

Abstract
Influenza A virus is one of the best-studied viruses and a model organism for the study of molecular evolution; in particular, much research has focused on detecting natural selection on influenza virus proteins. Here, we study the dynamics of the synonymous and nonsynonymous nucleotide composition of influenza A virus genes. In several genes, the nucleotide frequencies at synonymous positions drift away from the equilibria predicted from the synonymous substitution matrices. We investigate possible reasons for this unexpected behavior by fitting several regression models. Relaxation toward a mutation-selection equilibrium following a host jump fails to explain the dynamics of the synonymous nucleotide composition, even if we allow for slow temporal changes in the substitution matrix. Instead, we find that deep internal branches of the phylogeny show distinct patterns of nucleotide substitution and that these branches strongly influence the dynamics of nucleotide composition, suggesting that the observed trends are at least in part a result of natural selection acting on synonymous sites. Moreover, we find that the dynamics of the nucleotide composition at synonymous and nonsynonymous sites are highly correlated, providing evidence that even nonsynonymous sites can be influenced by selection pressure for nucleotide composition.
Influenza A virus is one the most significant causes of annual morbidity and mortality in humans (22) and has one of the largest databases of sequenced genes. There is a considerable body of work on the molecular evolution of influenza A virus in general (e.g., references 3, 8, 11, 23, 34, and 38) and on the nucleotide composition of the influenza A virus genes in particular (1, 13, 26, 41). Many of the recent studies have focused on detecting positive selection acting on the amino acid sequences of the influenza A virus proteins (3, 31, 37, 38). These studies utilize one or another variation of the dN/dS ratio test, which relies on the assumption of neutrality of synonymous mutations. Whether the synonymous mutations are selectively neutral is, therefore, not only an interesting question in itself but also important for understanding selection pressures on the protein level.
Here, we find evidence that selection for nucleotide composition shaped nucleotide usage at synonymous as well as at nonsynonymous sites in human influenza A viruses. Consistent with earlier observations (26), we observe that the nucleotide composition at the fourfold degenerate (FFD) and second codon position (SCP) sites of the 10 human influenza A virus genes (excluding PB1-F2) has been changing with time. This, in itself, is not surprising and not evidence for selection. Since all considered genes are known or suspected to have entered the genomes of human-adapted influenza viruses relatively recently (5, 33), it is natural to suppose that the mutational and/or selection pressure on these genes has changed. Consequently, one expects to observe a “relaxation” of the nucleotide composition toward a mutation-selection equilibrium determined by the substitution matrix. To test this, we have inferred the equilibrium nucleotide composition for each gene on the basis of the observed frequencies of different nucleotide substitutions on branches of the tree. Surprisingly, we observe that the frequencies of certain nucleotides in some genes drift away from the predicted equilibrium values. Seemingly paradoxical, this effect can be explained if the relative probabilities of different substitutions (substitution matrix) are changing with time. Such changes could be caused either by changes of the selection pressures or by changes in the mutation rates; it is difficult to discriminate between these two alternatives.
The divergence of the nucleotide composition away from the predicted equilibrium can also be explained by constant natural selection for nucleotide composition. Indeed, while natural selection is obviously involved in shaping the tree-specific substitution matrices, it also has a more subtle effect on the shape of the tree itself: variants that acquire deleterious mutations are more likely to go extinct and give rise to fewer offspring and, therefore, appear on branches that are close to the leaves of the phylogeny, while variants that acquire beneficial mutations are likely to produce more offspring and, therefore, appear on deep internal branches of the phylogeny. Thus, if selection played a role in shaping the phylogenies of the influenza A virus genes, then substitutions on deep internal branches would, on average, be more selectively advantageous than those on terminal branches (10, 25). Therefore, substitution patterns would be different between the more selection-driven substitutions on deep internal branches and the more mutation-driven substitutions on terminal branches, especially if the mutation and selection pressures happen to oppose each other. In fact, we observe such differences at both synonymous and nonsynonymous sites.
Influenza A virus gene phylogenies typically have distinct trunks, i.e., only one of the coexisting lineages survives in the long run. Since the variants on the trunk are the ancestors of all variants in future years (8), the long-term nucleotide composition dynamics is more strongly influenced by the more-adaptive substitutions on the trunk than by the less-adaptive substitutions on nontrunk branches. In other words, if a certain nucleotide is gained (lost) on the trunk, we expect to see an increase (decrease) in the corresponding nucleotide frequency over the years. To systematically test this hypothesis, we propose four linear regression models that predict the dynamics of the nucleotide frequencies. It turns out that the equilibrium inferred from the overall substitution matrix is a poor predictor for the nucleotide composition dynamics. If constant selection is explicitly taken into account, we can predict the evolution of the synonymous nucleotide composition significantly better. Prediction is further improved by allowing for slow changes of the substitution matrix through time. The latter is also true for the nonsynonymous nucleotide composition. Thus, without rejecting the hypothesis that the substitution processes change over time, we find strong evidence for selection for nucleotide composition influencing nucleotide usage at synonymous and at nonsynonymous sites.
MATERIALS AND METHODS
Data sets.
We downloaded all sequences of human influenza A H1N1, H2N2, and H3N2 viruses, from all viral segments available from the NCBI Influenza Virus Resource at the end of April 2006. Sequences were aligned using ClustalW version 1.83 (35), and coding regions were extracted. Occasional gaps were filled if more than 80% of sequences agreed on the symbol at the gap position; otherwise, the sequence with a gap was excluded from further analysis. In order to retain more sequences, we analyzed only the HA1 part of the HA(1) and HA(3) proteins. Regions overlapping between the M1 and M2 genes, and between the NS1 and NEP (also known as NS2) genes, were excluded from analyses.
The sequence accession numbers and/or sequence alignments we obtained are available upon request.
Phylogenetic trees.
We reconstructed the phylogenetic trees of all influenza A virus genes with PAUP version 4.0b10 (32). We reconstructed the topology of the phylogenetic trees using the neighbor-joining (NJ) method with the BreakTies RANDOM option. We used the NJ algorithm for reconstructing the tree topology because of its computational efficiency. We investigated the sensitivity of our results to this approach by reconstructing a maximum parsimony topology for the HA1 part of the HA(3) gene and for the PB2 gene and found that the synonymous nucleotide substitution matrices inferred from these trees are very similar to those inferred from NJ trees. Branch lengths and ancestral states were inferred using maximum parsimony (MP) with the ACCTRAN option. However, the results of our analysis were similar when the sequences at internal nodes were reconstructed using maximum likelihood with the GTR + I model (data not shown).
