

Abstract
Helicobacter hepaticus causes hepatitis in susceptible strains of mice. Previous studies indicated that A/JCr mice are susceptible and C57BL/6NCr mice are resistant to H. hepaticus-induced hepatitis. We used F1 hybrid mice derived from A/J and C57BL/6 matings to investigate their phenotype and determine their hepatic gene expression profile in response to H. hepaticus infection. F1 hybrid mice, as well as parental A/J and C57BL/6 mice, were divided equally into control and H. hepaticus-infected groups and euthanized at 18 months postinoculation. Hepatic lesions were evaluated histologically and the differential hepatic gene expression in F1 mice was determined by microarray-based global gene expression profiling analysis. H. hepaticus-infected parental strains including A/J and C57BL/6 mice, as well as F1 mice, developed significant hepatitis. Overall, hepatocellular carcinomas or dysplastic liver lesions were observed in 69% of H. hepaticus-infected F1 male mice and H. hepaticus was isolated from hepatic tissues of all F1 mice with liver tumors. Liver tumors, characterized by hepatic steatosis, developed in livers with high hepatitis scores. To identify gene expression specific to H. hepaticus-induced hepatitis and progression to hepatocellular carcinoma in F1 mice, a method using comparative group transcriptome analysis was utilized. The canonical pathway most significantly enriched was immunological disease. Fatty acid synthase and steaoryl-coenzyme A desaturase, the two rate-limiting enzymes in lipogenesis, were upregulated in neoplastic relative to dysplastic livers. This study suggests a synergistic interaction between hepatic steatosis and infectious hepatitis leading to hepatocellular carcinoma. The use of AB6F1 and B6AF1 mice, as well as genetically engineered mice, on a C57BL/6 background will allow studies investigating the role of chronic microbial hepatitis and steatohepatitis in the pathogenesis of liver cancer.
Hepatocellular carcinoma (HCC) is one of the most common causes of cancer death worldwide, and in some geographic areas it represents the primary cause of cancer-related death (2). Both genetic and environmental factors confer a high risk of developing HCC. In the United States, cirrhosis caused by hepatitis C virus, hepatitis B virus, or alcohol is the major predisposing lesion for the development of HCC. An epidemic of insulin resistance syndrome manifesting clinically as obesity and type 2 diabetes also has emerged as a risk factor for HCC (5, 11). The occurrence of fatty liver disease and its inherent propensity to progress to HCC highlights the link between lipid metabolism and liver inflammation (44). However, a significant proportion (up to 30%) of reported HCC cases do not have identifiable risk factors (8).
Chronic bacterial infection with accompanying inflammation also plays a role in the development of gastrointestinal cancer in humans. In the case of gastric cancer, the second most frequent cancer in the world, infection with Helicobacter pylori has been causally associated with its progression from gastritis to gastric adenocarcinoma (7). Infection with H. pylori is also recognized as a significant risk factor for the development of gastric mucosa-associated lymphoid tissue lymphoma (17). Helicobacter species DNA has been detected in human livers with primary HCC (1, 9, 16, 28, 42, 45). In addition, hepatic Helicobacter species DNA has been identified in liver tissue, bile, and/or gallbladder tissue from human patients with primary sclerosing cholangitis, primary biliary cirrhosis, chronic cholecystitis, and biliary tract malignancies (18, 35, 39). Increased serum antibodies to enterohepatic Helicobacter species have also been detected in humans with chronic liver diseases (61).
H. hepaticus was originally isolated from the livers and intestinal mucosa of A/JCr mice with chronic active hepatitis and HCC that were untreated controls in a long-term carcinogenesis study at the National Cancer Institute (19, 64). Chronic active hepatitis and liver tumors were also prevalent in other strains of mice, including C3H/HeNCr, SJL/NCr, BALB/cAnNCr, and SCID/NCr (63, 64). In contrast, no hepatic lesions were found in H. hepaticus-infected B6C3F1 and C57BL/6NCr mice, suggesting that these strains were genetically resistant to the development of liver disease (63, 64). Subsequently, 12 2-year carcinogenesis studies conducted by the National Toxicology Program indicated that infection with H. hepaticus, was associated with an increased incidence of HCC and hepatic hemangiosarcomas in B6C3F1 mice, implicating this bacterium as a confounding factor in those studies (4, 25). Further studies from our laboratory determined that the absence of hepatitis in C57BL/6NCr mice at 6 months postinoculation (p.i.) was associated with significantly higher levels of H. hepaticus in the cecum relative to those of A/JCr mice with hepatitis (65). Using AXB recombinant inbred (RI) strains indicated significant variation in hepatic inflammation, HCC, and hepatic hemangiosarcoma among these strains and highlighted a genetic susceptibility to inflammatory liver disease associated with H. hepaticus infection (29). More recently, selected enterohepatic Helicobacter species, but not H. pylori, have been shown to play a major role in the pathophysiology of cholesterol gallstone formation in C57L/J mice fed a lithogenic diet (36-38).
Previous studies determined that resistance to H. pylori-induced gastritis was dominantly inherited in F1 hybrid mice (59); however, the genetic susceptibility to H. hepaticus-induced liver cancer has not been experimentally investigated in F1 hybrid mice. We demonstrate here that F1 hybrid mice derived from A/J and C57BL/6 are susceptible to liver disease and cancer and that this codominantly inherited susceptibility is linked to hepatic expression of immune function-related genes and molecular networks associated with immune functions.
MATERIALS AND METHODS
Mice.
C57BL/6 and A/J mice were obtained from Emil Skamene (McGill University Health Centre, Montreal, Québec, Canada), embryo transfer re-derived, and housed in Helicobacter-free facilities. The first generation (F1) of hybrid mice was obtained by mating C57BL/6 and A/J mice.
Experimental design.
Forty A/J (20 males and 20 females), forty C57BL/6 (20 males and 20 females), and eighty F1 mice (40 males and 40 females) were equally divided into experimental and control groups. Mice in the experimental group were inoculated at 8 weeks of age with H. hepaticus (ATCC 51449). Inoculation was performed by oral gavage with 109 organisms in 0.2 ml of broth media every other day for a total of three doses. Control mice were inoculated in the same manner with sterile broth media. Fecal samples were collected prior to inoculation and at selected time points p.i. Mice were euthanized at 18 months p.i. Animals were housed in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care International. The MIT Institutional Animal Care and Use Committee approved this study.
H. hepaticus isolation.
Cecal and liver samples collected at necropsy were placed in vials containing brucella broth (Remel, Lenexa, KS) and 20% glycerol and were frozen at −80°C until they were processed for culture. The tissue was homogenized in 1 ml of brucella broth by using a glass tissue grinder. Approximately 100 μl of the homogenized sample was applied directly to medium impregnated with cefoperazone, vancomycin, and amphotericin B (Remel), and the remainder of the sample was filtered through a 0.45-μm-pore-size filter and plated onto Trypticase soy agar with 5% sheep blood (BAP; Remel). The plates were incubated at 37°C under microaerobic conditions for 7 to 10 days in vented jars containing N2, H2, and CO2 (80:10:10). Characteristic colonies were Gram stained, and bacteria were examined for morphology. Bacteria were assessed for catalase, urease, and oxidase activity, as well as for resistance to cephalothin and nalidixic acid.
DNA extraction.
