


Abstract
Genome-wide association (GWA) studies to map genes for complex traits are powerful yet costly. DNA-pooling strategies have the potential to dramatically reduce the cost of GWA studies. Pooling using Affymetrix arrays has been proposed and used but the efficiency of these arrays has not been quantified. We compared and contrasted Affymetrix Genechip HindIII and Illumina HumanHap300 arrays on the same DNA pools and showed that the HumanHap300 arrays are substantially more efficient. In terms of effective sample size, HumanHap300-based pooling extracts >80% of the information available with individual genotyping (IG). In contrast, Genechip HindIII-based pooling only extracts ∼30% of the available information. With HumanHap300 arrays concordance with IG data is excellent. Guidance is given on best study design and it is shown that even after taking into account pooling error, one stage scans can be performed for >100-fold reduced cost compared with IG. With appropriately designed two stage studies, IG can provide confirmation of pooling results whilst still providing ∼20-fold reduction in total cost compared with IG-based alternatives. The large cost savings with Illumina HumanHap300-based pooling imply that future studies need only be limited by the availability of samples and not cost.
INTRODUCTION
Genome-wide association (GWA) studies using arrays are now widely used to map loci contributing to complex disease in human populations [1–3]. However, high cost limits widespread use of GWA. One approach which substantially reduces the cost is DNA pooling. In pooling, instead of individually genotyping every person in the sample, the sample is genotyped in pools of individuals. In most previous applications of array-based pooling, researchers have focused upon Affymetrix arrays [4–10], with a number of groups publishing statistical and computational methods for analysing Affymetrix data [4,7,11–13].
Pooling was originally proposed for small-scale genotyping approaches (i.e. not array based) and results were mixed. This led to some scepticism about the value of pooling in practical applications. Several different groups have shown pooling to be tenable using large-scale microarrays [3–5,14,6–10,12]. Array-based pooling addresses concerns about practical applications because (i) the error introduced through pool construction is negligible [13] and (ii) the array error can be tightly controlled by using sufficient numbers of replicate arrays. Obtaining good overall pooling performance is therefore dependent upon achieving low levels of array-specific error.
Here we investigate the performance of the two main array platforms available for pooling, Affymetrix and Illumina, by applying arrays from both to the same pools. We describe new methods for analysis of individual bead level data on Illumina HumanHap300 arrays, including a quality control measure. Compared with Affymetrix Genechip HindIII arrays, we demonstrate substantial improvements in pooling efficiency. We show that when a sufficient number of arrays is used to control the pooling error, the results from pooling can be very similar to those obtained from individual genotyping (IG).
MATERIALS AND METHODS
DNA pools were constructed from 384 endometriosis cases and 384 ethnically matched controls. All samples were caucasian. Full details of the pool construction are given elsewhere (7,15). The same pools were typed using Illumina HumanHap300 arrays (∼317 000 SNPs) and Affymetrix Genechip HindIII arrays (∼57 000 SNPs). A total of 15 645 SNPs overlapped between the two array types. Three replicate arrays of each type were applied to each pool. The control pool data discussed in this publication have been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE9307.
Samples were individual genotyped (IG) on the Sequenom platform; full details are given elsewhere (16). Fifty-one SNPs were individually genotyped (IG) based on HumanHap300 pooling results. Out of the 51 SNPs, 41 were selected as a result of their being in the top 50 SNPs from pooling and a further 10 were selected on the basis of their being in the top 1000 SNPs from pooling as well as being good candidates on biological grounds. A further 53 SNPs that had previously been typed (independently of pooling results) also appeared on the HumanHap300 arrays. For comparison with pooling results, only SNPs that were genotyped on at least 350 of each 384 individual sample were used. The average number of individually genotyped individuals per SNP was 379 (i.e. on average, data were unavailable due to drop out for five individuals per SNP).
The method applied to the Illumina array data was a modification of a method described for Affymetrix arrays (7). The information available on HumanHap300 arrays differs from that available with Genechip HindIII arrays. Genechip HindIII arrays yielded multiple measures of fluorescent intensity for each allele and these were used to compute allele frequency estimates. To allow pooling analysis an option in the Illumina software was used to make raw two colour (green/red) beadscores available as output from array scans. A small number of SNPs had negative values for one or more beadscores and these SNPs were discarded. The beadscores required calibration because green beadscores tended to be larger than red beadscores. HumanHap300 arrays had 10 strands per array, each with ∼31 700 SNPs. Each strand on each array had a number of control SNPs to help assess array quality. The control SNPs assessed staining, allele extension, target removal, hybridization, stringency, non-specific binding and non-polymorphic SNP intensities. Preliminary analysis of both the control SNPs and the full data set indicated that the green/red ratio systematically differed by strand and by array. Calibration was done on a strand-by-strand basis by re-scaling the red beadscore to make the mean value of the pooling allele frequency (PAF) = 0.5 (over all SNPs on that strand); PAF was computed as the corrected red intensity divided by the total (corrected red plus green) intensity. On the HumanHap300 arrays, each SNP had up to 64 PAF estimates per array (mean 18); with Genechip HindIII arrays up to 10 PAF estimates were available per array (mean 8). A new quality control metric which took into account the variable number of PAF estimates per SNP was developed for the Illumina data.
