

Abstract
Lyssaviruses are highly neurotropic viruses associated with neuronal apoptosis. Previous observations have indicated that the matrix proteins (M) of some lyssaviruses induce strong neuronal apoptosis. However, the molecular mechanism(s) involved in this phenomenon is still unknown. We show that for Mokola virus (MOK), a lyssavirus of low pathogenicity, the M (M-MOK) targets mitochondria, disrupts the mitochondrial morphology, and induces apoptosis. Our analysis of truncated M-MOK mutants suggests that the information required for efficient mitochondrial targeting and dysfunction, as well as caspase-9 activation and apoptosis, is held between residues 46 and 110 of M-MOK. We used a yeast two-hybrid approach, a coimmunoprecipitation assay, and confocal microscopy to demonstrate that M-MOK physically associates with the subunit I of the cytochrome c (cyt-c) oxidase (CcO) of the mitochondrial respiratory chain; this is in contrast to the M of the highly pathogenic Thailand lyssavirus (M-THA). M-MOK expression induces a significant decrease in CcO activity, which is not the case with M-THA. M-MOK mutations (K77R and N81E) resulting in a similar sequence to M-THA at positions 77 and 81 annul cyt-c release and apoptosis and restore CcO activity. As expected, the reverse mutations, R77K and E81N, introduced in M-THA induce a phenotype similar to that due to M-MOK. These features indicate a novel mechanism for energy depletion during lyssavirus-induced apoptosis.
During coevolution with their hosts, viruses have developed many ways of manipulating the cellular machinery of infected cells. They inhibit or induce apoptosis for their own benefit, with the purpose of increasing viral replication and spread or subverting the host's immune response (4, 12, 51, 59).
Mitochondria have several functions in the cell, including energy production, calcium buffering, and regulation of cellular apoptosis. Death signals in the intrinsic pathway of apoptosis act directly on mitochondria, leading to their dysfunction and the release of proapoptotic factors responsible for the caspase-dependent and/or -independent death pathways (43). The process is tightly regulated positively or negatively by proteins from the Bcl-2 family (32). Caspase activation can be initiated in the extrinsic pathway of apoptosis by death receptors expressed at the cell surface; this later causes mitochondrial dysfunction (8, 20).
Lyssaviruses are highly neurotropic viruses associated with rabies, a fatal encephalomyelitis considered to be a reemerging zoonosis throughout most of the world (10). It has been suggested that lyssavirus-induced neuronal apoptosis (1), previously thought to be a principal cause of pathogenesis, is an important defense mechanism against lyssavirus infection (26, 34, 56). However, the molecular basis of lyssavirus-induced neuronal apoptosis is still poorly understood (16, 55). The involvement of the viral glycoprotein (G) in inducing neuronal apoptosis has been extensively shown (13, 38, 39, 45), whereas we have suggested that M is an inducer of neuronal cell death through a tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-dependent pathway (29). However, the molecular mechanism of apoptosis has not been precisely defined, and little is known about mitochondrial involvement during lyssavirus infections (46).
In this study, we take advantage of the fact that Mokola virus (MOK), a member of the genotype 3 lyssaviruses (5), is known to be less pathogenic than viruses of genotype 1 and, in particular, Thailand virus (THA) (3). We report for the first time the involvement of the mitochondrial machinery during MOK-induced apoptosis. We show that the MOK matrix protein (M-MOK), a previously described apoptogenic factor (29), interacts directly with cytochrome c (cyt-c) oxidase (CcO) subunit I (CcO1), the terminal component of the mitochondrial respiratory chain (MRC). This finding is of interest, as this interaction, which is not found with M-THA, may have a key role in controlling ATP synthesis and cellular respiration during lyssavirus-induced neuronal apoptosis and may contribute to the low pathogenesis of MOK infection.
MATERIALS AND METHODS
Materials.
The rabies virus nucleocapsid fluorescein isothiocyanate-conjugated antibody was obtained from Bio-Rad. ADP and staurosporine were purchased from Calbiochem and Sigma, respectively. cyt-c and CcO4 antibodies were purchased from Clontech Laboratories. Anti-CcO1 antibody (clone 1D6) and monoclonal anti-Flag M2 were obtained from Invitrogen and Sigma, respectively. Anti-enhanced green fluorescent protein (EGFP) monoclonal antibody (clone JL8) and Av peptide antibody against GFP were purchased from Clontech. Monoclonal anti-β-actin (clone AC-74) was obtained from Sigma. Protein A-labeled colloidal gold was obtained from the Cell Microscopy Center, AZU, Utrecht, The Netherlands. Horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies were obtained from Amersham Biosciences.
Cells and viruses.