DNA was extracted from feces and tissue (liver and cecum) by using a QIAamp DNA minikit (Qiagen, Inc., Valencia, CA) and a HighPure PCR template preparation kit (Roche Diagnostics GmbH, Mannheim, Germany), respectively.
PCR analyses.
PCR on fecal DNA was performed using H. hepaticus-specific primers C68 (B38) and C69 (B39) as previously described (51). PCR was performed by using an Expand High-Fidelity PCR system (Roche). PCR on liver DNA was performed using Helicobacter-specific primers C97 and C05 as previously described (18). These primers were used to generate 16S rRNA amplicons of approximately 1,200 bases that were subsequently used as templates for nested PCR using primers C68 and C69.
Genotyping of the X chromosome of F1 male mice was performed to ascertain the strain of the progenitor. PCR on liver DNA was performed with the primers DXMit64F (5′-GGATCAGTTAGCAGGGAAAGG-3′) and DXMit64R (5′-CACAGACTGAGAAGGCTGTCC-3′). These DXMit64 chromosomal markers amplify a 114-bp fragment in A/J mice and a 134-bp fragment in C57BL/6 mice. Reaction mixtures were prepared by using pureTaq Ready-To-Go PCR beads (Amersham Biosciences, Piscataway, NJ) containing approximately 100 to 250 ng of DNA to a total volume of 25 μl. PCRs were performed by using a Techne Genius (Techne, Inc., Princeton, NJ) thermal cycler under the following conditions: 40 cycles of denaturation (94°C, 1 min), annealing (56°C, 1 min), and extension (72°C, 1 min). A final extension (72°C, 8 min) completed the cycling protocol. PCR amplicons were visualized after electrophoresis in a 6% VisiGel separation matrix (Stratagene, La Jolla, CA) and staining with ethidium bromide.
Histopathologic evaluation.
Representative samples from the liver, including right lateral, median, left lateral, and caudate lobes, were collected, fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. Selected frozen liver sections were stained with Oil red O to determine the extent of lipid accumulation. Immunohistochemistry for cytokeratin and vimentin was performed on selected sections as previously described (46). Histopathological evaluation was performed by two veterinary pathologists blinded to the identity of the samples. Liver sections were scored as recently described (48). Briefly, a hepatitis index (HI) was generated by combining scores for lobular, portal, and interface hepatitis along with the number of liver lobes (of four) containing ≥5 inflammatory lesions. Mice with a histologic HI of ≥4 were defined as having hepatitis. Hepatocellular dyplasia/neoplasia was scored on a scale of 1 to 4.
Statistical analysis.
Statistical analyses for histopathologic scores were performed by using GraphPad Prism 4 (GraphPad Software, Inc.). The Mann-Whitney U test and the Fisher exact test were used to analyze the scores for hepatic inflammation. Nonhepatocellular tumors, including histiocytic sarcomas (33), were considered nonspecific age- or strain-related lesions and hence not considered for statistical analysis. Tests were considered significant at a P of <0.05.
Liver samples, RNA isolation, and quality assessment.
Liver was aseptically removed immediately after euthanasia, and sections of each liver lobe were collected and placed in a vial. Each vial was immediately placed in liquid nitrogen and then transferred to a −80°C freezer. Representative liver samples at 18 months p.i. from four F1 male mice were selected for microarray analysis based on known infection status and the presence of hepatitis, preneoplastic foci, and nodules of altered or dysplastic hepatocytes (i.e., foci of altered hepatocytes [FAH]), and/or liver tumors as demonstrated by histopathology. Samples were obtained from two H. hepaticus-infected F1 male mice with hepatitis and HCC and from two H. hepaticus-infected F1 male mice with hepatitis and FAH (without HCC). The liver of one age-matched uninfected control F1 male mouse with a single focal microscopic hepatic adenoma without hepatitis was also analyzed. Total RNA isolation from livers and quality assessment was performed as previously described (3).
Microarray data analyses.
Affymetrix murine genome arrays (i.e., the GeneChip Mouse Genome 430 2.0 Array) with 45,000 probesets representing over 39,000 transcripts and variants from over 34,000 well-characterized mouse genes were utilized. Expression intensities were calculated by using Robust MultiChip analysis (RMA) (30). Nonexpressed transcripts across all conditions were filtered prior to differential expression testing. Differentially expressed genes were identified based on two criteria: (i) an expression difference between groups of P < 0.05 (analysis of variance as calculated using the Partek Genomics Suite [10]) and (ii) an expression ratio of ≥1.5 or ≤−1.5. Unique genes were defined by reference sequence identifier numbers. Two comparisons were performed to determine the differentially expressed genes during H. hepaticus-induced hepatitis and progression to HCC: comparison 1 included livers from two infected F1 males with hepatitis and HCC versus liver from an uninfected F1 male with a single focal microscopic hepatic adenoma without hepatitis and comparison 2 included livers from the same two infected F1 males with hepatitis and HCC versus livers from two other infected F1 males with hepatitis and FAH.
To determine significantly enriched signaling Biocarta pathways based on gene ontology we used a web-based gene set analysis toolkit (WebGestalt) with a test of the hypergeometric mean (67). To determine significant networks based on molecular interactions, we used the Ingenuity Pathway Tool, where significant enrichment as determined with a Fisher exact test of protein-protein and protein-DNA interactions was identified. Raw microarray data have been deposited into the Gene Expression Omnibus database under accession number GSE9097.
Real-time quantitative PCR (TaqMan).
To confirm the microarray results, we performed real-time quantitative PCR. In addition to the samples used for microarray analyses, we included liver tissue from another uninfected control F1 male mouse without hepatitis, FAH, or liver tumors. Total RNA from each liver sample was isolated using TRIzol reagent according to the manufacturer's protocol (Invitrogen). For quantification of mRNA, approximately 5 μg of total RNA from each sample was converted into cDNA by using a High Capacity cDNA Archive kit following the manufacturer's protocol (Applied Biosystems). The cDNA levels of Scd1, Fasn, Elovl6, Ikbkg, Tgfb1, Stat1, and Ccl5 were determined by quantitative PCR using commercially available primers and probes (Applied Biosystems). Gene expression was normalized relative to the cDNA levels of the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene, a housekeeping gene, used as an endogenous control. Reactions were performed in duplicate containing a total volume of 25 μl that included 5 μl of cDNA, 1.25 μl of 20× primer-probe solution, 12.5 μl of 2× Master Mix (Applied Biosystems), and 6.25 μl of double-distilled water. The relative expression of mRNA was calculated as described by the TaqMan protocol. The comparisons were consistent with those performed for microarray analyses. The mRNA levels were expressed as the fold change relative to either the control uninfected group without hepatitis (comparison 1) or the hepatitis and FAH group (comparison 2).
RESULTS
Histopathologic evaluation. (i) H. hepaticus accentuates hepatic inflammation in parental C57BL/6 mice.