New quality control measure
An initial quality control step was applied to remove all SNPs that did not have a total of at least 20 PAF estimates per pool (i.e. per set of three arrays). The following quality control (QC) metric was then calculated to identify SNPs with PAF estimates that were very variable across arrays
where p_case denotes the PAF estimates and 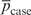 is the mean PAF estimate (similarly for controls). It is necessary to divide each variance on the right-hand side of the above equation to take into account the changes in variance when the frequency, p, changes; this follows because frequency estimates are distributed as binomial with variance p(1−p). The Q metric is defined as a ratio of case to control values to flag SNPs which vary in performance over replicate arrays. SNPs which have high variance on some arrays but low variance on others are unlikely to provide good results and these SNPs are hence flagged as outliers. Very high or very low values of Q denote outliers.
is the mean PAF estimate (similarly for controls). It is necessary to divide each variance on the right-hand side of the above equation to take into account the changes in variance when the frequency, p, changes; this follows because frequency estimates are distributed as binomial with variance p(1−p). The Q metric is defined as a ratio of case to control values to flag SNPs which vary in performance over replicate arrays. SNPs which have high variance on some arrays but low variance on others are unlikely to provide good results and these SNPs are hence flagged as outliers. Very high or very low values of Q denote outliers.
Given two independent random samples of observations from normal distributions, the ratio of the variances is known to have an F-distribution with degrees of freedom determined by the sizes of the two samples. Simulations of binomial random variates in R (17) showed (detailed data not shown) that Q will have a distribution that is close to F with degrees of freedom (number of case PAF estimates, number of control PAF estimates). That is, the assumption that the frequencies are approximately normal is reasonable for our purposes and we can use an F-distribution to evaluate the null distribution of Q. The simulations also showed that it is important to correct for p(1−p) in the numerator and denominator of Q; failing to do so leads to exaggerated Q values (because if for example the case pool has frequency estimate closer to 0.5 then the variance is expected to be higher than the control pool simply due to the fact that the binomial variance is higher). To assess quality control, the P-values from the relevant F-distribution were calculated—since the number of PAF estimates is entered in the degrees of freedom, this quality control routine appropriately takes into account the variation in number of available PAF estimates for any given SNP. Greater than 96% of SNPs had Q values in a narrow band (0.5–2) around 1. We opted to discard the 1006 SNPs (∼0.3% of total) which gave P-values <1/300 000 (i.e. we would expect to falsely discard one SNP that actually passed QC). This seemed a sensible balance between removing erroneous SNPs and retaining working SNPs. Using Q in this way, we successfully identified one false-positive SNP that was very significant in pools but non-significant with IG.
Statistical Model
After correction for green/red ratio variability, we applied a linear model-based approach (7). In the linear model, the response variable is the set of PAF estimates for each SNP. The predictor variable is case/control status. For the Affymetrix arrays, a general linear mixed model was used to account for the structure of the data (multiple replicates, sense/anti-sense information and probe measurements). For the Illumina HumanHap300 arrays, such a model was found to offer no advantage over a simpler model with only case/control status included. This was because (i) HumanHap300 arrays have no sense/anti-sense information and (ii) with HumanHap300 arrays, systematic variation between arrays (and array strands) was accounted for in the green/red calibration step.
For each array type, we estimated the pooling error across all SNPs on each array (7). Two estimates are possible. The first estimate, referred to here as pooling standard deviation [(PSD, this equals 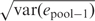 in Ref. (7)], gives a useful estimate of the pooling error across all SNPs but does not take into account the variable precision of the allele frequency estimates across different SNPs (precision varies by the number of beadscores that contribute to the allele frequency estimate for each SNP). The second estimate of pooling error, [var(e_pool − 2) in Ref. (7)] takes into account the variable precision of the allele frequency estimate for each individual SNP. The second estimate is used to construct a test statistic T2 − X for assessing the significance of each SNP [appendix 2 in Ref. (7)]. T2 − X was constructed to have the same null distribution (i.e. same number of false positives) as expected if IG was used (7) and it was used to prioritize SNPs for follow up with individual genotyping. Since the method is based upon contrasting case and control pools, the effect of unequal amplification of alleles is minimal as such effects ‘cancel out’.