We reconstructed a total of 11 trees: one tree for each of the PB2, PB1(1), PB1(3), PA, HA(1), HA(3), NP, NA(1), and NA(2) genes; one tree for the M1 and M2 genes together; one tree for NS1 and NEP genes together. When reconstructing the tree for M1 and M2, as well as the tree for NS1 and NEP, the full coding regions of the corresponding genes, including the overlapping parts, were used. The PB1(1), HA(1), and NA(1) trees were reconstructed using H1N1 sequences. Although the H1N1 variants of these genes are not present in the data set between 1957 and 1977, this gap should not affect our analyses because the 1977 variants are very similar to pre-1957 variants. The PB1(3) and the HA(3) trees were based on H3N2 sequences. The NA(2) tree was based on H2N2 and H3N2 sequences. The remaining five trees were based on data from all subtypes. We did not reconstruct trees for PB1(2) and HA(2) because the corresponding H2N2 data sets were too small for reliable inference.
The phylogenetic trees used in the analysis are available upon request.
Nucleotide substitution rates.
To characterize patterns of nucleotide replacement, we estimated the synonymous and nonsynonymous nucleotide substitution rates using a simple counting method similar to that developed by Nei and Gojobori (20), instead of maximum likelihood methods (40) such as those implemented in PAML (39). PAML is often superior to heuristic techniques because it accounts for all possibilities of multiple substitutions between the sequences under comparison (36). In the case of influenza virus data, however, adjacent nodes in the reconstructed phylogenies typically differ by no more than one mutation per codon site. Therefore, multiple mutations can be safely ignored, and the Nei-Gojobori-like method is expected to perform well (14, 19), while avoiding problems associated with numerical maximization over high-dimensional parameter spaces.
The following estimator for the synonymous substitution rate is constructed analogously to the Nei-Gojobori estimator (20).
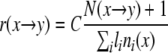 |
(1) |
Here, r(x→y) denotes the estimated synonymous nucleotide substitution rate from nucleotide x to nucleotide y (other than x); N(x→y) is the number of substitutions of x with y on the tree, at FFD sites; li is the branch length of branch i measured in total number of substitutions at FFD sites; ni(x) is the fraction of FFD sites occupied by nucleotide x in the parental sequence of the i-th branch; the sum in the denominator is taken over all branches. Thus, the denominator represents the opportunities for a substitution of a particular type to occur across the tree. The 1 is added to the numerator to avoid numerical instabilities that could arise when few (or no) events of a particular type are observed. The coefficient C is chosen so that the sum of all rates r(x→y) equals 1.
An analogous estimator was used for SCP sites.
Equilibrium nucleotide frequencies.
We use our estimated nucleotide substitution rates to calculate the corresponding predicted equilibrium nucleotide frequencies. Consider a four-by-four nucleotide substitution matrix R whose entries are Rxy = r(y→x) if x ≠ y and Rxx = −Σy:y≠xr(x→y) for x,y ∈{A, C, G, T}. Under the simplest model, the nucleotide frequency vector n (nucleotide composition) evolves according to the equation n˙ = Rn. The equilibrium nucleotide composition ne corresponding to substitution matrix R is the vector satisfying Rne = 0, and Σxne(x) = 1.
Distribution of substitutions on a tree.
To explore possible reasons for discrepancies between observed nucleotide composition and predicted equilibrium nucleotide composition, we tested whether the distribution of nucleotide substitutions was different between different parts of a phylogenetic tree.
(i) Trunk statistic.
First, we defined the trunk of a phylogenetic tree as the set of the internal branches connecting the root of the tree to the most recent common ancestor of all sequences sampled in and after the year 2003 for PB1(1), HA(1), and NA(1) and the year 2005 for the remaining trees. These years are the latest years in our data set that were represented by more than one sequence of the corresponding genes.
Next, we define the expected rate of change of nucleotide frequency along a branch. Consider a branch i and suppose that the number of FFD (or SCP) sites occupied by the nucleotide x in the ancestral sequence is Xai(x), while the number in the descendant sequence is Xdi(x). Thus, the fraction of nucleotide x in the ancestral node is ni(x) = Xai(x)/ΣyXai(y). Given the nucleotide substitution rates inferred from the whole tree, r(x→y), one can calculate the expected number of sites occupied by the nucleotide x in the descendant sequence: X^di(x) = Xai(x) + {li/[CΣzXai(z)]} Σy:y≠x[Xai(y)r(y→x) − Xai(x)r(x→y)]. This follows directly from equation 1. Thus, for each branch i, we can obtain the difference Yi(x) = Xdi(x) − X^di(x) between the expected and observed (or inferred) counts of nucleotide x in the descendant sequence. Using the sampled randomization test (30), we tested whether the two samples, St = {Yi(x):i is a trunk branch} and Snt = {Yi(x):i is a nontrunk branch}, come from identical distributions. As the test statistic, we used the difference between empirical mean values of Yi(x) over two samples: δtrunk(x) = 〈Yi(x)〉St − 〈Yi(x)〉Snt. We call this value the “trunk statistic.” Informally, a positive (negative) δtrunk value shows how many additional residues of a particular nucleotide are gained (lost) on a typical trunk branch compared to the rest of the tree.
(ii) Time statistic.
In order to test whether the nucleotide substitution rates change with time, we split the tree into two parts corresponding to the first and second half of the time period over which the viruses bearing the corresponding gene circulated. Subdivision into more than two sets would cause undersampling problems in the subsets corresponding to early years. However, if there were a clear long-term trend in the changes of the substitution matrix, we would expect to capture it even with this crude subdivision. To divide branches into two groups according to time, we exploited the single-trunk shape of the phylogenetic tree and the fact that, on such trees, the distance from the root to leaf nodes grows linearly with time (8). We measured the total height, hT, of the tree, i.e., the number of substitutions from the root to the most distant leaf. Then, for each branch i, the height hi is defined as the distance from the root to the child node of this branch. Analogously to the above case, we compared two samples, Searly = {Yi(x):hi ≤ (hT/2)} and Slate = {Yi(x):hi > (hT/2)}, using the time statistic δtime(x) = 〈Yi(x)〉Slate − 〈Yi(x)〉Searly. Informally, a positive (negative) value δtime shows how many additional residues of a particular nucleotide are gained (lost) on a typical branch in the second half of the tree compared to the first half of the tree.