A total of 12 control (3 males and 9 females) and 13 experimental (3 males and 10 females) mice were evaluated at 18 months p.i. (Table 1) . Multiple nonspecific background lesions of varying severity were present in both control and infected animals. These included both micro- and macrovesicular hepatocellular fatty change of various intensities, extramedullary hematopoiesis, and lobular and portal hepatitis in both groups and genders. The mean HI score of H. hepaticus-infected C57BL/6 mice was significantly different (P < 0.04) relative to that of uninfected controls (Fig. 1A). H. hepaticus-infected mice exhibited a slightly increased intensity of portal and lobular inflammation with more numbers of affected liver lobes compared to the background inflammation in the uninfected controls. The low numbers of males used in the present study did not allow genderwise contingency analysis of H. hepaticus-induced hepatitis using the Fischer's exact test. On analysis of HI scores of the infected and control groups with no gender stratification, there was no significant increase in risk or propensity for developing hepatitis (defined as a HI score of ≥4) in the infected group by contingency analysis using the Fisher exact test. The liver of a control male mouse exhibited nodular lesions consistent with HCC in addition to a focal adenoma. This HCC was characterized by severe hepatocellular fatty change without hepatic inflammation. Two H. hepaticus-infected female mice developed histiocytic sarcoma in the liver with extramedullary hematopoiesis and concurrent inflammation.
TABLE 1.
Number and gender of F1 and parental mice analyzed at 18 months p.i. with H. hepaticus or vehicle alone (control)
| Mouse strain | Gender | No. of H. hepaticus-infected mice | No. of control mice |
|---|---|---|---|
| F1 | Male | 13 | 16 |
| Female | 17 | 19 | |
| A/J | Male | 7 | 8 |
| Female | 9 | 7 | |
| C57BL/6 | Male | 3 | 3 |
| Female | 10 | 9 |
FIG. 1.

(A) HI scores in H. hepaticus-infected A/J mice were significantly higher (P < 0.0002) than in uninfected control A/J mice. HI scores in H. hepaticus-infected F1 mice were significantly higher (P < 0.0007) than in uninfected control F1 mice. HI scores in H. hepaticus-infected C57BL/6 mice were significantly higher (P < 0.04) than in uninfected control C57BL/6 mice. (B) HI scores in H. hepaticus-infected F1 male mice were significantly higher (P < 0.04) than in F1 female mice. (C) Dysplasia/neoplasia scores in H. hepaticus-infected F1 male mice were significantly higher (P < 0.002) than in F1 female mice. (D) High HI scores appeared to correlate with progression to dysplasia or neoplasia in infected F1 male mice. Hepatitis, HI ≥ 4.
(ii) H. hepaticus-induced hepatitis in parental A/J mice.
A total of 15 control (8 males and 7 females) and 16 experimental (7 males and 9 females) mice were evaluated at 18 months p.i. (Table 1). H. hepaticus-infected male mice had significant scores for HI (P < 0.0002) (Fig. 1A), lobular inflammation (P < 0.0006), portal inflammation (P < 0.0008), and number of liver lobes with ≥5 inflammatory foci (P < 0.007) than uninfected controls. Of the infected mice, 4 of 7 (57%) males and 3 of 9 (33%) females had an HI of ≥4. Although not statistically significant, infected males had higher mean scores for HI, portal inflammation, lobular inflammation, and interface inflammation than infected females. One infected male mouse had mild hepatocellular dysplasia. Adenoma and/or HCC or nonhepatocellular tumors were not observed in either the infected or the control groups. Hepatocellular fatty change was negligible in both groups of A/J mice.
(iii) High frequency of liver tumors in H. hepaticus-infected F1 male mice.
A total of 35 control (16 males and 19 females) and 30 experimental (13 males and 17 females) mice were evaluated at 18 months p.i. (Table 1). Gross hepatic tumors identified histologically as HCC were observed in 3 of 13 (23%) H. hepaticus-infected F1 male mice, including B6AF1 (n = 2) and AB6F1 (n = 1) mice (Fig. 2a). FAH, a premalignant liver lesion, was observed independent of HCC in 2 of 3 (67%) H. hepaticus-infected F1 male mice with HCC, including B6AF1 and AB6F1 mice. Dysplastic changes such as clear cell foci, increased atypia with oval cell proliferation and FAH were also observed in 6 of 13 (46%) H. hepaticus-infected F1 male mice with no obvious progression to HCC, including B6AF1 (n = 4) and AB6F1 (n = 1). One of these mice exhibited severe hepatocellular dysplasia and oval cell proliferation in association with multiple nodular portocentric round-cell tumor interpreted as lymphoma. Overall, 9 of 13 (69%) of H. hepaticus-infected F1 male mice, including B6AF1 (n = 6) and AB6F1 (n = 2) mice, developed dysplastic or neoplastic liver lesions. HCCs were not observed in infected F1 female mice or in control mice. Control mice exhibited histiocytic sarcoma (n = 2), focal adenoma without associated inflammation (n = 1), and focal hepatocellular atypia and peliosis hepatis (n = 1).
FIG. 2.

(a) Gross hepatic tumors in an H. hepaticus-infected AB6F1 male mouse at 18 months p.i.: HCC (arrows) and hemangiosarcoma-like lesion (arrowhead) (bar = 0.5 cm). (b) HCC shown in panel a with fat and glycogen vacuoles (bar = 800 μm). (c) Frozen section of HCC shown in panel b stained with Oil red O demonstrating microvesicular and macrovesicular steatosis (bar = 80 μm). (d) HCC in an H. hepaticus-infected B6AF1 male mouse at 18 months p.i. demonstrating basophilic-tigroid atypical hepatocytes (bar = 40 μm). (e) Mass with features of hemangiosarcoma as shown in panel a (bar = 800 μm). The inset shows higher magnification of atypical hepatocytes and proliferation of endothelial cell-lined vascular spaces with multifocal blood-filled dilatations (bar = 160 μm). (f) Portal hepatitis in an H. hepaticus-infected male mouse at 18 months p.i. (bar = 160 μm).
The occurrence of HCC was characterized by variants including solid forms containing large pleomorphic and highly anaplastic tumor cells, and trabecular tumors often comprised of small monomorphic hepatocytes. Both FAH and HCC were comprised of variably commingled clear, basophilic-tigroid, and eosinophilic cells (Fig. 2b, c, and d). One AB6F1 male mouse with HCC also displayed a dysplastic mass resembling hemangiosarcoma (Fig. 2a and e). This lesion was interpreted as atypical pelioid and nodular hyperplasia surrounding a central necrotic core caused by thromboembolic infarction. Another infected AB6F1 male mouse developed FAH/adenoma with hepatitis.
Both infected and uninfected F1 mice exhibited moderate to severe hepatocellular fatty change. This was characterized by centrilobular to midzonal microvesicular steatosis variably admixed with random macrovesicular steatosis. All three HCCs were characterized by both types of steatosis (Fig. 2b and c).
H. hepaticus-infected mice exhibited significantly higher scores for overall HI (P < 0.0007) (Fig. 1A), portal inflammation (P < 0.0004) (Fig. 2f), lobular inflammation (P < 0.0006), and number of lobes with ≥5 inflammatory foci (P < 0.009) than uninfected control mice. H. hepaticus-infected male mice had significantly higher values for HI (P < 0.04) (Fig. 1B) and number of lobes with ≥5 inflammatory foci (P < 0.04) than infected females. However, lobular and portal inflammation scores, although more prominent in males, were not statistically significant. Hepatitis (HI of ≥4) was present in 14 of 30 (46%) experimental mice with a gender distribution of 9 of 13 (69%) males and 5 of 17 (29%) females. Dysplasia/neoplasia scores in infected male mice were significantly higher (P < 0.002) than in control female mice (Fig. 1C). Eight of the nine (89%) infected males with hepatitis exhibited dysplastic or neoplastic progression (Fig. 1D). An apparent correlation with hepatitis and progression to dysplasia or neoplasia was observed in infected male mice. Infected male mice with adenoma (n = 1) or HCC (n = 3) typically also exhibited high HI scores. Infected male mice had a significant risk for developing hepatitis compared to the controls on contingency table analysis using the Fisher exact test (P < 0.001).