Mouse neuroblastoma cells (N2a), human carcinoma epithelial cells (HeLa), and BSR cells (clones of BHK-21) were cultured in Dulbecco's minimal essential medium supplemented with 0.2% glutamic acid from Gibco and 50 μg/ml gentamicin and 10% heat-inactivated fetal bovine serum from Eurobio. Two lyssaviruses, the THA of genotype 1 and the MOK of genotype 3, were used for cell infections with appropriate multiplicities of infection (MOI), as previously described (29). Infected cells were incubated at 37°C under 5% CO2.
Secondary structure prediction programs.
Secondary structure was predicted using Prof (41) and SAM-T02 (28) software. Prof classifies protein secondary structure prediction, formed by cascading together various types of classification using neural networks and linear discrimination (http://www.aber.ac.uk/∼phiwww/prof/). SAM-T02 uses a method for iterative Sequence Alignment and Modeling System hidden Markov model construction and remote BLAST analysis (http://www.soe.ucsc.edu/research/combio/HMM-apps/T02-query.html) (42).
Recombinant plasmid construction and site-directed mutagenesis.
DNA fragments coding for various M-MOK segments have been designed based on secondary structure prediction. Reverse transcription-PCRs on viral RNAs were performed and products were digested with the corresponding restriction enzymes as previously described (29) (see Data File S1 in the supplemental material). Cloning was carried out using the pEGFP-C1 plasmid (Clontech). In this system, the various segments under study are fused in frame to the C terminus of EGFP. Sets of sense and antisense primers (see Data File S1 in the supplemental material) were designed to amplify wild-type M-MOK (amino acids [aa]rsqb] 1 to 202) and its deletion mutants (M1, 1 to 110; M1.1, 1 to 48; M1.2, 46 to 110; M2, 106 to 202) by PCR. The products were then inserted into pEGFP-C1 plasmids, and these plasmids were used to transform Escherichia coli. The resulting clones were subsequently sequenced. For coimmunoprecipitation M-MOK and M-THA were expressed fused to a 3×Flag tag using vector pCiNeo (Promega) with an inserted Gateway cassette and a 3×Flag tag. PCR products obtained using primers described in Data File S1 in the supplemental material were inserted into this vector using Gateway technology. Mutations at designated residues in M-MOK and M-THA were created by the QuikChange II site-directed mutagenesis kit (Stratagene) (see Data File S1 in the supplemental material).
Cell viability assay and LDH release.
Adherent cells were collected by trypsin-EDTA (Gibco) (0.25%/1 mM) treatment and pooled with floating cells collected from the culture media. Cell viability was determined by a trypan blue (0.4% solution from Sigma) dye exclusion assay. For lactate dehydrogenase (LDH) release, cells were infected and aliquots of supernatants (300 μl) were assayed for LDH release at various times postinfection using the Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI). Minimum lysis was determined from cells cultured in medium alone. Cytotoxicity was expressed as a ratio of specific LDH release, which is determined by the following formula: (infected-cell release − background release obtained on cell culture media)/(noninfected-cell release − background release). All experiments were carried out in triplicate and conducted twice.
ATP synthesis measurement and nitric oxide determination.
For ATP measurements, cells were lysed with Reportasol extraction buffer (Novagen Inc.), normalized for protein content, and incubated with 1 mg/ml luciferin/luciferase (Sigma). Nitric oxide (NO) was measured in the cell culture medium (300 μl) using the Griess diazotization reaction (Molecular Probes). The absorbance of the nitrite formed subsequently in each sample was measured according to the manufacturer's instructions. The assay was calibrated using serial dilutions (0 to 50 μM) of a nitrite standard solution.
Caspase activity and TUNEL assay.
Caspase-9 activity was detected at the single-cell level using the CaspaTag activity kit (Q-Biogene), as previously described (29). Apoptosis was detected at the single-cell level by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) using an ApoDetect kit (Q-Biogen), according to the instructions of the manufacturer with minor modifications (29). Incubation with rhodamine-conjugated antibody was carried out for 45 min at room temperature. Cells were then washed twice with phosphate-buffered saline (PBS) for 5 min and treated with anti-rabies virus fluorescein isothiocyanate-conjugated antibody for 1 h at 37°C. For both assays, cells were fixed for 30 min at 4°C with 4% paraformaldehyde (PFA), mounted with a mixture of DAPI (4′,6′-diamidino-2-phenylindole) and Vectashield (Abcys), and observed under a fluorescence microscope using the corresponding band-pass filters.
Cotransfection and cellular localization of M-MOK and its deletion mutants.
Transfections were assessed with Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer's instructions. Plasmids carrying the full-length M protein and fragments of it fused to EGFP were cotransfected with the pDsRed2-Mito plasmid (Clontech). Transfected cells in multichamber slides were also treated with Hoechst stain for 5 min at 37°C to detect the nuclei. Cells were subsequently washed twice with PBS and fixed for 30 min at 4°C with 4% paraformaldehyde. Slides were mounted with Vectashield and analyzed using a Zeiss LSM 410 inverted laser scanning confocal microscope or a Zeiss Axioplan 2.2 equipped with the Zeiss ApoTome system.