in Ref. (7)], gives a useful estimate of the pooling error across all SNPs but does not take into account the variable precision of the allele frequency estimates across different SNPs (precision varies by the number of beadscores that contribute to the allele frequency estimate for each SNP). The second estimate of pooling error, [var(e_pool − 2) in Ref. (7)] takes into account the variable precision of the allele frequency estimate for each individual SNP. The second estimate is used to construct a test statistic T2 − X for assessing the significance of each SNP [appendix 2 in Ref. (7)]. T2 − X was constructed to have the same null distribution (i.e. same number of false positives) as expected if IG was used (7) and it was used to prioritize SNPs for follow up with individual genotyping. Since the method is based upon contrasting case and control pools, the effect of unequal amplification of alleles is minimal as such effects ‘cancel out’.
Estimating pooling error using both individual genotyping and pooling data
In addition to estimating the pooling error from just the pooling results, we can obtain an alternative estimate of PSD by contrasting the pooling and IG results. Denote the estimates of allele frequency from pools as 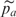 and
and 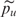 and the estimates from IG as pa and pu for cases and controls, respectively. We write the pool estimates as
and the estimates from IG as pa and pu for cases and controls, respectively. We write the pool estimates as
where e is the error due to pooling. We could obtain an estimate of the pooling error by calculating 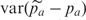 (which is an estimate of the PSD2 in this context since there is no binomial sampling variance) but such an estimate would be inflated by any unequal amplification of different alleles. Since in practice, we remove most of the effects of unequal amplification by focusing on the difference between cases and controls, the main interest is in calculating an estimate of the pooling error based on case–control differences. To do this we calculate the variance of the differences between the pooling and IG case–control differences. That is, we utilize
(which is an estimate of the PSD2 in this context since there is no binomial sampling variance) but such an estimate would be inflated by any unequal amplification of different alleles. Since in practice, we remove most of the effects of unequal amplification by focusing on the difference between cases and controls, the main interest is in calculating an estimate of the pooling error based on case–control differences. To do this we calculate the variance of the differences between the pooling and IG case–control differences. That is, we utilize 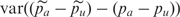 . is useful because
. is useful because
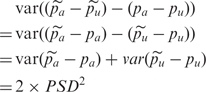 |
i.e. we can estimate PSD2 as half the variance of the difference between case–control frequency differences for pools and for IG.
Since the top 51 SNPs selected on the basis of pooling will give biased estimates of PSD, a simulation study was conducted to evaluate the effect of different levels of pooling error. A total of 384 cases and 384 controls, typed for 300 000 SNPs were simulated. SNPs were split into 10 groups with minor allele frequencies (MAFs) 0.06, 0.09, 0.11, 0.13, 0.16, 0.21, 0.26, 0.31, 0.35, 0.45 (MAFs selected to mimic those from individual genotyping results for top 50 SNPs). A random normal variate with variance PSD2 was added to each frequency to mimic the effects of pooling error. A test statistic comparing pooling case–control frequencies was computed, together with case–control allele frequency differences for pooling and for IG.
Two hundred data sets were simulated and analysed using R (17). To evaluate the results in our real data, we selected the top 50 SNPs on the basis of the pooling test statistic from each simulation replicate. These SNPs were examined to assess (i) The pooling error estimate from 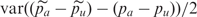 and (ii) The difference in frequency between cases and controls for pooling and for the same 50 SNPs with IG.
and (ii) The difference in frequency between cases and controls for pooling and for the same 50 SNPs with IG.
Partitioning pooling variance into array variance and pooling construction variance with Illumina
A previous analysis of Affymetrix Genechip HindIII arrays and these pools showed that the majority of pooling error was due to errors on the arrays rather than errors in pool construction (13). The same method was applied here with Illumina HumanHap300 arrays to partition the pooling variation into ‘technical’ errors (errors related to obtaining pooling allele frequencies from constituted pools, mainly driven by errors on arrays) and pooling construction errors (errors related to DNA preparation and pool construction). The method works by contrasting the variation within arrays on a single pool with the variation seen between pools. Here we have three arrays on each pool (case, control). Given separate estimates of var(e_pool − array − pairwise) (from all six pairwise combinations of arrays within single pools) and var(e_pool − total − pairwise) (from all nine pairwise combinations of arrays across pools), var(epool_construction) can be estimated by subtraction
For full details of the method see Ref. (13). There was some variation in the number of beadscores per HumanHap300 array so a weighted average was used over all pairwise combinations when the terms in the above equation were computed.