Statistical analysis of different mechanisms underlying the nucleotide composition dynamics.
Since we measured the trunk (time) statistics for all nucleotides of all genes, it is likely that some of our 4 by 11 (44) statistical values will show statistical significance due to random chance. However, since the statistical values for different nucleotides within a gene are not independent, the number of observed false positives is not distributed binomially with parameters of 44 and 0.05. We addressed this problem by noticing that, given that the null probability of observing a significant trunk (time) statistic for a particular nucleotide of a particular gene is 0.05, the total null probability of observing one or more significant values of the statistic in the gene cannot exceed 4 × 0.05, or 0.2. This directly follows from the inclusion-exclusion principle (6). Therefore, we used the exact binomial test with parameters of 13 and 0.2 to conservatively estimate a P value for the number of genes with at least one significant value of the trunk (time) statistic.
Next, we examined whether the time trend in nucleotide frequency (measured by the regression coefficient against time of isolation) correlates with (i) the distance of the nucleotide frequency (averaged over all sequences) to the equilibrium predicted by the nucleotide substitution matrix; (ii) the trunk statistic; and (iii) the time statistic. We fit four linear models.
For Model 1, r = α1d.
For Model 2, r = α2d + β2δtime.
For Model 3, r = α3d + γ3δtrunk.
For Model 4, r = α4d + β4δtime + γ4δtrunk.
Here, r is the vector of regression coefficients between the nucleotide frequency and time of isolation, d is the vector of distances to equilibria, i.e., differences between the average and the equilibrium nucleotide frequencies, and δtrunk and δtime are the vectors of the trunk and time statistic values, respectively. Before fitting the model, we normalized the data vectors to a mean of 0 and standard deviation of 1. Thus, αi, βi, and γi are standard partial regression coefficients.
To test the significance of the model fit, we fit the same linear model after permuting the entries of the distance to the equilibrium vector (for Model 1), time statistic vector (for Models 2 and 4), or trunk statistic vector (for Models 3 and 4) as described in the next subsection. To test whether Model 4 fits the data significantly better than either of the two-variable models, we performed two two-tailed permutation tests in which we permutated the entries of only one of the vectors, δtrunk or δtime. We called the obtained P values Ptrunk and Ptime, respectively.
Permutation test.
To conservatively test the significance of a correlation between vectors of statistics corresponding to all nucleotides of all genes, we employed a permutation test that preserves the nonindependence of the statistic values for different nucleotides within each gene. We permutated a statistic vector in the following way. First, we randomly permutated among each other the five groups of four values corresponding to nucleotides of different genes; then, within each of those groups, we permutated the values corresponding to different nucleotides of the same gene. Thus, the relationship between the statistic values corresponding to different nucleotides of the same gene was preserved in our permutation test. We use a two-tailed sampled permutation test to obtain the P values for the correlation coefficients.
RESULTS
We reconstructed the phylogenetic trees for all human influenza A virus genes, estimated the synonymous and nonsynonymous nucleotide substitution rates from all the branches of the trees, and inferred the corresponding equilibrium synonymous and nonsynonymous nucleotide compositions (see Materials and Methods).
To examine the temporal evolution of the synonymous nucleotide composition of the influenza virus genes at FFD sites, we calculated, for each gene, the linear regression coefficients between the nucleotide frequencies and the year of isolation. We found that the regression coefficients were significantly different from zero for at least one nucleotide in each gene, indicating that the synonymous nucleotide composition of influenza A virus has been changing during the course of virus evolution in the human host (Fig. 1; Table 1). Even though there is a strong variation in the number of sequences sampled in different years, by visually exploring the linear regression lines (Fig. 2) we observe that regression coefficients are not exclusively dominated by years with higher sample sizes but adequately describe the long-term temporal trends in the nucleotide frequency dynamics.
FIG. 1.

Absolute values of the linear regression coefficients between the nucleotide composition at FFD sites and the year of isolation of the sequence.
TABLE 1.
Dynamics of nucleotide frequencies at FFD sitesa
| Gene | Base | RC (10−4) | DE (10−2) | δtrunk (10−2) | δtime (10−2) |
|---|---|---|---|---|---|
| PB2 | A | 2.1** | 0.9 | −10.8 | 9.5 |
| C | 1.5** | 0.0 | 24.2* | −14.8** | |
| G | −6.9** | 0.2 | 2.1 | −19.1** | |
| T | 3.3** | −1.1 | −15.6 | 24.4** | |
| PB1(1) | A | −3.5** | −1.3 | −54.8 | 26.0 |
| C | 3.0** | −2.1 | −6.8 | 4.3 | |
| G | −1.2** | 3.3 | 42.7 | −27.5* | |
| T | 1.6** | 0.0 | 19.4 | −3.4 | |
| PB1(3) | A | −1.4** | 0.9 | −5.3 | −0.9 |
| C | 3.4** | −0.2 | 2.9 | 4.4 | |
| G | 1.5** | 0.5 | 14.5 | 0.2 | |
| T | −3.5** | −1.2 | −12.1 | −3.7 | |
| PA | A | 1.2** | −3.0 | −0.5 | 8.1 |
| C | 3.2** | 5.9 | 25.8* | 5.0 | |
| G | −0.4 | −2.2 | −4.4 | −12.0* | |
| T | −4.0** | −0.7 | −20.9* | −1.2 | |
| HA(1) | A | 3.6** | 1.4 | 14.6 | 13.5 |
| C | −2.5** | 4.5 | 12.2* | −28.2** | |
| G | −7.2** | −2.5 | −21.6* | −13.5* | |
| T | 6.1** | −3.4 | −5.2 | 28.2** | |
| HA(3) | A | 19.1** | 0.5 | 3.9 | −2.1 |
| C | 2.2** | −5.4 | 2.3 | 0.4 | |
| G | −24.1** | 5.5 | −4.6 | 1.1 | |
| T | 2.8** | −0.5 | −1.6 | 0.6 | |
| NP | A | 0.0 | −4.4 | −22.8* | 13.1** |
| C | −0.3** | −0.8 | −25.5** | −4.5 | |
| G | 0.4** | 5.6 | 64.0** | −7.9* | |
| T | −0.1 | −0.5 | −15.6* | −0.7 | |
| NA(1) | A | 8.9** | 2.8 | 61.5** | 14.3 |
| C | −2.0** | −1.9 | −37.8* | −3.8 | |
