescent labels attached at these sites should be highly influenced. One expects relatively low FRET values in the open-conformation, and a drastic change to higher FRET values in the closed conformation.
FRET measurements reveal changes in conformation during mechano-chemical cycle
Figure 3 shows the results of the fluorescence measurements of the Mutant 2×. Comparison of the fluorescence emission spectra of protein alone and in presence of DNA shows a drastic increase of fluorescence emission in the spectral region of the acceptor (670 nm) (Figure 3A). From these spectra one can determine the FRET efficiency (see Materials and Methods section). For the protein alone, the determined FRET value is rather low, as expected for the open conformation. Upon addition of DNA, the FRET efficiency increases dramatically (factor of 3 increase), consistent with the idea that the protein adopts a more closed conformation. This result is in contrast to crystallographic data which still showed an open conformation of the protein bound to a 25 bp DNA duplex (10).
Figure 3.

Increase of FRET efficiency upon binding to DNA (Mutant 2×). (A) Normalized emission spectra of labelled enzyme. The emission spectra of donor only labelled enzyme (black, used for correction of spectral cross-talk), as well as those of the double labelled protein alone (red) and in presence of DNA (green) are shown. (B) Overview of FRET efficiencies. Data has been normalized to the sample containing DNA (see Materials and Methods section); all bars represent the mean value of three different measurement series (shown as symbols), while each bar itself corresponds to a different sample composition.
The addition of saturating amounts of ATP as a nucleotide substrate does not lead to a further increase in FRET efficiency, which implies that the essential conformational change has already occurred. Instead in the presence of ATP the observed FRET efficiency decreases slightly with respect to the protein/DNA measurement. When using the nucleotide ADP, the FRET value remains (within our experimental errors) the same as for the ATP measurements and thus it is below the reference value of DNA and protein, and above the one for the protein alone. At first sight the data could indicate that there are three possible conformations of the protein that can be discerned by our FRET measurements: the unbound protein is in an open conformation, the DNA bound protein is in a closed conformation without additional nucleotides and in a semi-open conformation in the presence of ADP or ATP. However, there are several things that need to be tested in order to rule out other interpretations:
The nucleotide bound states could have lower affinity for the DNA. This would lead to more unbound protein and thus the mean FRET value would be reduced.
The protein undergoes a series of chemical and mechanical steps as part of its mechano-chemical cycle and the related transition rates are currently not known. Thus, the obtained results in presence of ATP and ADP might not be corresponding to the ATP or ADP bound state but rather to a downstream state that resembles the longest lived state within the cycle.
The existence of a third intermediate mechanical state is not strictly necessary. Due to the lack of time-dependent information the measured values could be explained simply by a change of the equilibrium constant for the closed-open transition.
In the following we will present further experiments that will address these issues directly.
The affinity of SsoRad54 towards DNA increases in the presence of ATP or ADP
We determined DNA dissociation constants in equilibrium using fluorescence anisotropy of dye labelled DNA oligomers (see Materials and Methods section). The measured data for the wildtype protein in absence of nucleotides is shown in Figure 2. DNA binding can be well described by Hill equation [Equation (3)] yielding a dissociation constant of 319 ± 31 nM. Our measurements show a significant increase of affinity upon addition of nucleotide, regardless of nature. Therefore the observed lower FRET values in the presence of ATP or ADP cannot be explained by an increased unbinding in the presence of nucleotides. The results also show an increased DNA affinity at higher temperature but again, affinities in presence of nucleotides are higher than for the DNA only sample, thus validating the low temperature results.
Re-arrangement of domains due to phosphate release and not due to ATP binding
In order to test the hypothesis that the ATP and/or ADP bound states could be short-lived and thus our experiments could probe not these states, but rather bottleneck states downstream in the reaction cycle, we performed measurements with a mutant that was able to bind, but not to hydrolyze ATP. To this end we introduced the previously characterized EQ mutation in the DExx box motif (10), which was applied in addition to the mutations of Mutant 2×. Moreover two native cysteines were deleted in order to reduce unspecific labelling (Mutant EQ). The computed FRET values for this protein are shown in Figure 4. For the protein alone we again find a low FRET efficiency and in the presence of DNA a high FRET efficiency, in agreement with our results of Mutant 2×. However, further addition of ATP does not lower the observed FRET value in contrast to the results for Mutant 2×. Moreover, even the transition state analogue ADP · AlFx shows the same FRET efficiency as the DNA bound protein in absence of nucleotides. Only the addition of ADP leads to a FRET value in between that of the DNA bound protein and that of the protein alone.