RESULTS
Using a method that estimates the pooling error across all SNPs on an array, we estimated the PSD for Affymetrix Genechip HindIII and Illumina HumanHap300 arrays. The PSD estimate was 0.024 for Genechip HindIII arrays but only 0.007 for HumanHap300 arrays. A similar disparity in pooling error is seen when the allele frequency differences between two arrays on the same pool (i.e. true difference is 0) is calculated for each platform (Figure 1). There are two possible explanations for the large decrease in pooling error with Illumina HumanHap300 arrays. First, the HumanHap300 arrays have more beadscores (i.e. PAF estimates) per SNP than the Genechip HindIII arrays have probe pairs (average numbers are 18 for HumanHap300 and 8 for Genechip HindIII). Second, whilst the HumanHap300 beadscores are true replicates across the array, the Genechip HindIII probe pair intensity scores consist of a central position for each SNP, together with six offset positions. These offset positions may not function as true replicates and may provide results for pooling which have reduced precision. To investigate if one poorly performing HindIII array was adversely affecting results, each array in turn was dropped from the analysis and PSD recalculated—the PSD estimates were consistent with what would be expected if all arrays performed similarly.
Fig. 1.

Affymetrix Genechip HindIII versus Illumina HumanHap300 array-specific error plots. The plots show the difference in allele frequency estimates for a pair of arrays for each type on the control pool (actual difference in frequency for each pool = 0). Affymetrix results are from a pair of 50K Genechip HindIII arrays and the Illumina results are from a pair of 300K HumanHap300 arrays. These results are for a single pair of arrays; in practice the array-specific error will be reduced through the use of multiple arrays.
Figure 2 shows pooling-derived allele frequency estimates for the 15 645 SNPs present on both array types plotted against the IG-derived frequencies from a sample of 271 publicly available caucasian controls (2). The Genechip HindIII results are more variable than the HumanHap300 results. The HumanHap300 results are closer to the line y=x, with the Genechip HindIII results showing systematic bias for small minor allele frequencies. Both Genechip HindIII and HumanHap300 pooling-based frequencies do not include correction for unequal amplification of alleles and this will account for some of the variation seen in Figure 2. Since the main interest is the difference between cases and controls it is instructive to compare the standard deviation of the differences (SDoD) between cases and controls. Concentrating on the difference between cases and controls (rather than say just the control frequencies) ensures that any effects of unequal amplification of alleles in pooling are minimized. SDoD differs from PSD in two ways; (i) SDoD addresses the difference in frequency between two samples rather than the single sample frequency and (ii) PSD consists solely of pooling error (i.e. array error plus pool construction error). Unlike PSD, SDoD comprises pooling error (i.e. array error plus pool construction error) plus the random sampling error resulting from sampling individuals from the population. With Affymetrix Genechip HindIII arrays, the SDoD for the 15 645 overlapping SNPs is 0.041; the analogous figure for Illumina HumanHap300 arrays is 0.023. To provide a benchmark from IG, we computed the SDoD in 271 individually genotyped publicly available controls and a matched set of 269 caucasian (Parkinson's;) cases (2); in this case the SDoD for the same 15 645 SNPs is 0.025 (due to random sampling of alleles from the population, assuming no association between most of the 15 645 SNPs and disease). With Affymetrix Genechip HindIII arrays, the SDoD is higher than in the publicly available (N=271 cases, 269 controls) IG-based sample, despite there being more (384) cases/controls in the pools (i.e. the random sampling error is smaller with N=384 pools but the moderately large pooling error increases the total SDoD). For Illumina HumanHap300-based pooling, the SDoD is smaller than in the N=∼270 IG-based sample, implying that the effective sample size (ESS) is >270. More precisely, the ESS estimates for Genechip HindIII and HumanHap300-based pooling are 0.0252/0.0412 × 270 = 103 and 0.0252/0.0232 × 270=309, respectively. Given the pool size, the HumanHap300-based estimate of the effective relative sample size is 309/384=0.80 for these 15 645 SNPs. The decrease in number of arrays used was 384/3-fold, implying a 0.8×384/3=102-fold decrease in cost compared with IG.
Fig. 2.

Publicly available caucasian control individual genotyping frequencies versus pooling frequencies for Affymetrix Genechip HindIII and Illumina HumanHap300 arrays. The data are the 15 645 SNPs in common between the Affymetrix Genechip HindIII and Illumina HumanHap300 arrays. The frequency of the sample of 271 publicly available caucasian controls is on the y-axis, with the pooling frequencies from the N=384 pooled case/controls on the x-axis. The broken line is y=x. The solid line is the regression line.