| G | −5.4** | −4.6 | −49.1* | −11.7 | |
| T | −1.5** | 3.7 | 25.3 | 1.2 | |
| NA(2) | A | 0.2** | −10.2 | −8.7 | 3.0 |
| C | 0.2** | 5.2 | 15.6** | −1.7 | |
| G | −0.3** | 0.3 | 3.3 | −2.5 | |
| T | −0.1** | 4.8 | −10.2 | 1.3 | |
| M1 | A | 0.9** | −0.6 | −0.2 | −2.4 |
| C | −2.2** | 2.1 | 0.5 | −0.2 | |
| G | 0.2 | 1.7 | 0.2 | 3.0 | |
| T | 1.1** | −3.1 | −0.5 | −0.4 | |
| M2 | A | 2.6** | −0.7 | 1.0 | −1.9* |
| C | 10.3** | −10.0 | −3.3 | −0.4 | |
| G | −0.1 | −3.8 | −4.2 | 1.5 | |
| T | −12.8** | 14.5 | 6.4* | 0.8 | |
| NS1 | A | 0.2 | 3.0 | 15.1** | −4.0 |
| C | −0.1 | 3.0 | −3.9 | −2.9 | |
| G | 0.2* | 0.6 | −6.3 | −0.5 | |
| T | −0.3** | −6.6 | −5.0 | 7.3** | |
| NEP | A | 4.6** | -8.2 | −6.1* | 0.0 |
| C | −8.0** | −1.4 | −0.5 | −1.4 | |
| G | 7.4** | 18.0 | 5.9* | −1.4 | |
| T | −4.1** | −8.4 | 0.6 | 2.9** |
RC, values of the linear regression coefficient between the nucleotide frequency and the time of isolation; DE, the difference between the average observed and the equilibrium nucleotide frequencies; δtrunk, the trunk statistic; δtime, the time statistic. Gene types are given in parentheses. Bold type indicates cases of divergence from the equilibrium. *, δtrunk or δtime statistical values significant at the 5% level; **, δtrunk or δtime statistical values significant at the 1% level (two-tailed test). A positive (negative) RC value implies that the frequency of the considered nucleotide is increasing (decreasing) with time. A positive (negative) DE value implies that the mean nucleotide frequency is above (below) the predicted equilibrium. If the product of RC and DE is positive (negative), the frequency of the considered nucleotide is diverging from (converging to) its equilibrium value. A positive (negative) δtrunk value shows how many additional residues of a particular nucleotide are gained (lost) on a typical trunk branch as compared to the rest of the tree. A positive (negative) δtime value shows how many additional residues of a particular nucleotide are gained (lost) on a typical branch in the second half of the tree as compared to the first half of the tree.
FIG. 2.

Selected cases of frequencies of nucleotides at FFD sites diverging away from the predicted equilibria. Best-fit lines (solid) are displayed for visual convenience only. Dashed lines indicate the equilibrium nucleotide frequencies as inferred from the full phylogenetic trees. Gray areas around these lines represent the 95% confidence intervals for the corresponding equilibria. Dash-dotted lines indicate the equilibrium nucleotide frequencies as inferred only from the trunk branches of the corresponding trees.
The frequencies of several nucleotides at FFD sites in several genes display unexpected dynamics: with time, they move away from the equilibria predicted from the synonymous substitution matrices (Fig. 2; Table 1). This apparent paradox implies that we cannot capture the correct dynamics of the synonymous nucleotide composition with just one set of nucleotide substitution rates. If rates differ substantially between different parts of the tree, the average rates we infer may lead to an incorrect prediction for the equilibrium nucleotide composition. Indeed, in several cases, divergence of the nucleotide frequency from the equilibrium coincides with large and significant differences in the synonymous substitution rates between parts of the tree (Table 1). As an example, consider the frequency of cytosine in the NA(2) gene. On the basis of the substitution matrix, it is expected to decrease (the equilibrium cytosine frequency is smaller than the observed frequency [Table 1]); in fact, however, it is increasing (Table 1; Fig. 2). Noticeably, cytosine is much more likely to be gained on a branch belonging to the tree trunk than elsewhere in the tree (Table 1), suggesting that the substitutions on the tree trunk are more important for its dynamics than the substitutions on nontrunk branches. Similarly, significant differences in the substitution patterns are observed between the early and the late parts of the tree, e.g., in the cytosine in PB2 and guanine in HA(1), as indicated by the significant values of the corresponding time statistics in Table 1. Significantly more genes in Table 1 have at least one significant trunk statistic value (P < 10−4) or time statistic value (P < 10−3) than expected randomly, which indicates that the significant values of the corresponding statistics in Table 1 are not just an artifact of multiple testing.
If the differences in substitution patterns between parts of the tree were, in fact, the cause of the observed discrepancies, we would expect that incorporating these differences into a model for the nucleotide frequency dynamics would lead to an improved fit to the data. To test this, we fit four linear regression models. The first model (Model 1) assumes a homogeneous substitution process; under this model, the frequency of each nucleotide always converges to the equilibrium predicted by the matrix of synonymous substitutions. The other three models (Models 2 to 4) incorporate variations in substitution rates within a tree, based on whether the nucleotide substitution rates differ between the “early” and the “late” halves of the tree (Model 2), the internal and the external branches of the tree (Model 3), or both (Model 4). We used the time and the trunk statistics as predictor variables to account for the inhomogeneity of substitution rates along the tree. In order to examine which of the four hypotheses explains the data better, we performed permutation analyses of the best-fit lines (see Materials and Methods) and found that the trunk statistic and the time statistic significantly improved model fit when considered together, and the trunk statistic significantly improved model fit even if considered separately from the time statistic (Table 2). Therefore, we can explain the discrepancy between expected and observed dynamics in nucleotide composition significantly better if we assume differences in substitution rates between different parts of the tree—in particular, between the trunk and the rest of the branches.