Assessment of colonization by H. hepaticus using culture and PCR analyses.
H. hepaticus was isolated from the cecal and/or liver tissue of 15 of 30 (50%) experimental F1 mice, including 11 males and 4 females. All three F1 mice with hepatic tumors had H. hepaticus isolated from their livers. Fecal and liver PCR analyses indicated that 25 of 30 (83%) experimental F1 mice were positive for H. hepaticus. Overall, both culture and PCR analyses indicated that 29 of 30 (97%) experimental F1 mice were positive for H. hepaticus either in the liver, cecum, or feces. All 36 controls were negative for H. hepaticus by culture or PCR.
H. hepaticus was isolated from the liver of 5 of 13 (38%) experimental C57BL/6 mice, including 1 male and 4 females. Liver and fecal PCR analyses indicated that 5 of 13 (38%) and 13 of 13 (100%) experimental C57BL/6 mice, respectively, were positive for H. hepaticus. In addition, H. hepaticus was isolated from the liver of 4 of 16 (25%) experimental A/J mice, including all four males. Liver and fecal PCR analyses indicated that 5 of 16 (31%) and 14 of 16 (88%) experimental A/J mice, respectively, were positive for H. hepaticus. Control C57BL/6 and A/J mice were negative for H. hepaticus by culture or PCR.
Genotyping for the X chromosome of F1 male mice.
A total of 27 F1 male mice from control (n = 15) and experimental (n = 12) groups were genotyped at 18 months p.i. A total of 25 of 27 (93%) were positive for the C57BL/6 X chromosome marker (B6AF1 strain) (Fig. 3). The two mice that were positive for the A/J X chromosome marker (AB6F1 strain) were in the experimental group (data not shown).
FIG. 3.

Genotyping of F1 male mice by PCR using primers for the X chromosome. The PCR primers amplified a 114-bp fragment in A/J mice and a 134-bp fragment in C57BL/6 mice. DNA samples from F1 male mice in lanes 5 to 9 were positive for the C57BL/6 X chromosomal marker and genotyped as B6AF1. The mice represented by DNA in lanes 5 and 8 developed hepatocellular carcinoma. The genotypes of F1 female mice represented by DNA in lanes 1 to 4 and lanes 10 to 13 were not determined. Lane A, A/J mouse control DNA; lane B, C57BL/6 mouse control DNA; lane M, 1-kb plus ladder; lane (−), no DNA.
Identifying genes specific to infection and tumor status.
In total, 416 genes were identified with differential expression in the present study (see Table S1 in the supplemental material). A total of 399 genes were significantly differentially expressed as a result of HCC relative to control (comparison 1; see Table S1 in the supplemental material). A comparison of infected mice with HCC to infected mice with FAH (dysplasia) identified 56 genes (comparison 2; see Table S1 in the supplemental material). Most of these genes (360 of 399) were unique to infection and HCC (Fig. 4a; see also Table S1 in the supplemental material), and 17 of 56 genes were unique to HCC relative to FAH (Fig. 4b; see also Table S1 in the supplemental material). A total of 39 shared genes were identified between the two comparisons, indicating specific modulation during infection and progression to HCC (Fig. 4c and Table S1 in the supplemental material).
FIG. 4.
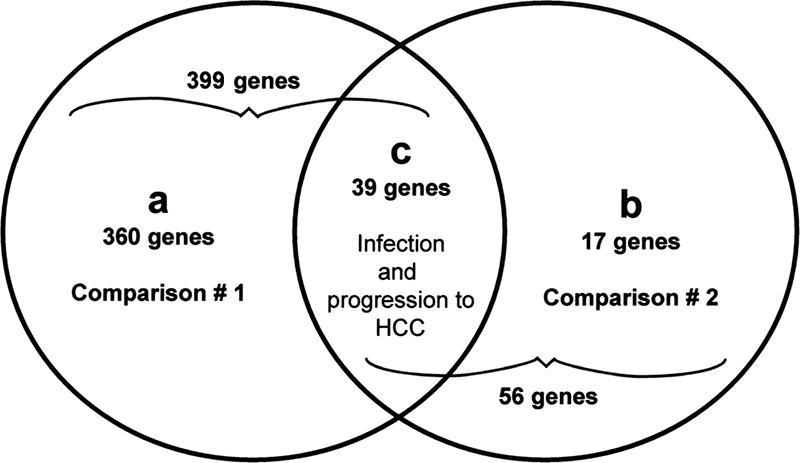
Venn diagram of the genes that were identified as differentially expressed using two criteria: (i) the expression difference between groups was P < 0.05 (analysis of variance) and (ii) the expression ratio was ≥1.5 or ≤−1.5. A total of 399 and 56 genes were identified as differentially transcribed in comparison 1 (infected mice with HCC versus uninfected control) and comparison 2 (infected mice with HCC versus infected mice with FAH), respectively. A total of 360 of 399 genes were unique to comparison 1 (a), and 17 of 56 genes were unique to comparison 2 (b). A total of 39 genes were common to both comparisons (c).
To identify putative biological pathways associated with infection and progression to HCC, we analyzed the gene lists for enrichment of Biocarta pathways (see Materials and Methods). For the 360 unique genes differentially transcribed in H. hepaticus-induced HCC (Fig. 4a and Table S1 in the supplemental material), gene ontology analysis identified significant associations with specific Biocarta pathways, including NF-κB, tumor necrosis factor receptor 2, mitogen-activated protein kinase (MAPK), acetylation and deacetylation of relA in the nucleus, ataxia telangiectasia mutated, and cell cycle (G2/M checkpoint) signaling genes and others (see Table S2 in the supplemental material).
For the 17 unique genes differentially transcribed in HCC relative to FAH (Fig. 4b and Table S1 in the supplemental material), no Biocarta pathways were enriched; however, 4 of the 17 genes play important roles in lipid metabolism. One of the most upregulated genes was the Scd1 gene, which encodes steaoryl-coenzyme A desaturase 1, the central lipogenic enzyme catalyzing in vivo reactions in the synthesis of monounsaturated fatty acids. Other lipogenic genes included the Fasn (fatty acid synthase), Pgd (phosphogluconate dehydrogenase), and Mod1 (malic enzyme) genes. Errfi1 (ERBB receptor feedback inhibitor 1), also known as Mig6, a novel tumor suppressor gene, was downregulated. For the 39 shared genes differentially transcribed in infection and progression to HCC (Fig. 4c and Table S1 in the supplemental material), significant associations were identified with MAPK and p38 MAPK signaling Biocarta pathways (see Table S2 in the supplemental material).