In addition to estimating the PSD from the pooling results, we obtained an alternative estimate of the PSD by contrasting the Illumina HumanHap300-based pooling and IG results. The PSD estimate was 0.016 for the top 51 SNPs selected from pooling. This PSD estimate is inflated because the top SNPs from pooling are likely to be the SNPs that happen to have an unusually large pooling error which influences case–control differences in the same direction as the actual difference (from IG). The inflated estimate was corrected by comparing it with simulation results; these showed the top 51 SNP-based estimate was compatible with a PSD of 0.01. Using both the IG and pooling data for the 53 SNPs selected independently of pooling yielded a PSD estimate of 0.011; since these SNPs are not selected on the basis of pooling, no correction is required to obtain a valid estimate of PSD. One SNP was an extremely discordant between pooling and IG (frequency difference ∼0.06); omitting this SNP from the 53 reduced the PSD estimate to 0.009.
Virtually all SNPs selected for follow up on the basis of pooling replicated with IG—49 out of 51 SNPs (>96%) achieved nominal significance (p<0.05) when individually genotyped. Figure 3 shows the allele frequency differences between cases and controls for the two sets of SNPs subjected to both IG and typing using HumanHap300 arrays on pooled DNA. For reasons given above, there is bias away from the line y=x in the follow-up SNP results (but not results for the SNPs selected independently of pooling). The average absolute case–control allele frequency difference was 0.060 (0.024 SD) for the top 51 SNPs when IG was used. With pooling the upwardly biased estimate of the average absolute difference was 0.097 (0.019 SD). Simulation indicated that this degree of bias was consistent with a PSD of 0.01.
Fig. 3.

Comparison of Illumina HumanHap300-based pooling and individual genotyping for 104 SNPs. The solid line is y=x. A total of 53 SNPs were selected independently of pooling results and 51 SNPs were selected on the basis of pooling results.
Previously we showed using Affymetrix Genechip HindIII arrays and these pools that the majority of pooling error was due to errors on the arrays rather than errors in pool construction (13). The same method was applied here with Illumina HumanHap300 arrays, resulting in the same conclusion; the estimate of pooling construction error was very close to zero (the estimate of pooling construction variance was slightly negative, −0.000026). This is an important result because it implies that the overall pooling error can be readily controlled by the number of arrays. The method for estimating pool construction variance will be useful in other studies because it allows researchers to perform an additional check on pool construction accuracy before follow-up is done with individual genotyping.
The best overall pooling study design depends upon whether the priority is either (i) obtaining results that are essentially indistinguishable from IG or (ii) cost efficiency. Figure 4 shows the power for different designs with the number of cases and number of controls fixed at 2000. Distributing a fixed number of arrays across several subpools gives equivalent power to the same arrays with one large pool (7) (assuming negligible pooling construction error) so the exact pool size will not be critical. Since we have empirical evidence that pools of ∼400 individuals are effective, we split each 2000 into five pools. If obtaining results close to those from IG is required then using 15 or possibly 30 Illumina HumanHap300 arrays is required (i.e. the case where either 3 or 6 arrays are used per pool of 400 individuals). If cost efficiency is paramount, 10 HumanHap300 arrays per 2000 individuals still gives reasonable power; the cost of arrays is 200 times less than with IG. If the number of cases/controls is not fixed, then applying ∼15 arrays to 3000 cases/controls would give the same power as IG applied to 2000 (in practice this might involve using two arrays for each of eight pools with 375 individuals/pool). That is, if a 50% larger sample were available for pooling the power would be the same as shown for IG in Figure 4 but with a 2000/15=133-fold reduction in array cost. Because of the large PSD, Affymetrix Genechip HindIII arrays yield substantially lower power; ∼150 Genechip HindIII arrays are required to obtain results similar to those seen with 15 Illumina HumanHap300 arrays.
Fig. 4.

Power curves for individual genotyping and pooling. Power is for 2000 cases, 2000 controls. ‘30x HumanHap300’ assumes 6 Illumina HumanHap300 arrays per N=400 pool. ‘15x HumanHap300’ assumes 3 Illumina HumanHap300 arrays per N=400 pool. ‘10x HumanHap300’ assumes 2 Illumina HumanHap300 arrays per N=400 pool. ‘15x Genechip HindIII’ assumes 3 Affymetrix Genechip HindIII arrays per N=400 pool. PSD is taken to be 0.009 for Illumina HumanHap arrays, 0.024 for Affymetrix Genechip HindIII arrays. Assumptions for power calculation are a multiplicative disease model, marker allele frequency and disease allele frequency both =0.4, complete linkage disequilibrium between marker and disease alleles, alpha = 0.0000001 (i.e. 500 000 tests), disease prevalence 0.01.