TABLE 2.
Standard partial regression and correlation coefficients for the four linear modelsa
| Model | αi | βi | γi | Ri2 |
|---|---|---|---|---|
| 1 | −0.16 | 0.02 | ||
| 2 | −0.12 | 0.20 | 0.07 (P = 0.12) | |
| 3 | −0.28 | 0.34 | 0.12 (P < 0.05) | |
| 4 | −0.26 | 0.27 | 0.39 | 0.19 (Ptime < 0.01, Ptrunk < 0.05) |
Standard partial regression and correlation coefficients (described in Materials and Methods) for predicting dynamics of the nucleotide composition at FFD sites. P values for Models 2 and 3 are for comparison with model 1. P values for Model 4 are for comparison with Models 2 (Ptrunk) and 3 (Ptime).
We performed an analogous analysis for the nucleotide composition at SCP sites. SCP sites are fully nondegenerate, i.e., each nucleotide change at the second codon position leads to an amino acid change and, therefore, the nucleotide composition at such sites is expected to be under stronger constraint than the nucleotide composition at FFD sites. Indeed, the nonsynonymous nucleotide composition changes much slower than the synonymous nucleotide composition: the regression coefficients between the nucleotide frequencies at SCP sites and the time of virus isolation are about 1 order of magnitude smaller than the corresponding regression coefficients for the synonymous nucleotide composition (Tables 1 and 3). Nevertheless, they are significantly different from zero for all genes (Table 3). Moreover, synonymous and nonsynonymous nucleotide compositions change in a strongly correlated manner, as measured by the corresponding regression coefficients between the nucleotide frequencies and the time of virus isolation: the standard regression coefficient between the vectors of regression coefficients at SCP and FFD sites is 0.43 (R2 = 0.19, permutation test [see Materials and Methods]; P < 0.05). Analogously to the synonymous nucleotide composition, we found that significantly more genes in Table 3 have at least one significant trunk statistic value (P < 0.01) or time statistic value (P < 0.01) than expected by chance, indicating that there are significant differences in the substitution patterns at SCP sites between the trunk and nontrunk branches, as well as between the early and the late parts of the phylogenetic trees. We also fit four linear regression models for predicting the nonsynonymous nucleotide composition dynamics and found that Model 4 explains the data significantly better than either Model 2 or Model 3, suggesting that the trunk effect as well as changes in the substitution patterns over time are important in determining the nonsynonymous nucleotide composition (Table 4).
TABLE 3.
Dynamics of nucleotide frequencies at SCP sitesa
| Gene | Base | RC (10−5) | DE (10−2) | δtrunk (10−2) | δtime (10−2) |
|---|---|---|---|---|---|
| PB2 | A | −5.8** | 4.4 | −8.9 | −2.0 |
| C | 0.0 | −1.4 | −2.8 | −1.6 | |
| G | 4.7** | 10.2 | 14.6* | 2.7 | |
| T | 1.1** | −13.3 | −2.9 | 0.9 | |
| PB1(1) | A | 5.7** | 13.5 | 24.9 | −8.7 |
| C | −0.1 | −6.0 | 2.7 | −13.6 | |
| G | −5.7** | 7.5 | −11.7 | −6.5 | |
| T | 0.0 | −14.9 | 2.7 | 8.4 | |
| PB1(3) | A | 4.4** | 9.3 | 7.9* | −9.6** |
| C | −1.5** | 1.6 | 0.4 | 2.7* | |
| G | −4.4** | −3.0 | −7.6 | 9.3** | |
| T | 1.4** | −7.9 | −0.8 | −2.4* | |
| PA | A | 11.4** | 15.4 | 21.8** | 3.6 |
| C | −1.9** | −4.1 | −3.5 | −7.2** | |
| G | −8.7** | 0.8 | −18.1** | −3.0 | |
| T | −0.8** | −12.1 | −0.2 | 6.5** | |
| HA(1) | A | −22.7** | 11.3 | 28.3* | −61.0** |
| C | −17.1** | 8.0 | −15.0* | 23.4** | |
| G | 16.4** | 1.2 | −11.0 | 26.2** | |
| T | 23.4** | −20.5 | −2.3 | 11.5* | |
| HA(3) | A | 55.5** | 2.2 | 8.5 | −1.6 |
| C | −4.9** | 2.1 | −1.1 | 0.9 | |
| G | −27.8** | 6.0 | 5.5 | −5.1* | |
| T | −22.8** | −10.3 | −12.9** | 5.8** | |
| NP | A | −1.0 | 2.5 | 31.2** | 0.3 |
| C | 0.3 | 5.6 | 4.9 | −4.5** | |
| G | 4.5** | −7.3 | −26.7** | 4.1* | |
| T | −3.9** | −0.7 | −9.4** | 0.0 | |
| NA(1) | A | 16.3** | −1.6 | −2.4 | −8.4 |
| C | 14.2** | −9.5 | 16.1 | 3.4 | |
| G | −27.7** | 3.8 | −12.5 | −3.2 | |
| T | −2.8** | 7.3 | −1.2 | 8.2 | |
| NA(2) | A | 0.8** | −4.0 | 5.5 | 2.0 |
| C | −0.1 | 0.6 | −5.3 | −3.6 | |
| G | −1.7** | −3.8 | −16.1** | 1.1 | |
| T | 1.0** | 7.3 | 15.9** | 0.5 | |
| M1 | A | 7.7** | 6.4 | −0.4 | 0.8 |
| C | 3.0** | 0.8 | 4.0 | −1.5 | |
| G | −7.4** | 3.7 | 3.3 | −3.4 | |
| T | −3.3** | −10.9 | −6.9 | 4.1** | |
| M2 | A | 8.7** | 9.7 | −0.4 | 0.8 |
| C | 6.7** | −10.7 | 4.0 | −1.5 | |
| G | −13.3** | 4.5 | 3.3 | −3.4 | |
| T | −2.1 | −3.5 | −6.9 | 4.1** | |
| NS1 | A | 28.0** | −0.6 | 7.0 | −1.4 |
| C | −18.6** | −3.9 | −5.9 | −1.3 | |
| G | −19.3** | 1.3 | −3.7 | 3.2 | |
| T | 9.9** | 3.2 | 2.7 | −0.4 | |
| NEP | A | −18.6** | 4.2 | 7.0 | −1.4 |
| C | 22.3** | −8.9 | −5.9 | −1.3 | |
| G | 18.7** | −2.6 | −3.7 | 3.2 | |
| T | −22.5** | 7.3 | 2.7 | −0.4 |
TABLE 4.