Subcellular fractionation and Western blot (WB) analysis.
Total protein extraction and subcellular fractionation were performed using radioimmunoprecipitation assay lysis buffer (Santa Cruz Biotechnology) and the ApoAlert cell fractionation kit (Clontech), according to instructions of the manufacturers. Protein concentration was determined using bicinchoninic acid protein assay reagent (Pierce), and proteins (5 to 20 μg) were analyzed by the NuPAGE system, run on a 4 to 12% gradient or 10% gels (Invitrogen) and blotted to polyvinylidene difluoride membranes by XCell SureLock Mini-Cell (Invitrogen), based on the manufacturer's instructions. Chemiluminescent detection (SuperSignal West Pico or West Dura chemiluminescent substrates; Pierce) on Biomax-MR film (Kodak) was followed by densitometric quantification of digitized images using Scion Image software.
Yeast two-hybrid screening.
The human brain Matchmaker cDNA library (Clontech) was screened with M-MOK fused to the GAL4 DNA binding domain (BD-M-MOK). The Y187::CG1945 diploid Saccharomyces cerevisiae strain (GAL1-LacZ and GAL1-HIS3) was transformed with a plasmid coding for BD-M-MOK and with pACTII, harboring the GAL4 activating domain (AD) containing the human brain cDNA library, using the lithium acetate procedure (17). The transformed yeasts were plated on synthetic dextrose medium lacking Trp, Leu, and His (SD/-Trp/-Leu/-His medium). One and a half million interactions were screened; 750 His-positive colonies were streaked 4 to 5 days later on SD/-Trp/-Leu/-His plates for assaying β-galactosidase activity (15). The cDNA of the selected colonies was amplified by PCR and sequenced.
Pairwise two-hybrid interaction assay.
cDNA of relevant proteins was used as bait and prey in two-hybrid assays (CcO1 interacting clone, M-THA, and M-MOK); the cDNA was fused to GAL4-BD inserted in pGBKT7 (Clontech) and to GAL4-AD inserted in pACTII (Clontech). The two plasmids were modified to allow cloning using the Gateway cloning technology (Invitrogen). All constructs were obtained by PCR amplification of cDNA using primers with convenient recombination sites. Yeast SFY526 was transformed with two plasmids (pGBKT7-X and pACTII-Y or pGBKT7-Y and pACTII-X) using the lithium acetate procedure (17) to study the interaction between X and Y proteins. Transformed cells were plated on SD/-Trp/-Leu minimal medium plates, and colonies were streaked 4 to 5 days later on SD/-Trp/-Leu plates for assaying β-galactosidase activity (15).
Immunoprecipitation.
HeLa cells were transfected with M-MOK or M-THA fused to a 3×Flag tag. Cells were lysed in TNE buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA) supplemented with 1% Triton X-100 and 1× protease inhibitor cocktail (Complete; Roche) 24 h posttransfection. Magnetic beads coated with goat anti-mouse antibody (Dynabeads; Invitrogen) were washed in TNE buffer. Particles (2 × 107) of washed beads were incubated with 2 μg of monoclonal anti-CcO1 antibody (Invitrogen) for 3 h. Cell lysates were clarified by centrifugation at 4°C for 5 min at 5,000 × g. One hundred micrograms of clarified lysate was precleared by incubation with 2 × 107 particles of washed beads without CcO1 antibody in TNE buffer. One hour later, beads coupled with anti-CcO1 were incubated with precleared cell lysate and incubated for 2 h on a wheel mixer. Immunopurified CcO1-matrix complexes were washed with TNE buffer three times. All washes and incubations were performed at 4°C. Precipitated proteins were eluted in 1× lithium dodecyl sulfate sample buffer supplemented with reducing agent (Invitrogen) and were analyzed by Western blotting.
Quantitation of CcO activity.
We measured CcO activity with a CcO assay kit (Sigma). The measurements were performed on total cellular protein of infected and transfected cells and were expressed as a percentage of activity relative to that for control cells. The activity was presented as a ratio of the change in activity to protein quantity.
Immunogold labeling and electron microscopy.
Transfected HeLa cells were fixed for 15 min by the direct addition of a volume of 4% PFA in 0.1 M Sorensen buffer, pH 7.4, equal to the volume of the culture medium. The cells were then incubated for 3 h at room temperature in 4% PFA. Fixed cells were washed with PBS, after which free aldehyde groups were quenched with 50 mM glycine in PBS. Cells were scraped in PBS containing 1% gelatin and pelleted in 12% gelatin. The cell pellets were solidified on ice and cut into small blocks, which were incubated overnight at 4°C in 2.3 M sucrose for cryoprotection. The blocks were subsequently mounted on aluminum pins and were frozen in liquid nitrogen. Cryoultramicrotome FC6 Leica and anti-GFP polyclonal antibody (BD Biosciences) and protein A-colloidal gold (47) were used for ultrathin cryosectioning and immunogold labeling. Image acquisition was performed by a JEOL 1010 transmission electron microscope (accelerating voltage, 80 kV) equipped with a Keen View charge-coupled-device camera and AnalySIS Pro Software, version 5.0 (Eloïse SARL).