The most efficient designs for genome-wide association are multi-stage designs (18–21). Since pooling offers a low cost per genotype, the optimal two-stage design (relative to an IG-based study) will have a larger proportion of samples in stage 1 and a smaller proportion of markers in stage 2. Consider a specific example with the same assumptions as in Figure 4 and effect size GRR=1.35. Assuming that the stage 1: stage 2 cost ratio (per genotype) is 30 with IG, the best design that gives 80% power involves using 34% of the sample in stage 1 with 0.44% of markers in stage 2 (1700 markers); calculations were done using CaTS (19). Assuming that ∼500 000 SNP arrays cost $US800 each, and that stage 2 genotyping costs ∼$US0.05 per genotype, this IG-based study costs ∼$US1.3 million. With pooling, it is advantageous to use a high proportion of the sample in stage 1, with a small proportion of markers in stage 2. If three pools of 400 (three arrays per pool) are used in stage 1, the effective sample size (in terms of equivalent number of individually genotyped individuals) is 960. To ensure the same power as with IG, 215 markers should be followed up in the remaining 800 individuals in stage 2. This power calculation is based upon ‘joint analysis’ in the sense defined by Ref. (19); this can be achieved in two ways. First, the whole N=2000 sample can be genotyped for the 215 markers and a test of association calculated on the IG data for the 215 markers; in this case the cost of the pooling and IG components are ∼$US14000 and ∼$US43000, respectively. Here, since IG is done on stage 1, the pooling results can be confirmed with IG before markers are chosen for follow-up. Second, the stage 2 sample alone can be genotyped for the 215 markers (with IG done only in stage 2, IG costs are $US17000), with the results from the pooling used for the joint analysis of the 215 markers. The results from the pooling can be used in a joint analysis by combining the corrected test statistic (T2 − X from methods section and (7)) with the test statistic from the IG-based stage 2 sample; this form of joint analysis is valid because the statistic T2 − X has the appropriate false positive rate (7). The overall pooling-based two stage cost is therefore in the range $US50000 (no IG on stage 1 samples) to $US80000 (with IG on stage 1 samples); we assume ∼$20000 pool construction costs. An overall cost of $US50000-80000 implies a decrease in overall (two-stage) cost compared with IG of ∼20-fold. The main component of the pool construction cost is labour. This cost has to be balanced against the additional labour cost with IG of running large numbers of arrays in stage 1 and of running substantially more SNPs in stage 2. In practice, the costs are unlikely to be markedly different in either case, with the associated costs unlikely to exceed a few tens of thousands of dollars.
If either a larger proportion of the sample is used for pooling or if more arrays are used per pool, the most cost-efficient design in terms of statistical power involves using fewer SNPs in stage 2. However, in such circumstances, some practical issues arise. It will generally not be efficient to genotype a very small number of SNPs (<25 on the Sequenom platform, say). Also, although our novel quality control method managed to identify the one SNP that performed poorly when individually genotyped, in practice some redundancy should be built into the list of SNPs chosen for follow-up to cover cases where poorly performing SNPs are not identified. In the specific case of Sequenom, up to 250 SNPs can be efficiently genotyped in a moderate time frame and samples of this size seem a sensible balance between cost efficiency and robustness to the occasional pooling artefact. Increasing the number of markers in stage 2 above ∼300 is not an attractive option; this is because of (i) decreased cost efficiency and (ii) the fact that medium-scale platforms such as Illumina OPAs are only cost effective with much larger sets of SNPs (>1500).
DISCUSSION
Estimates of the variation in allele frequencies due to pooling errors were derived both from information across many SNPs on an array and from comparing pooling and IG results. The results were broadly similar in either case, with PSD estimates ranging from 0.007 to 0.011 for Illumina HumanHap300 arrays. The pooling error was substantially larger for the Affymetrix Genechip HindIII arrays (PSD estimate 0.024), with the error appearing to systematically worsen for small minor allele frequencies (Figure 2). For the purposes of calculating the number of arrays required to maintain the pooling error at a given level, the relevant measure of pooling error is the pooling variance, not the standard deviation. On the variance scale, Genechip HindIII arrays show 5–10 times increased variance compared with HumanHap300 arrays. That is, to achieve equivalent results, a 5–10-fold increase in the number of arrays will be required if Genechip HindIII rather than HumanHap300 arrays are used. We have not attempted to account for the cost of the different array types. The price of arrays changes so quickly that any conclusions drawn would very rapidly become out of date. At the time of writing Affymetrix Genechip HindIII arrays cost less than Illumina HumanHap 300 arrays and this may allow researchers to simply use more arrays to offset the decreased efficiency of the Genechip HindIII arrays.
The Affymetrix 100k array set includes the HindIII array, along with another array, the XBA (∼50 000 SNPs per array). We applied six Affymetrix XBA arrays to the pools described above (three arrays per pool). The estimates of pooling error were similar to those seen for the HindIII arrays, with a PSD estimate for the XBA arrays of 0.029. This result was expected because the XBA arrays contain a similar number of probes per SNP as the HindIII arrays.