Standard partial regression and correlation coefficients for the four linear modelsa
| Model | αi | βi | γi | Ri2 |
|---|---|---|---|---|
| 1 | −0.19 | 0.04 | ||
| 2 | −0.15 | 0.15 | 0.06 (P = 0.13) | |
| 3 | −0.28 | 0.25 | 0.09 (P = 0.06) | |
| 4 | −0.25 | 0.28 | 0.36 | 0.15 (Ptime < 0.05, Ptrunk < 0.05) |
DISCUSSION
We predicted the gene-specific equilibrium nucleotide compositions on the basis of the substitution matrices inferred from substitutions at FFD and SCP sites. If the substitution probabilities were fully described by a constant nucleotide substitution matrix, then the nucleotide composition at the corresponding sites would eventually tend to the corresponding equilibrium. In contrast, we found that in several genes the frequencies of some nucleotides drift away from the predicted equilibria. This discrepancy between the expected and observed dynamics is apparently not due to artifacts of phylogenetic reconstruction, since different reconstruction methods produce similar results (see Materials and Methods). Within-subtype reassortment events have recently been shown to be common in influenza A virus (11) but cannot cause these discrepancies, because our analysis treats each gene separately. Within-segment recombination is a potential cause for concern but has not been observed for influenza A virus. Thus, there remain three potential causes for the observed discrepancies: the origin of the sequences, temporal changes in the substitution matrix, and natural selection for nucleotide composition. We discuss these scenarios below.
Origin of sequences.
Some, possibly all, human influenza A virus genes came relatively recently from avian influenza viruses (5, 33). Whether the genes came through reassortment events or a complete avian virus switched hosts, the mutation and selection pressures on the nucleotide composition of the gene are likely to have changed. For an individual gene, a host jump is similar to a horizontal gene transfer event, for example, in bacteria, where one organism acquires a new gene from another not necessarily closely related. If the donor and acceptor organisms have different equilibrium nucleotide frequencies due to differences in mutation biases, the nucleotide content in the newly acquired genes relaxes to the new equilibrium. This process is called amelioration (17). Amelioration is almost certain to have played a role in the dynamics of nucleotide composition in the human influenza A virus. However, by definition, it cannot lead to a steady drift of the nucleotide composition away from the predicted equilibrium.
Time-dependent mutation biases.
The equilibrium defined by the substitution matrix can be dynamic if the properties of the polymerase and/or selection pressure slowly change over time. This may lead to divergence of the nucleotide frequency from the calculated “average” equilibrium and, thus, potentially can explain the anomalous behavior of certain nucleotide frequencies for influenza A virus. Indeed, patterns of substitution of cytosine in the PB2 gene at FFD sites, guanine in the PB1(3) gene at SCP sites, etc., significantly differ between the “early” and the “late” halves of the trees (Tables 1 and 3). However, we observe no significant differences between the two halves of the tree in other anomalous cases [e.g., cytosine in NA(2) at FFD sites] and, in general, time-dependent changes in the substitution process do not substantially improve our ability to predict the synonymous or nonsynonymous nucleotide composition dynamics (Tables 2 and 4).
Natural selection and the “trunk effect.”
Mutations that have a selective advantage are more likely to be fixed in a population. This fact is reflected in the reconstructed phylogeny: one expects to find more selectively advantageous substitutions on branches that give rise to a large number of descendant branches and fewer on branches with fewer descendants (10, 21). Influenza A virus gene phylogenies have distinct trunks (2, 8). The sequences on the trunk are, on average, more fit than the sequences on the terminal branches (3, 25), and therefore the substitutions on the trunk (nontrunk) branches can be expected to be more beneficial (more deleterious).
If influenza virus genes evolved under constant selection for nucleotide usage, we would expect to find differences between the nucleotide substitution matrices inferred from internal versus external branches. Substitutions found on internal branches will, on average, be more advantageous and, thus, we expect the substitution matrix inferred from internal branches to be different from the substitution matrix inferred from external branches. We term the discrepancy between the two matrices the “trunk effect.” Lacking sufficient data to accurately infer trunk-specific synonymous nucleotide substitution matrices, we detected the trunk effect using the trunk statistic.
In order to infer the equilibrium nucleotide frequencies, we relied on the overall substitution matrix that is determined by substitutions on all branches of the tree. Since the trunk accounts for less than 10% of all branches, the (more-beneficial) substitutions that happen on trunk branches contribute relatively little to this matrix. However, since these substitutions happen in the sequences that produce more descendants, their influence on the nucleotide composition of future individuals is disproportionately high, potentially explaining the observed discrepancy between the equilibrium nucleotide frequencies inferred from the substitution matrix, influenced by more-deleterious mutations, and the largely selection-driven temporal dynamics of the nucleotide content.
To test this scenario, we assessed the differences in the substitution patterns between the trunk and nontrunk branches. Consistent with this hypothesis, we found that patterns of synonymous and nonsynonymous nucleotide substitutions in multiple genes are significantly different between the trunk and nontrunk branches (Tables 1 and 3).
Conceivably, the trunk effect could also be caused by the physical linkage between slightly deleterious and strongly beneficial mutations. Indeed, some influenza A virus genes, especially HA and NA, evolve under strong amino acid-level positive selection to evade the human immune response (3, 7, 31). Frequent selective sweeps associated with positive selection could possibly drive to fixation the hitchhiking, slightly deleterious mutations (9), including those disrupting the favored nucleotide composition. Conversely, negative selection could keep such weakly deleterious mutations at low frequencies on branches not experiencing the sweeps (i.e., nontrunk branches), possibly leading to a difference in the substitution matrix between the trunk and nontrunk branches. Although the combination of these factors could potentially lead to the observed trunk effect, this scenario appears to be less parsimonious, and it is also inconsistent with the observed correlation between the nucleotide dynamics at the FFD and SCP sites. Under either scenario, the observed trunk effect implies natural selection on nucleotide composition.