Statistical analysis.
Statistical analyses were performed using Stata 4.0 (Statacorp Software, College Station, TX). The Mann-Whitney (nonparametric and unpaired) test, Kruskal-Wallis test, Student t test, and χ2 test were performed to determine the differences between the values. Statistical significance is identified as P values less than 0.05 (95% confidence interval).
RESULTS
MOK induces aponecrosis and mitochondrial dysfunction in neuronal cells.
MOK, an attenuated lyssavirus strain (3), is an early inducer of neuronal cell death in comparison to THA, the neurovirulent prototype of rabies virus (29); thus, N2a cells were infected with MOK and THA (MOI, 1) and a comparison of kinetics was performed with a TUNEL assay to confirm the results of these studies (Fig. 1A). A minimum of 300 cells were counted, and those with intense chromatin clumping and nuclear fragmentation were considered apoptotic. As expected, apoptosis was induced as early as 48 h postinfection (p.i.) in MOK-infected neuronal cells and increased to reach a maximum level at 96 h p.i. (approximately 37%) (Fig. 1A). By contrast, TUNEL-positive cells induced by THA infection remained at a level similar to that of uninfected cells during the 72-h-p.i. period (Fig. 1A) and slightly increased at 96 h p.i. (13%) (Fig. 1A). MOK-infected cells, in contrast to THA-infected cells, showed a rapid decrease in cell viability, as determined by a trypan blue exclusion assay (Fig. 1B); this confirms the time-dependent difference in cell death activation and cytotoxicity between the two isolates.
FIG. 1.

Mitochondrial involvement in lyssavirus-induced apoptosis. Neuroblastoma cells (N2a) were infected with THA and MOK lyssaviruses (MOI, 1). After incubation at 37°C for the indicated intervals, cells were processed. Results are expressed as averages of data obtained from three independent experiments. Error bars indicate standard deviations of these values. Significant effects are indicated by asterisks (P < 0.05). (A) A minimum of 300 cells were counted at 24, 48, 72, and 96 h p.i., and TUNEL-positive cells were considered apoptotic. (B) Viability of infected cells was assessed using a trypan blue exclusion assay. The results are expressed as the percentages of viable cells relative to control. (C) Representative immunoblots of fractionated cells into soluble (CYTO) and membrane fractions containing mitochondria (MITO) were analyzed by Western blotting at 24 and 48 h p.i. for the detection of cyt-c, CcO4, and β-actin with anti-cyt-c, anti-CcO4, and anti-β-actin antibodies, respectively. (D) Graphical presentation of cyt-c release due to viral infections. Bars represent the cytosolic-to-mitochondrial ratio of cyt-c, measured by densitometry of two independent WB experiments. (E) Caspase-9 activation in infected N2a cells was quantified at 48, 72, and 96 h p.i. A minimum of 300 cells were counted, and results were expressed as the percentages of cells exhibiting positive fluorescence, indicative of the presence of active caspase-9. (F) N2a cells were either mock infected (Ctrl) or infected with MOK (MOI, 10) in the presence of ADP at the indicated concentrations. At 72 h p.i., the percentage of lyssavirus-infected cells undergoing apoptosis was measured by TUNEL assay. (G) ATP was measured in N2a cells with a luciferase assay, and the ATP level was expressed in arbitrary units, relative to protein content. (H) NO measurements were performed using the Griess diazotization reaction at the indicated times. (I) Lysis of infected cells was assessed using LDH release. The results are expressed as ratios of infected/uninfected cells.
We then further examined whether the intrinsic mitochondrial pathway is active during lyssavirus-induced cell death. In the first set of experiments, N2a cells were infected with MOK and THA (MOI, 1) and the release of cyt-c from mitochondria into the cytosol was assessed by Western blotting after cell fractionation. The ratio of cytosolic to mitochondrial cyt-c in cells infected with MOK at 48 h p.i. was threefold greater than at 24 h p.i. and threefold greater than that observed in control cells; however, in the case of THA infection, cyt-c remained sequestered in the mitochondria (Fig. 1C and D). Cytosolic cyt-c leads to the recruitment and activation of the initiator procaspase-9. Thus, we next determined the level of caspase-9 processed during infection by fluorescence microscopy and by using the specific fluorescein-labeled caspase-9 inhibitor 5-carboxyfluorescein-LEHD-fluoromethyl ketone. The results were consistent with caspase-9 being activated as early as 48 h p.i. in MOK-infected cells, in contrast to THA-infected cells (Fig. 1E). Moreover, treatment with ADP, a potent inhibitor of the mitochondrial megachannel and cyt-c release (18), revealed a dose-dependent inhibition of caspase-9 processing (data not shown) and inhibited apoptosis in MOK-infected cells (Fig. 1F). In the second set of experiments, we employed colorimetric and luminometric assays to monitor NO levels in cell culture medium and ATP produced by mitochondria during infection, respectively. A decrease of ATP synthesis was concomitant with an increase in NO levels in the supernatant of MOK-infected neuronal cells at 72 h p.i. (Fig. 1G and H). By contrast, the level of cellular ATP and nitrite/nitrate in the supernatant of THA-infected cells remained stable during this time (Fig. 1G and H). Finally, the early release of LDH in the supernatant of MOK-infected cells was greater than that in the supernatant of THA-infected cells (Fig. 1I), suggesting the involvement of an aponecrotic mechanism during MOK infection.