Here we consider only Affymetrix 100k and Illumina HumanHap300 arrays. There are several other array types that would be potentially suitable for pooling. Affymetrix 500k arrays are similar to their 100k arrays; the main difference is that the number of probe pairs available per array is reduced from 10 to 6. Since the overall pooling error appears to be strongly related to number of available probe pairs, it seems likely that, compared with Affymetrix 50K arrays, more Affymetrix 500k arrays would need to be used to maintain the pooling error at a suitably low level. Illumina offer 550K arrays; these arrays contain a superset of the SNPs on the Illumina 300k arrays and have the same constellation of bead scores. This means that results would be expected to be similar to those seen here. Both Illumina and Affymetrix have released 1 million SNP arrays. Such arrays are potentially very useful for pooling for two reasons. First, the increased redundancy means that, providing pooling error is essentially random across SNPs, the chance of pooling missing a real signal is decreased because several SNPs will tag any variant of interest. As long as at least one of these tagging SNPs have the expected (small) level of pooling error then the pooling will detect any true associations that are detectable with IG. Second, using these very dense arrays with pooling may offer an inexpensive means of ‘filling the gaps’ from a previous IG-based GWA study based on 100k/300k arrays.
The pooling approach here is designed for simple case–control analysis. If there are subgroups of cases/controls (e.g. sex, age), then in IG-based approaches these can be dealt with by fitting them as covariates. This is not possible with pools but providing pools are suitably designed in advance this is not a problem. For example, since studies large enough for powerful GWA analysis are typically large (say N=1000 cases, N=1000 controls, ideally larger), separate pools can be constructed for say males and females (with N=250 or 500 per pool, with different sexes in separate pools). For quantitative covariates, separate pools can be made with say high, medium or low covariate values. The factor limiting how much information is lost compared with an IG-based design would be how many separate pools can efficiently be made based on covariate information. Above we used 3 Illumina HumanHap300 arrays per ∼400 individual pool; since the main determinant of error in pooling appears to be in array-specific errors instead of pooling construction errors, similar results to the 3 arrays per N=400 case should be possible with 3 N=133 with one array per pool. In this case since each pool is relatively small, the available sample could be divided into several pools, each with a different covariate grouping (e.g. bottom 10th percentile, 10th–20th percentile, etc.). Another limitation of pooling is that only allelic tests (rather than genotypic or haplotypic) of association are available. Allelic tests are the most widely reported in the literature for IG-based studies. Because allelic tests are powerful for a range of disease models, most researchers focus their efforts on tests of this type (these tests typically only have 1 degree of freedom, compared with >1 for tests of genotypes/haplotypes).
For analysis of quantitative traits (instead of disease traits), pooling may also be advantageous because much of the information for association is contained in the tails of the distribution and pools can be constructed using individuals from the extremes of the distribution. Detailed design and analysis of such studies is beyond the scope of this article and is discussed elsewhere (22).
In summary, a number of sources of information have been used to estimate the magnitude of pooling error with two array types on the same pool. Illumina HumanHap300 arrays offer substantial increases in accuracy for pooling and this greatly extends the usefulness of pooling. Concordance between individual genotyping and pooling is expected to be excellent. With HumanHap300-based pooling, very few arrays are required to extract the majority of information on association from a sample and the limiting factor in future genome-wide association studies is likely to be available sample size and not cost.
ACKNOWLEDGEMENTS
The QIMR Molecular and Genetic Epidemiology Laboratories provided expert assistance in collection and preparation of the DNA pools. Susan Treloar's; pioneering work enabled the establishment of the QIMR Endometriosis study. The study and sample collections were partly supported by grants 339430, 339446, 389892, 496674 and 496675 from the National Health and Medical Research Council (NHMRC) and by the Co-operative Research Centre for the Discovery of Genes for Common Human Diseases established and supported by the Australian Government's; Co-operative Research Centre's; Program.