Forces affecting the nucleotide composition in influenza A virus.
Our results indicate that both effects, the effect of the time-varying substitution matrix and the trunk effect, are significant in several genes at both FFD and SCP sites (Tables 1 and 3). To test whether these effects can explain the anomalous nucleotide composition dynamics we observed, we fit four regression models and found that the trunk statistic significantly improved the prediction of the nucleotide frequency dynamics at FFD sites (Table 2). Moreover, the fit was further improved for the nucleotide composition at FFD and SCP sites if both the time and the trunk statistics were taken into account (Tables 3 and 4).
In all of our models, we observed a negative correlation between the direction of change of the nucleotide frequency (as described by the linear regression coefficient against time of isolation) and the distance to the overall equilibrium (as described by the difference between the observed and the equilibrium nucleotide frequencies), as would be expected if frequencies tended to move toward their equilibria. We also observed a positive correlation between the direction of change of the nucleotide frequency and the trunk statistic. This conforms with our explanation of how the trunk effect influences the dynamics of the nucleotide composition: if more residues of a particular nucleotide are gained on the trunk (the trunk statistic is positive), then the corresponding nucleotide frequency increases over time. In this sense, substitutions on the trunk are “more important,” as expected. We did not have a prior expectation as to which half of the tree is more important when the effect of the time-varying substitution matrix is considered. Our models reveal a positive correlation between the direction of change of the nucleotide frequency and the time statistic, implying that the later half of the tree is more important; this may have to do with the fact that there are many more sequences in the later halves of the phylogenetic trees than in the earlier halves.
Since the models were fit to normalized data, the corresponding partial regression coefficients indicate the relative importance of the effects that determine the direction of change of the nucleotide composition. The trunk effect appears to be the strongest force driving the synonymous and nonsynonymous nucleotide compositions, since the corresponding regression coefficients are the largest (Tables 2 and 4). This suggests that selection plays a significant role in the evolution of the synonymous and nonsynonymous nucleotide compositions of the influenza A virus genes. Although the observed trunk effect at the SCP sites may be a consequence of protein-level selection, the strong correlation between the nucleotide composition dynamics at synonymous and nonsynonymous sites suggests that both dynamics are governed by common forces, in particular by natural selection for nucleotide composition.
Mechanisms of selection for nucleotide composition.
Our results provide a strong case for natural selection for nucleotide composition at synonymous and nonsynonymous sites in genes with discrepancies between the expected and observed dynamics of the nucleotide composition. Moreover, we can pinpoint the role of selection in specific cases of observed divergence of nucleotide dynamics from equilibrium (Tables 1 and 3). Since we would not expect such selection to produce sign discrepancies in all cases, it is likely that selection is affecting the nucleotide composition dynamics in some other genes as well.
It is worth noting that two of the genes with the most rapidly changing synonymous nucleotide compositions (HA and NA) (Fig. 1) are the most important targets for the human immune system and are also known to be under the strongest selection at the protein level. Since many conventional methods of detecting natural selection rely on synonymous substitutions as the neutral “standard,” the estimates for the role of selection in the protein evolution of influenza virus (3, 31, 37, 38) may be affected by selection on synonymous substitutions. Several recent studies (15, 18) have already raised concerns about the application of dN/dS methods for detecting genes and sites under positive selection, although in a different context: these studies were concerned with the heterogeneity of synonymous substitution rates along the genetic sequence. In particular, it has been shown that the synonymous substitution rates in HA(3) are significantly nonuniform (15). Since synonymous substitution rate heterogeneity is likely to be an indicator of selection for nucleotide usage, it would be instructive to perform such an analysis in other influenza A virus genes as well, specifically, in those in which our analysis revealed a significant trunk effect.
We can think of several mechanisms of selection for nucleotide composition. It is known that different viral genes are expressed in an infected cell at different rates, at different instances, and in different quantities (27, 29). In those viral proteins that need to be expressed in large quantities (such as the nucleoprotein) or fast and early in the infection phase (such as the NS1 protein and the NEP), certain codons may be preferred to facilitate expression. At least three mechanisms are known by which nucleotide composition could affect expression efficiency. First, it is well established that some codons are more translationally efficient than others (12, 28). Second, it has been discovered recently that the nucleotide composition of a gene also influences its transcriptional efficiency (16). Third, nucleotide composition affects the secondary structure of mRNA and hence its stability and degradation rates (4). Finally, selection on synonymous sites could act through the secondary structure of the viral genomic RNA, which is known to interact with the nucleoprotein during the packaging and replication processes (24). Which of these or, perhaps, other processes influence the nucleotide composition of the influenza A virus genes remains an important open question.
Acknowledgments
We thank Wilfred Ndifon and Joshua Plotkin for valuable discussions in the process of manuscript preparation.
S.K. gratefully acknowledges financial support by the Burroughs Wellcome Fund Training Program in Biological Dynamics (1001782) and by DARPA grant HR0011-05-1-0057. G.A.B. gratefully acknowledges fellowships from the Pew Charitable Trusts, award 2000-002558, and the Burroughs Wellcome Fund, award 1001782, both to Princeton University, and the Molecular and Cellular Biology Program of the Russian Academy of Sciences. J.D. gratefully acknowledges financial support by NIH grant P50 GM071508.
Footnotes
Published ahead of print on 5 March 2008.
REFERENCES
- 1.Ahn, I., B.-J. Jeong, S.-E. Bae, J. Jung, and H. Son. 2006. Genomic analysis of influenza A viruses, including avian flu (H5N1) strains. Eur. J. Epidemiol. 21511-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buonagurio, D. A., S. Nakada, J. D. Parvin, M. Krystal, P. Palese, and W. M. Fitch. 1986. Evolution of human influenza A viruses over 50 years: rapid, uniform rate of change in NS gene. Science 232980-982. [DOI] [PubMed] [Google Scholar]
- 3.Bush, R. M., W. M. Fitch, C. A. Bender, and N. J. Cox. 1999. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol. Biol. Evol. 161457-1465. [DOI] [PubMed] [Google Scholar]
- 4.Chamary, J. V., J. L. Parmley, and L. D. Hurst. 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 798-108. [DOI] [PubMed] [Google Scholar]
- 5.Cox, N. J., and K. Subbarao. 2000. Global epidemiology of influenza: past and present. Annu. Rev. Med. 51407-421. [DOI] [PubMed] [Google Scholar]
- 6.Erickson, M. J. 1996. Introduction to combinatorics. Wiley-Interscience, New York, NY.