Figure 4.

Overview of FRET measurements of the Mutant EQ sample. Normalized FRET efficiencies of protein alone (red), in the presence of DNA (green), DNA and ATP (blue), DNA and ADP (cyan) and DNA, ADP and AlFx (magenta) are shown. The values for three separate sets of measurements are indicated as circles, triangles and squares.
These results indicate that in the case of an active protein ATP binding is followed by a rapid hydrolysis and product release. In the performed ensemble experiments we only measure the time average of the catalytic cycle and thus the short lived ATP bound state does not influence our result in case of the active protein. In contrast Mutant EQ can bind but not hydrolyse ATP and thus in presence of saturating amounts of ATP we are truly probing the ATP bound state.
As a test for these claims we measured the ATPase activity for all investigated proteins as described in the Materials and Methods section. The results are summarized in Table 2. The introduction of two non-native cysteines in Mutant 2× slightly reduces its activity compared with the wildtype. The activity of Mutant 4× which in addition has two of the native cysteines removed from the vicinity of the active center is already 30-fold below that of Mutant 2×. Nevertheless its activity is still above background level. Interestingly we observed FRET values for this mutant that are almost identical to those of the Mutant EQ just discussed (data shown in Supplementary Data), further supporting the idea that the observed difference in the FRET efficiency in presence of ATP is caused by the different times needed to hydrolyze ATP. The ATPase activity of Mutant EQ was at the level of our experimental background, thus confirming the expected effect of the EQ mutation.
Table 2..
Comparison of ATPase-activities of SsoRad54cd wildtype and its mutants
| Mutant | Activity
|
|---|---|
| Wildtype | 1.1 × 103 ± 0.1 × 103 |
| Mutant 2× (labelled) | 7 × 102 ± 0.1 × 102 |
| Mutant 4× (labelled) | 2.2 × 101 ± 0.1 × 101 |
| Mutant EQ | <3 × 100 ± 2 × 100 |
Therefore not ATP binding, but hydrolysis or phosphate release results in a conformational change that leads to the observed intermediate FRET values in the presence of DNA and ADP. However, the question still remains if the observed FRET value is due to a stable conformation at an intermediate distance or due to a change of equilibrium constant between open and closed conformation. We therefore used single-molecule FRET experiments in order to address this question.
Single-pair FRET measurements reveal dynamic equilibrium between open and closed states
We employed single-molecule fluorescence microscopy of individual dye labelled SsoRad54cd molecules alone, or bound to DNA and immobilized to a fused silica surface (see Materials and Methods section for details). By measuring the fluorescence intensities for both donor and acceptor emission as a function of time we were able to calculate the time varying FRET efficiencies for each individual molecule. With this technique we are able to observe directly the dynamic opening and closing of the protein molecule. Figure 5 shows a representative time trajectory.
Figure 5.

Single-pair FRET data reveals conformational changes in real time. The detected fluorescence intensity of the donor (green) and acceptor (red) show strong fluctuations with time, resulting in variations of FRET efficiency (black). At the same time the total intensity (blue) is rather constant. Direct excitation of the acceptor molecule (magenta) furthermore shows that the dynamics in the FRET signal are not caused by transient acceptor dark states. After 65 frames the acceptor photobleaches and therefore no more FRET values are computed beyond this time point. After 130 frames, the donor bleaches as well.
The data clearly show strong fluctuations in FRET efficiencies over time for a single complex. In our experimental scheme, we immobilized the DNA molecules using biotin-neutravidin linkers. In a separate experiment, we employed a different fluorescent dye (ATTO647N) that was more photostable in order to investigate if some of the observed dynamics were actually caused by protein unbinding and rebinding. The ATTO647N labelled protein detaches from the DNA with a rate <0.1/s (data shown in Supplementary Data). Therefore our data clearly show that, while the protein is bound to DNA, it is free to open and close during the experimental time frame consistent with an open conformation found in the DNA bound crystal form (10).
For a more quantitative assessment of the observed behaviour, we recorded many single-molecule FRET traces and computed histograms shown in Figure 6. The time-average over many molecules should resemble the results of our ensemble measurements, namely a clear increase in FRET efficiencies upon DNA binding. We therefore also investigated the FRET efficiency of the protein alone, by immobilizing the protein directly in the absence of DNA.
Figure 6.