This study used data from the SNP Database at the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds/); the original genotyping was performed in the laboratory of Dr Singleton and Dr Hardy (NIA, LNG), Bethesda, MD USA. Funding to pay the Open Access publication charges for this article was provided by NHMRC grants.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 2.Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert M, et al. Genome-wide genotyping in parkinson's; disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 3.Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, Swan GE, Rutter J, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum. Mol. Genet. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brohede J, Dunne R, McKay JD, Hannan GN. PPC: an algorithm for accurate estimation of SNP allele frequencies in small equimolar pools of DNA using data from high density microarrays. Nucleic Acids Res. 2005;33:e142. doi: 10.1093/nar/gni142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butcher LM, Meaburn E, Knight J, Sham PC, Schalkwyk LC, Craig IW, Plomin R. Snps, microarrays and pooled DNA: identification of four loci associated with mild mental impairment in a sample of 6000 children. Hum. Mol. Genet. 2005;14:1315–1325. doi: 10.1093/hmg/ddi142. [DOI] [PubMed] [Google Scholar]
- 6.Kirov G, Nikolov I, Georgieva L, Moskvina V, Owen MJ, O'D;onovan MC. Pooled DNA genotyping on affymetrix snp genotyping arrays. BMC Genomics. 2006;7:27. doi: 10.1186/1471-2164-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macgregor S, Visscher PM, Montgomery G. Analysis of pooled DNA samples on high density arrays without prior knowledge of differential hybridization rates. Nucleic Acids Res. 2006;34:e55. doi: 10.1093/nar/gkl136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meaburn E, Butcher LM, Schalkwyk LC, Plomin R. Genotyping pooled DNA using 100K SNP microarrays: a step towards genomewide association scans. Nucleic Acids Res. 2006;34:e27. doi: 10.1093/nar/gnj027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, Huynh KD, Brunner F, et al. Common KIBRA alleles are associated with human memory performance. Science. 2006;314:475–478. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- 10.Spinola M, Leoni VP, Galvan A, Korsching E, Conti B, Pastorino U, Ravagnani F, Columbano A, et al. Genome-wide single nucleotide polymorphism analysis of lung cancer risk detects the klf6 gene. Cancer Lett. 2007;251:311–316. doi: 10.1016/j.canlet.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 11.Pearson JV, Huentelman MJ, Halperin RF, Tembe WD, Melquist S, Homer N, Brun M, Szelinger S, Coon K, et al. Identification of the genetic basis for complex disorders by use of pooling-based genomewide single-nucleotide-polymorphism association studies. Am. J. Hum. Genet. 2007;80:126–139. doi: 10.1086/510686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steer S, Abkevich V, Gutin A, Cordell HJ, Gendall KL, Merriman ME, Rodger RA, Rowley KA, et al. Genomic DNA pooling for whole-genome association scans in complex disease: empirical demonstration of efficacy in rheumatoid arthritis. Genes Immun. 2007;8:57, 68. doi: 10.1038/sj.gene.6364359. [DOI] [PubMed] [Google Scholar]
- 13.Macgregor S. Most pooling variation in array-based DNA pooling is attributable to array error rather than pool construction error. Eur. J. Hum. Genet. 2007;15:501–504. doi: 10.1038/sj.ejhg.5201768. [DOI] [PubMed] [Google Scholar]
- 14.Craig DW, Huentelman MJ, Hu-Lince D, Zismann VL, Kruer MC, Lee AM, Puffenberger EG, Pearson JM, Stephan DA. Identification of disease causing loci using an array-based genotyping approach on pooled DNA. BMC Genomics. 2005;6:138. doi: 10.1186/1471-2164-6-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao ZZ, Nyholt DR, James MR, Mayne R, Treloar SA, Montgomery GW. A comparison of DNA pools constructed following whole genome amplification for two-stage snp genotyping designs. Twin Res. Hum. Genet. 2005;8:353–361. doi: 10.1375/1832427054936718. [DOI] [PubMed] [Google Scholar]
- 16.Zhao ZZ, Nyholt DR, Le L, Martin NG, James MR, Treloar SA, Montgomery GW. KRAS variation and risk of endometriosis. Mol. Hum. Reprod. 2006;12:671–676. doi: 10.1093/molehr/gal078. [DOI] [PubMed] [Google Scholar]
- 17.R Development Core Team R: A language and environment for statistical computing R Foundation for Statistical Computing Vienna. Austria (2004) ISBN 3-900051-00-3. [Google Scholar]
- 18.Satagopan JM, Verbel DA, Venkatraman ES, Offit KE, Begg CB. Two-stage designs for gene-disease association studies. Biometrics. 2002;58:163–170. doi: 10.1111/j.0006-341x.2002.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 20.Zehetmayer S, Bauer P, Posch M. Two-stage designs for experiments with a large number of hypotheses. Bioinformatics. 2005;21:3771–3777. doi: 10.1093/bioinformatics/bti604. [DOI] [PubMed] [Google Scholar]
- 21.Zuo Y, Zou G, Zhao H. Two-stage designs in case-control association analysis. Genetics. 2006;173:1747–1760. doi: 10.1534/genetics.105.042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knight J, Sham P. Design and analysis of association studies using pooled dna from large twin samples. Behav. Genet. 2006;36:665–677. doi: 10.1007/s10519-005-9016-9. [DOI] [PubMed] [Google Scholar]