- 7.Fanning, T. G., A. H. Reid, and J. K. Taubenberger. 2000. Influenza a virus neuraminidase: regions of the protein potentially involved in virus-host interactions. Virology 276417-423. [DOI] [PubMed] [Google Scholar]
- 8.Fitch, W. M., R. M. Bush, C. A. Bender, and N. J. Cox. 1997. Long term trends in the evolution of H(3)NA1 human influenza type A. Proc. Natl. Acad. Sci. USA 947712-7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillespie, J. H. 2001. Is the population size of a species relevant to its evolution? Evolution 552161-2169. [DOI] [PubMed] [Google Scholar]
- 10.Golding, G. B. 1987. The detection of deleterious selection using ancestors inferred from a phylogenetic history. Genet. Res. 4971-82. [DOI] [PubMed] [Google Scholar]
- 11.Holmes, E. C., E. Ghedin, N. Miller, J. Taylor, Y. Bao, K. St. George, B. T. Grenfell, S. L. Salzberg, C. M. Fraser, D. J. Lipman, and J. K. Taubenberger. 2005. Whole-genome analysis of human influenza a virus lineages reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol. 3e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikemura, T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 213-34. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins, G. M., and E. C. Holmes. 2003. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 921-7. [DOI] [PubMed] [Google Scholar]
- 14.Kosakovsky Pond, S. L., and S. D. W. Frost. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 221208-1222. [DOI] [PubMed] [Google Scholar]
- 15.Kosakovsky Pond, S. L., and S. V. Muse. 2005. Site-to-site variation of synonymous substitution rates. Mol. Biol. Evol. 222375-2385. [DOI] [PubMed] [Google Scholar]
- 16.Kudla, G., L. Lipinski, F. Caffin, A. Helwak, and M. Zylicz. 2006. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 4e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence, J. G., and H. Ochman. 1997. Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 44383-397. [DOI] [PubMed] [Google Scholar]
- 18.Mayrose, I., A. Doron-Faigenboim, E. Bacharach, and T. Pupko. 2007. Towards realistic codon models: among site variability and dependency of synonymous and non-synononymous rates. Bioinformatics 23i319-i327. [DOI] [PubMed] [Google Scholar]
- 19.Muse, S. V. 1996. Estimating synonymous and nonsynonymous substitution rates. Mol. Biol. Evol. 13105-114. [DOI] [PubMed] [Google Scholar]
- 20.Nei, M., and T. Gojobori. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3418-426. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen, R., and D. M. Weinreich. 1999. The age of nonsynonymous and synonymous mutations in animal mtDNA and implications for the mildly deleterious theory. Genetics 153497-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palese, P. 2004. Influenza: old and new threats. Nat. Med. 1082-87. [DOI] [PubMed] [Google Scholar]
- 23.Plotkin, J., J. Dushoff, and S. A. Levin. 2002. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proc. Natl. Acad. Sci. USA 996263-6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Portela, A., and P. Digard. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 83723-734. [DOI] [PubMed] [Google Scholar]
- 25.Pybus, O. G., A. Rambaut, R. Belshaw, R. P. Freckleton, A. J. Drummond, and E. C. Holmes. 2007. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 24845-852. [DOI] [PubMed] [Google Scholar]
- 26.Rabadan, R., A. J. Levine, and H. Robins. 2006. Comparison between avian and human influenza A virus reveals a mutational bias on the viral genomes. J. Virol. 8011887-11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shapiro, G. I., T. J. Gurney, and R. M. Krug. 1987. Influenza virus gene expression: control mechanisms at early and late times of infection and nuclear-cytoplasmic transport of virus-specific RNAs. J. Virol. 61764-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharp, P. M., and W. H. Li. 1986. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 2428-38. [DOI] [PubMed] [Google Scholar]
- 29.Smith, G. L., and A. J. Hay. 1982. Replication of the influenza virus genome. Virology 11896-108. [DOI] [PubMed] [Google Scholar]
- 30.Sokal, R. R., and F. J. Rohlf. 1995. Biometry: the principles and practice of statistics in biological research, 3rd ed. W. H. Freeman and Co., New York, NY.
- 31.Suzuki, Y. 2006. Natural selection on the influenza A virus genome. Mol. Biol. Evol. 231902-1911. [DOI] [PubMed] [Google Scholar]
- 32.Swofford, D. L. 2002. PAUP*: phylogenetic analysis using parsimony (* and other methods), version 4. Sinauer Associates, Sunderland, MA.
- 33.Taubenberger, J. K., A. H. Reid, and T. G. Fanning. 2000. The 1918 influenza virus: a killer comes into view. Virology 274241-245. [DOI] [PubMed] [Google Scholar]
- 34.Taubenberger, J. K., A. H. Reid, R. M. Lourens, R. Wang, G. Jin, and T. G. Fanning. 2005. Characterization of the 1918 influenza virus polymerase genes. Nature 437889-893. [DOI] [PubMed] [Google Scholar]
- 35.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakeley, J. 1996. The excess of transitions among nucleotide substitutions: new methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 11158-163. [DOI] [PubMed] [Google Scholar]
- 37.Wolf, Y. I., C. Viboud, E. C. Holmes, E. V. Koonin, and D. J. Lipman. 2006. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol. Direct. 134-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang, Z. 2000. Maximum likelihood estimation on large phylogenies and analysis of adaptive evolution in human influenza A. J. Mol. Evol. 51423-432. [DOI] [PubMed] [Google Scholar]
- 39.Yang, Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 241586-1591. [DOI] [PubMed] [Google Scholar]
- 40.Yang, Z., and J. P. Bielawski. 2000. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15496-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou, T., W. Gu, J. Ma, X. Sun, and Z. Lu. 2005. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. BioSystems 8177-86. [DOI] [PubMed] [Google Scholar]