Histograms of single-molecule FRET values. (A) Protein bound to DNA. The histogram of FRET efficiencies for more than 140 molecules can be described by two Gaussians of similar amplitude centred at 92% and 66%, respectively. (B) Protein without DNA. The FRET efficiency of 80 molecules can also be described by a double Gaussian centred at 32 and 81%, respectively. Both the insets of (A) and (B) show a fit (red) to the cumulative probability distributions (black) using the error function.
The distribution of FRET efficiencies in the presence of DNA (Figure 6A) is extremely broad. The observed distribution is incompatible with a single Gaussian but can be fitted with two Gaussians, one centred at ∼92% and the other at ∼66% FRET efficiency. The peak centred at ∼92% FRET efficiency is presumably due to the closed conformation of the complex. For an open complex we expect to see extremely low FRET values below 20%. Thus the broad peak at lower FRET values (centred at ∼66% FRET efficiency) cannot be attributed to the open conformation, but rather to fast conformational changes of the protein that cannot be resolved in our experiments. Single-molecule data also allows for the analysis of the switching kinetics between different states. Our analysis (shown in the Supplementary Data) yields a characteristic switching time between open and closed conformation in the order of τ = 0.1 s. In comparison the histogram for the protein only sample (Figure 6B) looks very different. Again, the distribution is extremely broad. Fitting the observed histogram with two Gaussian populations we find peaks located at ∼32% and ∼81%. The predominant first peak we interpret as the open conformation of the enzyme. The extremely broad second peak is probably caused by the dynamic transitions to the closed state. In comparison to the results in presence of DNA, now both peaks are fairly broad caused by the high flexibility of the protein in absence of DNA. In order to rule out that some of the observed effects are caused by the choice of dye labels, we repeated the experiments using Cy3 and Alexa647 as donor-acceptor pair and found similar results (data not shown). We also tried to investigate this dynamic process with a higher temporal resolution (30 ms), but this only lead to even broader distributions of FRET efficiencies (data not shown). This indicates that conformational dynamics must also occur on an even faster time scale. Moreover, we repeated the experiments for molecules diffusing freely in solution using burst analysis (data shown in Supplementary Data). Again we find broad histograms, indicative of large fluctuations. Unfortunately we were not able to observe any changes in the presence of ATP (as compared with the DNA bound complexes), presumably due to the low activity of the investigated protein at the experimental temperature (25°C).
The results of both our ensemble as well as of our single molecule experiments lead to the same conclusion. The protein in solution exhibits two different conformational states in the DNA bound and unbound form. The results are independent of the particular choice of the donor-acceptor pair, since measurements with a different acceptor yielded similar results (Supplementary Data). The conformation is however not static, but our data allows us to infer frequent opening and closing events.
DISCUSSION
As a model system for the conformational changes that a Swi2/Snf2 protein undergoes during its mechano-chemical cycle we investigated the archaeal protein SsoRad54cd using FRET. We show that the protein exists in an open conformation in its unbound form in accordance with the crystal structure obtained by Dürr et al. (10). Binding to DNA leads to a conformational change resulting in a drastic increase of measured FRET efficiency. A rotation of domain 2 with respect to domain 1 by ∼180° as proposed from the crystallographic analysis (10) (Figure 1B) would both lead to an active conformation of the protein consistent with other helicases and in accord with the observed FRET changes. Other studies have shown that DNA translocation of eukaryotic Rad54 is quite processive (7,21). Therefore the binding of the protein to the DNA is strictly speaking not part of the regular mechano-chemical cycle and it remains to be seen if such a DNA induced large conformational change is a unique feature of the SsoRad54 protein, or happens in other Swi2/Snf2 proteins as well. For instance, the structure of the zebrafish Rad54 homologue displays a more closed conformation of the domains 1 and 2, even in the absence of DNA (27). However, our single-molecule FRET studies show that even the bound protein undergoes strong and rapid conformational changes. The ability of the protein to undergo large conformational changes is likely of importance for DNA translocation.
Moreover we investigated the conformational changes upon ATP binding, hydrolysis and ADP release. Our results can be summarized in a model for DNA translocation shown in Figure 7 which besides our own data takes into account structural comparisons to helicases (9), previous single-molecule (7,21) and biochemical data of related Swi2/Snf2 enzymes (8).
Figure 7.

Mechanistic model for DNA translocation of a Swi2/Snf2 enzyme. The initial binding positions of domain 1 and 2 to DNA are indicated by coloured spheres. The contact region on the protein is represented as a small cylinder. The ‘open’ conformation of the protein in the absence of DNA is not shown.