Network analysis was then used to identify all known molecular interactions among the gene products of the transcripts modulated by infection and progression to HCC. Using the Ingenuity database, we identified significantly enriched molecular networks. For the 360 genes differentially transcribed in H. hepaticus-induced HCC (Fig. 4a), 16 significantly enriched networks were identified. The top two networks were associated with organismal injury, inflammation, and cancer (P < 10−57) (see Table S3 in the supplemental material). For the 17 genes differentially transcribed in HCC relative to FAH (Fig. 4b), a single significantly enriched network (P < 10−38) was identified. This network was associated with cancer, cell cycle, cellular growth and differentiation, lipid metabolism, and small molecule biochemistry (Fig. 5). For the 39 genes in common differentially transcribed in infection and progression to HCC (Fig. 4c), two highly significantly enriched networks (P < 10−34 and P < 10−29) were identified (Fig. 6a and b, respectively). The canonical pathway enriched within these two networks was immunological disease. Genes involved in immune response included the major histocompatibility genes H2-Aa, H2-Eb1, and H2-Ab1. Other genes included the Ikbkg, Stat1, Tgfb1, Ccl5, Cd44, Pla2g7, Cyba, and Rac2 genes.
FIG. 5.

Pathway mapping analysis identified a highly significant network between the proteins encoded by the transcripts differentially modulated (P < 10−38). Red indicates a protein encoded by a transcript significantly upregulated, and green indicates downregulation.
FIG. 6.

Pathway mapping analysis identified two highly significant networks between the proteins encoded by the transcripts differentially modulated (P < 10−34 and P < 10−29). Red indicates a protein encoded by a transcript significantly upregulated.
Real-time quantitative PCR.
Modulation of these genes was confirmed in comparisons 1 and 2, respectively, and is expressed as the relative fold change (Fig. 7): Scd1 (1.96 and 3.0), Fasn (1.35 and 2.25), Ikbkg (1.42 and 1.30), Tgfb1 (1.61 and 1.10), Stat1 (2.16 and 1.37), and Ccl5 (5.77 and 1.61). The expression of Elovl6 in comparison 1 (−2.05) was not consistent with the microarray analysis; however, upregulation was validated in comparison 2 (1.23).
FIG. 7.

Quantitative PCR analysis of selected genes in comparison 1 (infected mice with HCC versus uninfected control) and comparison 2 (infected mice with HCC versus infected mice with foci of altered or dysplastic hepatocytes). The results are expressed as the relative fold change. *, Validated genes (13 of 14 genes).
DISCUSSION
H. hepaticus is the prototype carcinogenic enterohepatic Helicobacter species and a close relative of H. pylori (12, 58). H. hepaticus infection in susceptible mouse strains has been used as a model to study the pathogenesis of liver, colon, and breast cancer (13, 20, 43, 47). H. hepaticus infection of A/J mice is associated with a Th1 cell-mediated immune response and results in chronic hepatitis (20, 46, 66). The cytolethal distending toxin of H. hepaticus appears to play a role in promoting dysplastic changes in the livers of H. hepaticus-infected A/JCr mice (23). The genetic susceptibility to H. hepaticus-induced hepatic inflammation and proliferation using AXB RI strains suggested a polygenic basis of disease resistance and susceptibility (29). The severity of hepatitis correlated with the development of liver tumors (29). Quantitative trait analysis linked susceptibility of AXB mice to H. hepaticus-induced HCC to loci on chromosome 19 (29). In the present study, we demonstrated that liver cancer susceptibility is codominantly inherited in the F1 generation of hybrid male mice derived from C57BL/6 and A/J (B6AF1 or AB6F1) chronically infected with H. hepaticus. Previous studies by the National Toxicology Program using naturally H. hepaticus-infected mice determined that B6C3F1 mice were susceptible to hepatitis, HCC, and hemangiosarcoma (4, 25).
Our study also demonstrates for the first time that H. hepaticus induces significant hepatic inflammation in C57BL/6 mice at 18 months p.i. (20 months of age). Previous epidemiological studies suggested that C57BL/6NCr mice were resistant to H. hepaticus-induced hepatitis at 12 months of age (64). The apparent differences in susceptibility to H. hepaticus-induced hepatitis may be due to the fact that in previous studies, mice had been evaluated at earlier time points and/or that there are host genetic differences between these substrains. As expected, A/J mice developed significant hepatitis; however, no liver tumors were observed. Previous studies using naturally H. hepaticus-infected A/JCr male mice documented a high prevalence of liver tumors at 19 months of age (64). The experimentally infected parental A/J mice in our study could have developed liver tumors at a later time point. Significant inflammation and cancer were observed in H. hepaticus-infected F1 male mice. Hepatitis in F1 mice appeared to correlate with the development of HCC. This association of inflammation leading to liver cancer has been previously observed in infected AXB RI mice (29). Male predominance of H. hepaticus-associated liver disease has also been documented in infected A/J and B6C3F1 mice but not in infected AXB RI mice (20, 29, 40, 46, 48). Similarly, a high susceptibility to gastric adenocarcinoma has been observed in H. pylori-infected male INS-GAS mice (21, 22, 41). These findings are consistent with the high incidence of HCC and gastric adenocarcinoma in men (2, 57).
In the present study, we used a method of analysis consisting of comparative transcriptome analysis between two group comparisons to determine that immunological disease was the canonical pathway enriched in the livers during H. hepaticus infection and progression to liver cancer. Hepatocarcinogenesis was characterized by upregulation of a tumor suppressor gene known as Ikbkg (NEMO), an essential regulatory component of the IKK complex required for NF-κB activation (34). Consistent with a p38 MAPK pathway, we observed upregulation of Stat1 and Tgfb1. Although Stat1 upregulation suggested an interferon-dependent mechanism of liver inflammation and injury, Tgfb1 upregulation suggested a compensatory mechanism to suppress inflammation and gamma interferon-induced STAT1 activation (32, 50, 60). The hepatic inflammatory profile included the upregulation of proinflammatory genes such as Ccl5 (RANTES) and Cd44. Ccl5 is involved in CD4+ T-cell and monocyte chemotaxis, whereas Cd44 functions as the signaling component of the macrophage inhibitory factor-CD74 receptor complex which activates innate immunity and plays a role in chronic inflammation and tumorigenesis (53). Increased expression of specific major histocompatibility complex genes was consistent with our studies in H. hepaticus A/JCr mice and suggested their possible role in host susceptibility (3). Major histocompatibility complex class II genes also influence the susceptibility of humans to chronic hepatitis C (27).
Oxidative stress may be involved in H. hepaticus-induced hepatocarcinogenesis. Of the 39 genes in common between the two group comparisons, 3 included upregulation of Pla2g7, Cyba, and Rac2. Pla2g7 plays a role in the generation of F2-isoprostanes, specific products of lipid peroxidation (56). Cyba and Rac2 encode subunits of NADPH oxidase involved in the generation, by phagocytic cells, of reactive oxygen species directed against bacteria (52). As a consequence, the presence of H. hepaticus in the liver induces inflammation in the surrounding hepatic tissue. Pla2g7 and Cyba have also been observed to be upregulated in A/JCr mice with preneoplastic liver lesions associated with H. hepaticus infection (3). Our findings are consistent with previous studies indicating increased oxidative damage in H. hepaticus-induced hepatitis and lipid peroxidation in H. hepaticus- and H. pylori-induced liver and gastric cancer, respectively (54, 55, 62).