M-MOK induces apoptosis by a mechanism involving the mitochondrial pathway.
Among the five proteins of lyssaviruses, we have previously shown that M is involved in the early induction of apoptosis in HeLa cells (29). We generated various constructs and truncated M-MOK proteins fused downstream to EGFP to study the role of M-MOK in the induction of apoptosis in more detail (Fig. 2A and B). The truncated-protein designs were based on a sequence comparison and structure predictions for M-MOK and M-THA proteins (Fig. 2A). M-MOK presents a 79% sequence similarity to M-THA. The N terminus is composed of two β-sheets (β1 and β2) and two α-helices (α1 and α2). The C terminus comprises four β-sheets (β3 to β6). A central region of 28 aa, without any defined secondary structure and linking β2 and β3, connects the N and C termini. Based on this prediction, M was first divided in two parts: (i) M1, comprising the 110 aa at the N-terminal position and encompassing α1-β1-α2-β2, and (ii) M2, comprising the four β-sheets (β3 to β6) in the C-terminal half of the protein. M1 was further subdivided into M1.1, encompassing α1 and the late-budding domain (LBP) (24), and M1.2, encompassing two β-sheets (β1 and β2) and one α-helix (α2).
FIG. 2.

Schematic representation of M-MOK deletion constructs fused to EGFP. (A) Sequence alignments of THA and MOK matrix proteins (GenBank accession numbers AY540348 and AY540347, respectively). The late-budding domain (LBP) is indicated (24). (B) Representation of EGFP fusion proteins, including subfragments of M-MOK. Putative α and β domains, as determined by Prof and SAM-T02, are gray.
HeLa cells were transfected with various M-MOK constructs, and the level of apoptosis was assessed by TUNEL assay. As expected, M-MOK induced apoptosis. Approximately 20% of cells were TUNEL positive, whereas the level of apoptosis in cells transfected with the M-THA plasmid and the control expressing EGFP remained very low (Fig. 3A). These data suggest possible apoptogenic activity specific to M-MOK but not to M-THA.
FIG. 3.

M-MOK-, M1-, and M1.2-induced caspase-9 activation and apoptosis, a process inhibited by ADP. HeLa cells were transfected with plasmids expressing EGFP alone (V), full-length M (EGFP-M-THA and EGFP-M-MOK), or truncated forms of M (EGFP-M1, EGFP-M2, EGFP-M1.1, and EGFP-M1.2). After incubation at 37°C for 24 h, cells were processed. (A) The percentage of transfected cells undergoing apoptosis was measured by TUNEL assay in the absence (control) or presence of ADP (200 μM). A minimum of 300 transfected cells were counted to detect the percentage of TUNEL-positive cells. Data are means of three independent experiments ± standard errors of the means. ND, not done. Significant effects are indicated by asterisks, pound signs, and plus signs (P < 0.05). (B) Representative immunoblot with anti-caspase 9, anti-β-actin, and anti-EGFP antibodies. The cytoplasmic fraction of cells expressing full-length and truncated M-MOK were analyzed 24 h posttransfection. Bands from caspase-9 cleavage (37 and 35 kDa) and β-actin, as well as bands of related protein constructs, are displayed. Molecular masses are indicated. Nontransfected cells (T−) and cells treated for 1 hour with 1 μM staurosporine (T+) were used as controls.
Next, we examined whether any of the M truncations were sufficient to induce apoptosis. The results showed that the truncated forms M1, M2, and M1.2 induced apoptosis in more than 25% of the cells (Fig. 3A). This is in contrast to M1.1, in which the level of induced apoptosis remained below 5%. We investigated the effect of M on the mitochondrial intrinsic pathway; thus, we analyzed transfected cells for caspase-9 activation (Fig. 3B). Interestingly, we observed greater levels of the caspase-9 cleaved forms (35 and 37 kDa) in M-MOK-, M1-, M2-, M1.1-, and M1.2-transfected cells than in cells transfected with the vector alone. M1 induces more caspase-9 cleavage than M2 despite a lower expression (Fig. 3B). Similarly, M1.2, although expressed in a smaller amount, demonstrates more caspase-9 cleavage than M1.1, in which the amount of cleaved caspase-9 after transfection remained the lowest of all four M fragments (Fig. 3B). As controls, β-actin was used to normalize the various cellular fractions, whereas staurosporine, an inhibitor of protein kinase C, was used as a strong inducer of apoptosis (Fig. 3B). Finally, M-MOK- and M1-induced apoptosis was partially inhibited by treatment with ADP, whereas M2-induced apoptosis was not (Fig. 3A). This indicates that there is an alternate mechanism for inducing apoptosis in the case of M2. The N-terminal fragments of M1 and M1.2 may be implicated in activating the mitochondrial megachannel and apoptotic activity induced by M.