The results from our FRET studies as well as structural comparisons to related superfamily 2 helicases provide strong evidence that when bound to the DNA, the protein is in a closed conformation (Figure 1B). A model of this closed conformation is obtained by taking into account the structurally observed DNA contacts to domain 1 and motif Ia, and by comparison with related superfamily 2 helicases crystallized in the presence of nucleic acids (12–14,28). The latter indicates that the conserved helicase motif IV is a likely candidate for DNA contact of domain 2 in the ATPase competent conformation (10,27) (Figure 7A). ATP binding presumably leads to a further compaction of the binding pocket and as a result the binding site on domain 1 slides on the DNA in the 3′->5′ direction (Figure 7B), resulting in a structure where motifs Ia and IV are positioned as observed in the AMP-PNP bound crystal structures of VASA and exon junction complex RNA helicases (12–14). The resulting model for nucleotide binding and hydrolysis induced directional translation of DNA is similar to the proposed inchworm model of superfamily 1 and 2 helicases (29,30). Such a small change of conformation during this processive translocation cannot be detected by our FRET measurements. Binding of ATP is followed by a rapid hydrolysis and the transition state persists still in the closed conformation as indicated by our measurements in presence of the transition-state analogue ADP · AlFx (Figure 7C). Hydrolysis or phosphate release leads to a conformational change that causes unbinding of domain 2 from the DNA (Figure 7D). In the ADP bound form we therefore measure an intermediate FRET value caused by either a semi-open conformation or a shift in the dynamic equilibrium between open and closed state. Once ADP is released domain 2 binds again with motif IV positioned downstream from the binding site on domain 1. Thus in the whole cycle the protein translocates in the 3′->5′ direction with respect to the strand bound to motifs Ia and IV. Recent single-molecule data on a related superfamily 2 helicase shows that DNA translocates by 1 bp per enzymatic cycle (17). The high affinity DNA binding site on domain 1 remains attached to the protein at all times. In contrast, domain 2, which showed much less affinity for DNA (10), unbinds transiently leading to a partial opening of the protein.
It should be pointed out that the described mechano-chemical cycle applies for an enzyme that functions as a single subunit DNA translocase. There is experimental evidence, that Swi2/Snf2 DNA translocases can operate also as multimers (7,31,32), however in the experiments described in this article we see no evidence for multimerization under the chosen experimental conditions. Non-specific large protein aggregates were observed only at very high protein concentrations but it is unclear whether this aggregation influences the mechanism of DNA translocation. For the case of Swi2/Snf2 helicases that operate in multimeric forms it will be interesting to study if and how the mechano-chemical cycles of the individual ATPases are coordinated.
In summary our data allows us to formulate a working model for the mechano-chemical cycle of a Swi2/Snf2 DNA translocase (Figure 7). For comparison, single-molecule force spectroscopy has in the past been applied successfully in order to reveal details of the mechano-chemical cycle of a related DNA translocase (33). Interestingly these studies also revealed that translocation occurs at a step following ATP binding and hydrolysis. Moreover, based upon biochemical studies of nucleosome remodelling by Isw2 (8) a reaction cycle has been proposed for the movement of the nucleosome in which the ADP bound form has less affinity to the substrate. Again, conformational changes following ATP hydrolysis and phosphate release are thought to be responsible for translocation. Therefore the model described in this article shares similarities between a large class of proteins that translocate along DNA.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
ACKNOWLEDGEMENTS
The project was funded by the Deutsche Forschungsgemeinschaft (SFB 646). K.-P.H. acknowledges financial support by the Integrated Project ‘DNA Repair’ of the European Commission. We would like to thank Katrin Gutsmiedl, Tanvi Shah, Markus Horn and Juan Torreno Pina for help with the ensemble FRET measurements, Matthias Höller and Don Lamb for help with the burst analysis, Axel Kirchhofer for help with protein expression and purification and Christoph Bräuchle for fruitful discussions. Funding to pay the Open Access publication charges for this article was provided by Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
REFERENCES
- 1.Svejstrup JQ. Chromatin elongation factors. Curr. Opin. Genet. Dev. 2002;12:156–161. doi: 10.1016/s0959-437x(02)00281-2. [DOI] [PubMed] [Google Scholar]