The increased expression of lipogenic genes, including Fasn and Scd1, measured in H. hepaticus-induced liver cancer compared to FAH (dysplasia) is consistent with previous studies showing that certain cancers, including liver, colon, breast, prostate, lung, bladder, and stomach cancer, have high levels of lipid synthesis and expression of fatty acid synthase and steaoryl-coenzyme A desaturase (SCD1) (6). Fatty acid synthase and SCD1 are the two rate-limiting enzymes in lipogenesis and are induced by insulin (6). Overexpression of Fasn, which encodes the key enzyme of de novo fatty acid synthesis, has been previously demonstrated in glycogenotic FAH and HCC in rats (14). Overexpression of Scd1 has also been found in the livers of mouse (including C3H/He and B6C3F1) and rat strains genetically susceptible to hepatocarcinogenesis (15). Scd1 located on chromosome 19 may represent a downstream target of hepatocellular tumor-modifier loci in mice and rats (15). Scd1 controls proliferation and survival in human transformed cells (49). Scd1 also plays a crucial role in lipid metabolism, body weight control, and diet-induced hepatic insulin resistance (24, 31). The increased expression of genes involved in lipid metabolism correlated with the steatotic changes observed in HCC, suggesting that the dominant phenotypic background of hepatic steatosis coupled with the increased hepatic inflammation in infected F1 mice synergistically leads to liver tumors. In humans, fatty liver disease has an inherent propensity to progress to HCC (44).
In conclusion, the present study fulfills Koch's postulates that H. hepaticus is a hepatocarcinogen by illustrating the oncogenic potential of H. hepaticus in two susceptible F1 mouse strains. This novel mouse model provides an alternative for studies investigating the role of chronic microbial hepatitis and fatty liver in the pathogenesis of liver cancer. In addition, our findings suggest that H. hepaticus-infected genetically engineered mouse strains on a C57BL/6 background at 18 months p.i. can be used to study the pathogenesis of chronic hepatitis. The transcriptional profiling of hepatic genes in H. hepaticus-infected F1 male mice with liver cancer indicated that an immunological mediated disease process is associated with hepatocarcinogenesis. Likewise, immune effector mechanisms mediate liver injury in various liver diseases, including human viral hepatitis (26). Future studies exploring the role of Helicobacter spp. in human chronic hepatitis and liver cancer are warranted.
Supplementary Material
Acknowledgments
This study was supported by grants R01CA67529 (to J.G.F.), R01AI50952 (to J.G.F.), T32RR07036 (to J.G.F.), P01CA26731 (to J.G.F.), and P30ES02109 (to L.D.S. and J.G.F.) from the National Institutes of Health.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 19 February 2008.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Avenaud, P., A. Marais, L. Monteiro, B. Le Bail, P. Bioulac Sage, C. Balabaud, and F. Megraud. 2000. Detection of Helicobacter species in the liver of patients with and without primary liver carcinoma. Cancer 891431-1439. [PubMed] [Google Scholar]
- 2.Bosch, F. X., J. Ribes, R. Cleries, and M. Diaz. 2005. Epidemiology of hepatocellular carcinoma. Clin. Liver Dis. 9191-211. [DOI] [PubMed] [Google Scholar]
- 3.Boutin, S. R., A. B. Rogers, Z. Shen, R. C. Fry, J. A. Love, P. R. Nambiar, S. Suerbaum, and J. G. Fox. 2004. Hepatic temporal gene expression profiling in Helicobacter hepaticus-infected A/JCr mice. Toxicol. Pathol. 32678-693. [DOI] [PubMed] [Google Scholar]
- 4.Bucher, J. R., J. R. Hailey, J. R. Roycroft, J. K. Haseman, R. C. Sills, S. L. Grumbein, P. W. Mellick, and B. J. Chou. 1999. Inhalation toxicity and carcinogenicity studies of cobalt sulfate. Toxicol. Sci. 4956-67. [DOI] [PubMed] [Google Scholar]
- 5.Caldwell, S. H., D. M. Crespo, H. S. Kang, and A. M. Al-Osaimi. 2004. Obesity and hepatocellular carcinoma. Gastroenterology 127S97-S103. [DOI] [PubMed] [Google Scholar]
- 6.Chang, Y., J. Wang, X. Lu, D. P. Thewke, and R. J. Mason. 2005. KGF induces lipogenic genes through a PI3K and JNK/SREBP-1 pathway in H292 cells. J. Lipid Res. 462624-2635. [DOI] [PubMed] [Google Scholar]
- 7.Correa, P. 2004. The biological model of gastric carcinogenesis. IARC Sci. Publ. 2004301-310. [PubMed] [Google Scholar]
- 8.Davila, J. A., N. J. Petersena, H. A. Nelson, and H. B. El-Serag. 2003. Geographic variation within the United States in the incidence of hepatocellular carcinoma. J. Clin. Epidemiol. 56487-493. [DOI] [PubMed] [Google Scholar]
- 9.Dore, M. P., G. Realdi, D. Mura, D. Y. Graham, and A. R. Sepulveda. 2002. Helicobacter infection in patients with HCV-related chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Dig. Dis. Sci. 471638-1643. [DOI] [PubMed] [Google Scholar]
- 10.Downey, T. 2006. Analysis of a multifactor microarray study using Partek genomics solution. Methods Enzymol. 411256-270. [DOI] [PubMed] [Google Scholar]
- 11.El-Serag, H. B. 2004. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology 127S27-S34. [DOI] [PubMed] [Google Scholar]
- 12.Eppinger, M., C. Baar, G. Raddatz, D. H. Huson, and S. C. Schuster. 2004. Comparative analysis of four Campylobacterales. Nat. Rev. Microbiol. 2872-885. [DOI] [PubMed] [Google Scholar]
- 13.Erdman, S. E., V. P. Rao, T. Poutahidis, M. M. Ihrig, Z. Ge, Y. Feng, M. Tomczak, A. B. Rogers, B. H. Horwitz, and J. G. Fox. 2003. CD4+ CD25+ regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 636042-6050. [PubMed] [Google Scholar]
- 14.Evert, M., R. Schneider-Stock, and F. Dombrowski. 2005. Overexpression of fatty acid synthase in chemically and hormonally induced hepatocarcinogenesis of the rat. Lab. Investig. 8599-108. [DOI] [PubMed] [Google Scholar]
- 15.Falvella, F. S., R. M. Pascale, M. Gariboldi, G. Manenti, M. R. De Miglio, M. M. Simile, T. A. Dragani, and F. Feo. 2002. Stearoyl-CoA desaturase 1 (Scd1) gene overexpression is associated with genetic predisposition to hepatocarcinogenesis in mice and rats. Carcinogenesis 231933-1936. [DOI] [PubMed] [Google Scholar]
- 16.Fan, X. G., X. N. Peng, Y. Huang, J. Yakoob, Z. M. Wang, and Y. P. Chen. 2002. Helicobacter species ribosomal DNA recovered from the liver tissue of Chinese patients with primary hepatocellular carcinoma. Clin. Infect. Dis. 351555-1557. [DOI] [PubMed] [Google Scholar]
- 17.Farinha, P., and R. D. Gascoyne. 2005. Helicobacter pylori and MALT lymphoma. Gastroenterology 1281579-1605. [DOI] [PubMed] [Google Scholar]