M-MOK is imported to mitochondria.
We investigated the subcellular localization of M-MOK and its truncated forms. Therefore, HeLa cells were cotransfected with various chimeric forms of M fused to the C terminus of EGFP and the reporter plasmid DsRed2-Mito, coding for CcO7, which is used as a specific mitochondrial marker. Cells were then observed using ApoTome fluorescence microscopy (Fig. 4A). The results indicated a partial colocalization between the EGFP-M-MOK fluorescence signal and that of CcO7-DsRed2 (Fig. 4A), suggesting a mitochondrial localization of M-MOK in transfected cells. As a control, EGFP alone produces a diffuse signal in HeLa cells, reflecting its lack of intracellular targeting (Fig. 4A). Fragments M1, M1.2, and, to a far lesser extent, M2, were found to share coincident signals with DsRed2-Mito by ApoTome microscopy (Fig. 4A). By contrast, fragment M1.1 did not appear to localize at the mitochondrial level. This localization was also confirmed with another type of tag (i.e., Flag) (data not shown). HeLa transfected cells were fractionated to document further the localization of the various fragments. The resulting cytoplasmic and mitochondrion-enriched fractions were subjected to WB analysis and probed for M (Fig. 4B), using actin as a cytoplasmic marker and CcO4 as a mitochondrial marker. This subcellular fractionation also confirmed that M and fragments M1, M2, and M1.2 are imported to the mitochondria. The fractionation also demonstrates that the presence of M1 and M1.2 in the mitochondrion-enriched fraction is more important than that of M2 (Fig. 4B). In an attempt to further determine the localization of M and fragment M1 within the mitochondrial compartment, we transfected HeLa cells with these constructs fused to EGFP. Specific staining of these cells using gold-labeled anti-GFP antibodies indicated that M-MOK and M1, when targeted to the mitochondria, are associated with the inner membrane or with the intermembrane space (Fig. 4C). Taken together, these data established that M-MOK and the fragments M1 and M1.2 are targeted to the mitochondria and more specifically to the inner membranes and intermembrane zones of the mitochondria.
FIG. 4.

M-MOK and fragments M1 and M1.2 are localized in the mitochondria. HeLa cells were transfected with plasmids expressing EGFP-M or its truncated forms M1, M2, M1.1, and M1.2, as well as EGFP alone (V), and were cotransfected with plasmid vectors expressing DsRed2-Mito. (A) The subcellular localization of these various constructions was monitored 24 h posttransfection with a Zeiss Axioplan 2.2 fluorescence microscope. The microscope was equipped with a Zeiss ApoTome system, and representative images revealing mitochondrial localization are shown. Images in the right column (merge) show overlay images between various chimeric constructs of the left column (EGFP) and those of the middle column, which display the mitochondrial pattern of the cell (DsRed2). (B) WB analysis of the cytoplasmic (c) and mitochondrion-enriched (m) fractions of HeLa cells expressing various protein constructs. (Top) Ratio of the mitochondrion-enriched fraction to the cytosolic fraction. Values are obtained by densitometry of WB experiments. Error bars and average values were obtained from two experiments. (Bottom) Representative immunoblot with anti-EGFP, anti-CcO4, and anti-β-actin antibodies. Different times of exposure to the film are used to show EGFP-tagged constructs. However, CcO4 and β-actin bands are obtained from single exposure times. (C) Ultrastructures of mitochondria of HeLa cells transfected with plasmids expressing vector alone (EGFP), the EGFP-tagged full-length molecule (M-MOK), and its M1 fragment after immunogold labeling are shown. Ultrathin cryosections were labeled with polyclonal antibodies against GFP and protein A-colloidal gold. Representative images detailing the localization of M-MOK and M1 in the inner membrane and intermembrane space of the mitochondria are shown. Bars, 0.5 μm for all photos. In the case of M-MOK, the magnification of the framed region of the mitochondria is increased twofold (2×). (D) Representative images acquired by laser scanning confocal microscopy showing the colocalization (merge) of expressed EGFP-M1 and CcO1 (×60 objective, twofold zoom).
Matrix protein forms an in vivo complex with subunit I of the CcO.