- 2.Becker PB, Horz W. ATP-dependent nucleosome remodeling. Annu. Rev. Biochem. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- 3.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 4.Svejstrup JQ. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 2002;3:21–29. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- 5.Sprouse RO, Brenowitz M, Auble DT. Snf2/Swi2-related ATPase Mot1 drives displacement of TATA-binding protein by gripping DNA. EMBO J. 2006;25:1492–1504. doi: 10.1038/sj.emboj.7601050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan TL, Kanaar R, Wyman C. Rad54, a Jack of all trades in homologous recombination. DNA Repair (Amst.) 2003;2:787–794. doi: 10.1016/s1568-7864(03)00070-3. [DOI] [PubMed] [Google Scholar]
- 7.Amitani I, Baskin RJ, Kowalczykowski SC. Visualization of Rad54, a chromatin remodeling protein, translocating on single DNA molecules. Mol. Cell. 2006;23:143–148. doi: 10.1016/j.molcel.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Fitzgerald DJ, DeLuca C, Berger I, Gaillard H, Sigrist R, Schimmele K, Richmond TJ. Reaction cycle of the yeast Isw2 chromatin remodeling complex. EMBO J. 2004;23:3836–3843. doi: 10.1038/sj.emboj.7600364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hopfner KP, Michaelis J. Mechanisms of nucleic acid translocases: lessons from structural biology and single-molecule biophysics. Curr. Opin. Struct. Biol. 2007;17:87–95. doi: 10.1016/j.sbi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Durr H, Korner C, Muller M, Hickmann V, Hopfner KP. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell. 2005;121:363–373. doi: 10.1016/j.cell.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 11.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 12.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 13.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 14.Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Meth. Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 16.Lilley DM, Wilson TJ. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 2000;4:507–517. doi: 10.1016/s1367-5931(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 17.Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513–516. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanchard SC, Kim HD, Gonzalez R.L., Jr, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc. Natl Acad. Sci. USA. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science. 2006;314:1144–1147. doi: 10.1126/science.1131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hugel T, Michaelis J, Hetherington CL, Jardine PJ, Grimes S, Walter JM, Falk W, Anderson DL, Bustamante C. Experimental test of connector rotation during DNA packaging into bacteriophage phi29 capsids. PLoS Biol. 2007;5:e59. doi: 10.1371/journal.pbio.0050059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad TK, Robertson RB, Visnapuu ML, Chi P, Sung P, Greene EC. A DNA-translocating Snf2 molecular motor: Saccharomyces cerevisiae Rdh54 displays processive translocation and extrudes DNA loops. J. Mol. Biol. 2007;369:940–953. doi: 10.1016/j.jmb.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 2005;88:2939–2953. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeTilly V, Royer CA. Fluorescence anisotropy assays implicate protein-protein interactions in regulating trp repressor DNA binding. Biochemistry. 1993;32:7753–7758. doi: 10.1021/bi00081a021. [DOI] [PubMed] [Google Scholar]
- 24.Andrecka J, Lewis R, Bruckner F, Lehmann E, Cramer P, Michaelis J. Single-molecule tracking of mRNA exiting from RNA polymerase II. Proc. Natl Acad. Sci. USA. 2008;105:135–140. doi: 10.1073/pnas.0703815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabanayagam CR, Eid JS, Meller A. Long time scale blinking kinetics of cyanine fluorophores conjugated to DNA and its effect on Forster resonance energy transfer. J. Chem. Phys. 2005;123:224708. doi: 10.1063/1.2136157. [DOI] [PubMed] [Google Scholar]
- 26.Caruthers JM, McKay DB. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 27.Thoma NH, Czyzewski BK, Alexeev AA, Mazin AV, Kowalczykowski SC, Pavletich NP. Structure of the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54. Nat. Struct. Mol. Biol. 2005;12:350–356. doi: 10.1038/nsmb919. [DOI] [PubMed] [Google Scholar]
- 28.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 29.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 30.Hopfner KP, Tainer JA. DNA mismatch repair: the hands of a genome guardian. Structure. 2000;8:R237–R241. doi: 10.1016/s0969-2126(00)00545-1. [DOI] [PubMed] [Google Scholar]
- 31.Ristic D, Wyman C, Paulusma C, Kanaar R. The architecture of the human Rad54-DNA complex provides evidence for protein translocation along DNA. Proc. Natl Acad. Sci. USA. 2001;98:8454–8460. doi: 10.1073/pnas.151056798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiianitsa K, Solinger JA, Heyer WD. Terminal association of Rad54 protein with the Rad51-dsDNA filament. Proc. Natl Acad. Sci. USA. 2006;103:9767–9772. doi: 10.1073/pnas.0604240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chemla YR, Aathavan K, Michaelis J, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Mechanism of force generation of a viral DNA packaging motor. Cell. 2005;122:683–692. doi: 10.1016/j.cell.2005.06.024. [DOI] [PubMed] [Google Scholar]