- 18.Fox, J. G., F. E. Dewhirst, Z. Shen, Y. Feng, N. S. Taylor, B. J. Paster, R. L. Ericson, C. N. Lau, P. Correa, J. C. Araya, and I. Roa. 1998. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 114755-763. [DOI] [PubMed] [Google Scholar]
- 19.Fox, J. G., F. E. Dewhirst, J. G. Tully, B. J. Paster, L. Yan, N. S. Taylor, M. J. Collins, Jr., P. L. Gorelick, and J. M. Ward. 1994. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 321238-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox, J. G., X. Li, L. Yan, R. J. Cahill, R. Hurley, R. Lewis, and J. C. Murphy. 1996. Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection: a model of Helicobacter-induced carcinogenesis. Infect. Immun. 641548-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox, J. G., A. B. Rogers, M. Ihrig, N. S. Taylor, M. T. Whary, G. Dockray, A. Varro, and T. C. Wang. 2003. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 63942-950. [PubMed] [Google Scholar]
- 22.Fox, J. G., T. C. Wang, A. B. Rogers, T. Poutahidis, Z. Ge, N. Taylor, C. A. Dangler, D. A. Israel, U. Krishna, K. Gaus, and R. M. Peek, Jr. 2003. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 1241879-1890. [DOI] [PubMed] [Google Scholar]
- 23.Ge, Z., A. B. Rogers, Y. Feng, A. Lee, S. Xu, N. S. Taylor, and J. G. Fox. 2007. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 92070-2080. [DOI] [PubMed] [Google Scholar]
- 24.Gutierrez-Juarez, R., A. Pocai, C. Mulas, H. Ono, S. Bhanot, B. P. Monia, and L. Rossetti. 2006. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Investig. 1161686-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hailey, J. R., J. K. Haseman, J. R. Bucher, A. E. Radovsky, D. E. Malarkey, R. T. Miller, A. Nyska, and R. R. Maronpot. 1998. Impact of Helicobacter hepaticus infection in B6C3F1 mice from twelve National Toxicology Program two-year carcinogenesis studies. Toxicol. Pathol. 26602-611. [DOI] [PubMed] [Google Scholar]
- 26.Herkel, J., M. Schuchmann, G. Tiegs, and A. W. Lohse. 2005. Immune-mediated liver injury. J. Hepatol. 42920-923. [DOI] [PubMed] [Google Scholar]
- 27.Hohler, T., G. Gerken, A. Notghi, P. Knolle, R. Lubjuhn, H. Taheri, P. M. Schneider, K. H. Meyer zum Buschenfelde, and C. Rittner. 1997. MHC class II genes influence the susceptibility to chronic active hepatitis C. J. Hepatol. 27259-264. [DOI] [PubMed] [Google Scholar]
- 28.Huang, Y., X. G. Fan, Z. M. Wang, J. H. Zhou, X. F. Tian, and N. Li. 2004. Identification of Helicobacter species in human liver samples from patients with primary hepatocellular carcinoma. J. Clin. Pathol. 571273-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ihrig, M., M. D. Schrenzel, and J. G. Fox. 1999. Differential susceptibility to hepatic inflammation and proliferation in AXB recombinant inbred mice chronically infected with Helicobacter hepaticus. Am. J. Pathol. 155571-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irizarry, R. A., B. M. Bolstad, F. Collin, L. M. Cope, B. Hobbs, and T. P. Speed. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang, G., Z. Li, F. Liu, K. Ellsworth, Q. Dallas-Yang, M. Wu, J. Ronan, C. Esau, C. Murphy, D. Szalkowski, R. Bergeron, T. Doebber, and B. B. Zhang. 2005. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Investig. 1151030-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim, H. S., and M. S. Lee. 2007. STAT1 as a key modulator of cell death. Cell Signal. 19454-465. [DOI] [PubMed] [Google Scholar]
- 33.Lacroix-Triki, M., L. Lacoste-Collin, S. Jozan, J. P. Charlet, C. Caratero, and M. Courtade. 2003. Histiocytic sarcoma in C57BL/6J female mice is associated with liver hematopoiesis: review of 41 cases. Toxicol. Pathol. 31304-309. [DOI] [PubMed] [Google Scholar]
- 34.Luedde, T., N. Beraza, V. Kotsikoris, G. van Loo, A. Nenci, R. De Vos, T. Roskams, C. Trautwein, and M. Pasparakis. 2007. Deletion of NEMO/IKKγ in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 11119-132. [DOI] [PubMed] [Google Scholar]
- 35.Matsukura, N., S. Yokomuro, S. Yamada, T. Tajiri, T. Sundo, T. Hadama, S. Kamiya, Z. Naito, and J. G. Fox. 2002. Association between Helicobacter bilis in bile and biliary tract malignancies: H. bilis in bile from Japanese and Thai patients with benign and malignant diseases in the biliary tract. Jpn. J. Cancer Res. 93842-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurer, K. J., M. M. Ihrig, A. B. Rogers, V. Ng, G. Bouchard, M. R. Leonard, M. C. Carey, and J. G. Fox. 2005. Identification of cholelithogenic enterohepatic Helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology 1281023-1033. [DOI] [PubMed] [Google Scholar]
- 37.Maurer, K. J., V. P. Rao, Z. Ge, A. B. Rogers, T. J. Oura, M. C. Carey, and J. G. Fox. 2007. T-cell function is critical for murine cholesterol gallstone formation. Gastroenterology 1331304-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurer, K. J., A. B. Rogers, Z. Ge, A. J. Wiese, M. C. Carey, and J. G. Fox. 2006. Helicobacter pylori and cholesterol gallstone formation in C57L/J. mice: a prospective study. Am. J. Physiol. Gastrointest. Liver Physiol. 290G175-G182. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson, H. O., J. Taneera, M. Castedal, E. Glatz, R. Olsson, and T. Wadstrom. 2000. Identification of Helicobacter pylori and other Helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary sclerosing cholangitis or primary biliary cirrhosis. J. Clin. Microbiol. 381072-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nyska, A., R. R. Maronpot, S. R. Eldridge, J. K. Haseman, and J. R. Hailey. 1997. Alteration in cell kinetics in control B6C3F1 mice infected with Helicobacter hepaticus. Toxicol. Pathol. 25591-596. [DOI] [PubMed] [Google Scholar]
- 41.Ohtani, M., A. Garcia, A. B. Rogers, Z. Ge, N. S. Taylor, S. Xu, K. Watanabe, R. P. Marini, M. T. Whary, T. C. Wang, and J. G. Fox. 2007. Protective role of 17β-estradiol against the development of Helicobacter pylori-induced gastric cancer in INS-GAS mice. Carcinogenesis 282597-2604. [DOI] [PubMed] [Google Scholar]
- 42.Pellicano, R., V. Mazzaferro, W. F. Grigioni, M. A. Cutufia, S. Fagoonee, L. Silengo, M. Rizzetto, and A. Ponzetto. 2004. Helicobacter species sequences in liver samples from patients with or without hepatocellular carcinoma. World J. Gastroenterol. 10598-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao, V. P., T. Poutahidis, Z. Ge, P. R. Nambiar, C. Boussahmain, Y. Y. Wang, B. H. Horwitz, J. G. Fox, and S. E. Erdman. 2006. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 667395-7400. [DOI] [PubMed] [Google Scholar]