We performed a yeast two-hybrid screening of a human brain cDNA library to understand the molecular mechanism with which M-MOK induces cell death via the mitochondrial pathway. We tested 1.5 × 106 interactions during this screening, using M-MOK as bait. Among these, 750 His-positive colonies were obtained and analyzed for β-galactosidase activity. The 72 LacZ-positive colonies were sequenced and identified using BLAST (www.ncbi.nlm.nih.gov/BLAST/BLAST.cgi). Four of the 72 positive colonies displayed high similarity (>99%) with the Homo sapiens CcO1 gene (GenBank accession number AY963578.2). Due to the mitochondrial genetic code and within the context of the two-hybrid assay, the CcO1 insert encodes a very small fragment corresponding to aa 104 to 125 of CcO1.
In the second step, we tested the interaction of the CcO1 fragment with M-MOK and M-THA by a pairwise two-hybrid analysis (Fig. 5A). Yeast was transformed with plasmids encoding M-MOK and M-THA fused to GAL4-AD and GAL4-BD, respectively, and a plasmid encoding the CcO1 fragment fused to either GAL4-BD or GAL4-AD. Only yeast coexpressing M-MOK and CcO1 showed strong β-galactosidase activity. This transactivation of the LacZ reporter gene was not due to autotransactivation by M-MOK and/or CcO1, as yeast expressing only one of the two proteins failed to display β-galactosidase activity (Fig. 5A). Moreover, this interaction is specific for M-MOK, as M-THA is unable to interact with CcO1, as demonstrated by the white color of the corresponding yeast colonies in the X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) overlay assay (Fig. 5A).
FIG. 5.
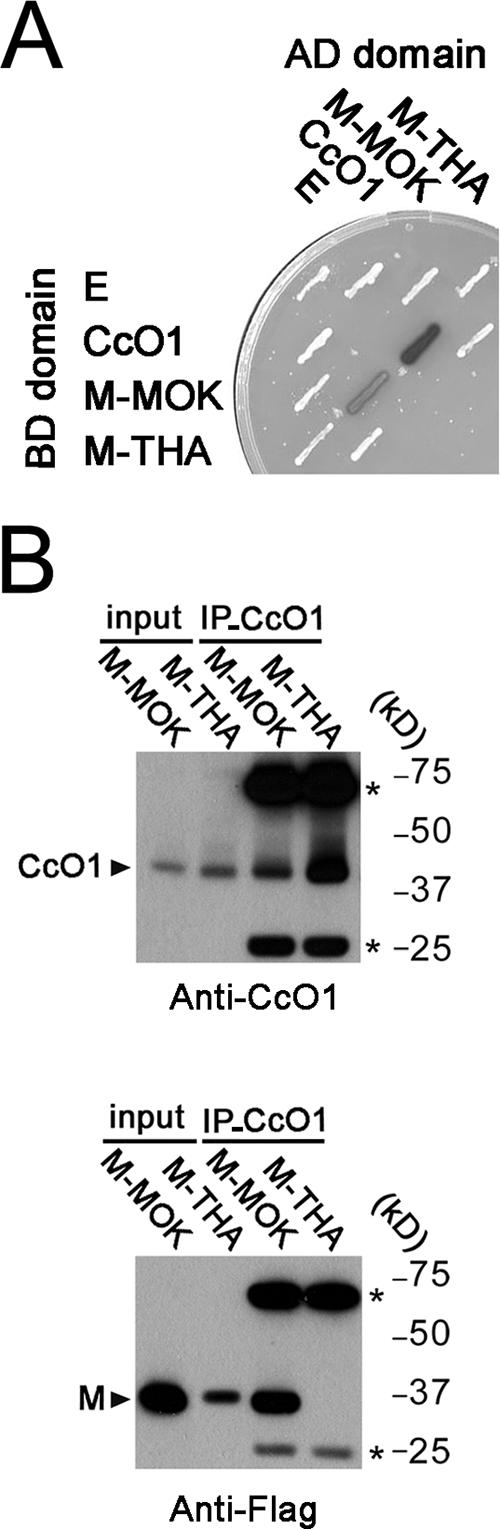
Physical interaction between M-MOK and CcO1. (A) Yeast two-hybrid analysis of the CcO1 interaction with M-MOK and M-THA. β-Galactosidase activity of Y187::CG1945 S. cerevisiae transformed with the indicated bait (AD) and prey (BD) plasmids is revealed by an X-Gal overlay assay. An empty vector (E) is used as a control. (B) CcO1-matrix complexes were immunoprecipitated with anti-CcO1 antibody from transfected HeLa cells expressing M-MOK or M-THA fused to 3×Flag and immunoblotted with anti-CcO1 and anti-Flag antibodies. (Top) Input lysates containing M-MOK and M-THA, as well as their corresponding immunoprecipitation (IP-CcO1) complexes after treatment with anti-CcO1 antibody. Bands corresponding to antibody heavy and light chains are defined by asterisks. (Bottom) Duplicate of the same blot in the top panel that was developed after treatment with anti-Flag antibody.