- 44.Reddy, J. K., and M. S. Rao. 2006. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest Liver Physiol. 290G852-G858. [DOI] [PubMed] [Google Scholar]
- 45.Rocha, M., P. Avenaud, A. Menard, B. Le Bail, C. Balabaud, P. Bioulac-Sage, D. M. de Magalhaesq Ueiroz, and F. Megraud. 2005. Association of Helicobacter species with hepatitis C cirrhosis with or without hepatocellular carcinoma. Gut 54396-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rogers, A. B., S. R. Boutin, M. T. Whary, N. Sundina, Z. Ge, K. Cormier, and J. G. Fox. 2004. Progression of chronic hepatitis and preneoplasia in Helicobacter hepaticus-infected A/JCr mice. Toxicol. Pathol. 32668-677. [DOI] [PubMed] [Google Scholar]
- 47.Rogers, A. B., and J. G. Fox. 2004. Inflammation and Cancer I. Rodent models of infectious gastrointestinal and liver cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 286G361-G366. [DOI] [PubMed] [Google Scholar]
- 48.Rogers, A. B., E. J. Theve, Y. Feng, R. C. Fry, K. Taghizadeh, K. M. Clapp, C. Boussahmain, K. S. Cormier, and J. G. Fox. 2007. Hepatocellular carcinoma associated with liver-gender disruption in male mice. Cancer Res. 6711536-11546. [DOI] [PubMed] [Google Scholar]
- 49.Scaglia, N., and R. A. Igal. 2005. Stearoyl-CoA desaturase is involved in the control of proliferation, anchorage-independent growth, and survival in human transformed cells. J. Biol. Chem. 28025339-25349. [DOI] [PubMed] [Google Scholar]
- 50.Schramm, C., M. Protschka, H. H. Kohler, J. Podlech, M. J. Reddehase, P. Schirmacher, P. R. Galle, A. W. Lohse, and M. Blessing. 2003. Impairment of TGF-β signaling in T cells increases susceptibility to experimental autoimmune hepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 284G525-G535. [DOI] [PubMed] [Google Scholar]
- 51.Shames, B., J. G. Fox, F. Dewhirst, L. Yan, Z. Shen, and N. S. Taylor. 1995. Identification of widespread Helicobacter hepaticus infection in feces in commercial mouse colonies by culture and PCR assay. J. Clin. Microbiol. 332968-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheppard, F. R., M. R. Kelher, E. E. Moore, N. J. McLaughlin, A. Banerjee, and C. C. Silliman. 2005. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 781025-1042. [DOI] [PubMed] [Google Scholar]
- 53.Shi, X., L. Leng, T. Wang, W. Wang, X. Du, J. Li, C. McDonald, Z. Chen, J. W. Murphy, E. Lolis, P. Noble, W. Knudson, and R. Bucala. 2006. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25595-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh, R., C. Leuratti, S. Josyula, M. A. Sipowicz, B. A. Diwan, K. S. Kasprzak, H. A. Schut, L. J. Marnett, L. M. Anderson, and D. E. Shuker. 2001. Lobe-specific increases in malondialdehyde DNA adduct formation in the livers of mice following infection with Helicobacter hepaticus. Carcinogenesis 221281-1287. [DOI] [PubMed] [Google Scholar]
- 55.Sipowicz, M. A., P. Chomarat, B. A. Diwan, M. A. Anver, Y. C. Awasthi, J. M. Ward, J. M. Rice, K. S. Kasprzak, C. P. Wild, and L. M. Anderson. 1997. Increased oxidative DNA damage and hepatocyte overexpression of specific cytochrome P450 isoforms in hepatitis of mice infected with Helicobacter hepaticus. Am. J. Pathol. 151933-941. [PMC free article] [PubMed] [Google Scholar]
- 56.Stafforini, D. M., J. R. Sheller, T. S. Blackwell, A. Sapirstein, F. E. Yull, T. M. McIntyre, J. V. Bonventre, S. M. Prescott, and L. J. Roberts, 2nd. 2006. Release of free F2-isoprostanes from esterified phospholipids is catalyzed by intracellular and plasma platelet-activating factor acetylhydrolases. J. Biol. Chem. 2814616-4623. [DOI] [PubMed] [Google Scholar]
- 57.Stoicov, C., R. Saffari, X. Cai, C. Hasyagar, and J. Houghton. 2004. Molecular biology of gastric cancer: Helicobacter infection and gastric adenocarcinoma: bacterial and host factors responsible for altered growth signaling. Gene 3411-17. [DOI] [PubMed] [Google Scholar]
- 58.Suerbaum, S., C. Josenhans, T. Sterzenbach, B. Drescher, P. Brandt, M. Bell, M. Droge, B. Fartmann, H. P. Fischer, Z. Ge, A. Horster, R. Holland, K. Klein, J. Konig, L. Macko, G. L. Mendz, G. Nyakatura, D. B. Schauer, Z. Shen, J. Weber, M. Frosch, and J. G. Fox. 2003. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. USA 1007901-7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sutton, P., T. Kolesnikow, S. Danon, J. Wilson, and A. Lee. 2000. Dominant nonresponsiveness to Helicobacter pylori infection is associated with production of interleukin 10 but not gamma interferon. Infect. Immun. 684802-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takaki, H., Y. Minoda, K. Koga, G. Takaesu, A. Yoshimura, and T. Kobayashi. 2006. TGF-β1 suppresses IFN-γ-induced NO production in macrophages by suppressing STAT1 activation and accelerating iNOS protein degradation. Genes Cells 11871-882. [DOI] [PubMed] [Google Scholar]
- 61.Vorobjova, T., I. Nilsson, S. Terjajev, M. Granholm, M. Lyyra, T. Porkka, T. Prukk, R. Salupere, H. I. Maaroos, T. Wadstrom, and R. Uibo. 2006. Serum antibodies to enterohepatic Helicobacter spp. in patients with chronic liver diseases and in a population with high prevalence of H. pylori infection. Dig. Liver Dis. 38171-176. [DOI] [PubMed] [Google Scholar]
- 62.Wang, G., Y. Hong, M. K. Johnson, and R. J. Maier. 2006. Lipid peroxidation as a source of oxidative damage in Helicobacter pylori: protective roles of peroxiredoxins. Biochim. Biophys. Acta 17601596-1603. [DOI] [PubMed] [Google Scholar]
- 63.Ward, J. M., M. R. Anver, D. C. Haines, and R. E. Benveniste. 1994. Chronic active hepatitis in mice caused by Helicobacter hepaticus. Am. J. Pathol. 145959-968. [PMC free article] [PubMed] [Google Scholar]
- 64.Ward, J. M., J. G. Fox, M. R. Anver, D. C. Haines, C. V. George, M. J. Collins, Jr., P. L. Gorelick, K. Nagashima, M. A. Gonda, R. V. Gilden, et al. 1994. Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J. Natl. Cancer Inst. 861222-1227. [DOI] [PubMed] [Google Scholar]
- 65.Whary, M. T., J. Cline, A. King, Z. Ge, Z. Shen, B. Sheppard, and J. G. Fox. 2001. Long-term colonization levels of Helicobacter hepaticus in the cecum of hepatitis-prone A/JCr mice are significantly lower than those in hepatitis-resistant C57BL/6 mice. Comp. Med. 51413-417. [PubMed] [Google Scholar]
- 66.Whary, M. T., T. J. Morgan, C. A. Dangler, K. J. Gaudes, N. S. Taylor, and J. G. Fox. 1998. Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect. Immun. 663142-3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang, B., S. Kirov, and J. Snoddy. 2005. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33W741-W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.