Interaction between CcO1 and M-MOK was confirmed by coimmunoprecipitation experiment. CcO1 was immunoprecipitated from a total cell lysate of HeLa cells expressing M-MOK or M-THA fused to a 3×Flag tag. CcO1-matrix complexes were analyzed by Western blotting with anti-CcO1 and anti-Flag antibodies. Results showed that M-MOK was coimmunopurified with CcO1 (Fig. 5B). A parallel experiment with M-THA showed no coprecipitation with CcO1. As there was a higher expression for M-MOK than for M-THA, a fourfold increase in the quantity of input M-THA was used in the experiment. Although the quantity of the recovered CcO1 was proportionally increased, no interaction with CcO1 was detected for M-THA, confirming the specificity of the interaction of M-MOK with CcO1 (Fig. 5B). The interaction of M-MOK with CcO1 was further confirmed by pull down experiment (see Fig. SA1 in the supplemental material). EGFP-M1 and CcO1 signals appeared to colocalize in the mitochondria by immunofluorescence and confocal microscopy (Fig. 4D). All these results are consistent with a physical interaction between M-MOK and CcO1.
M-MOK reduces CcO activity.
CcO is located on the inner mitochondrial membrane. It is the principal terminal oxidase in the aerobic metabolism of all animals (9). Interestingly, MOK-infected HeLa cells displayed progressive and significantly greater reduction of CcO activity than THA-infected and noninfected cells (Fig. 6A). We further characterized the biological roles of M-MOK and its truncated forms. These proteins are mostly imported into the mitochondria (M1 and M1.2) on CcO1. Therefore, we next determined the CcO activity in transfected cells after 24 h. HeLa cells transfected with M-MOK displayed a greater decrease in CcO activity (fourfold reduction) than cells transfected with the vector expressing the EGFP tag alone (Fig. 6B). Fragments M1 and M1.2 also induced a greater reduction in CcO activity than M1.1 despite a lower expression level (Fig. 6C). This indicates that M-MOK and its deletion mutant M1.2, encompassing residues 46 to 110, induced a significant reduction in CcO activity.
FIG. 6.
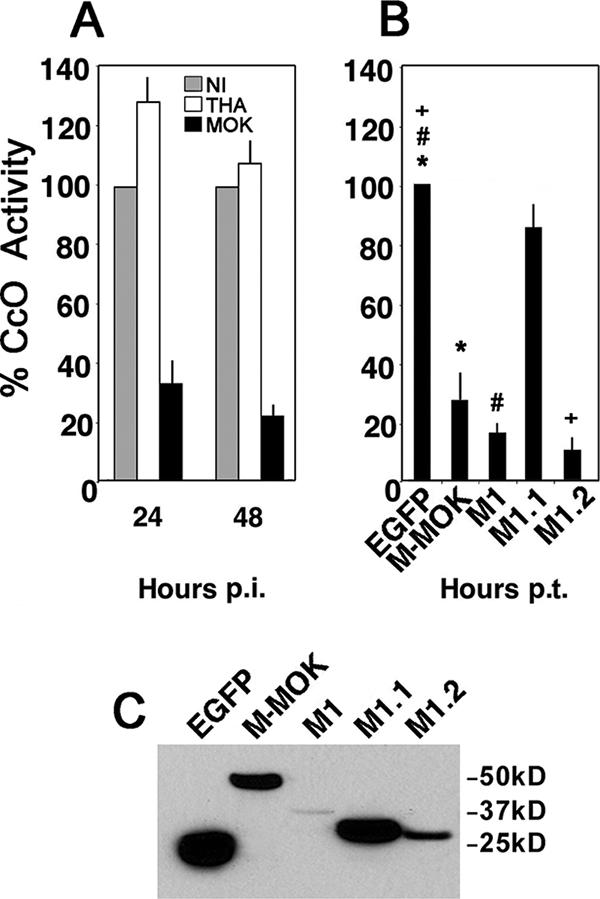
M-MOK and fragments M1 and M1.2 inhibit CcO activity. (A) CcO activity was evaluated in noninfected HeLa cells (NI) as well as HeLa cells infected with THA and MOK by spectrocolorimetry at 24 h and 48 h p.i. The measurements were performed after protein normalization and were expressed as the percentages of activity relative to that for NI. All values were obtained from three independent experiments. (B) HeLa cells were transfected with plasmids expressing EGFP alone, full-length M fused to EGFP (M-MOK), or its truncated forms (M1, M1.1, and M1.2). CcO activity was evaluated 24 h posttransfection (p.t.). The measurements were performed after protein normalization and were expressed as the percentages of activity relative to CcO activity in cells expressing EGFP alone. All values were obtained from three independent experiments. Significant effects are indicated by asterisks, pound signs, and plus signs (P < 0.05). (C) Immunoblot experiment with anti-EGFP showing expression of different protein constructs at 24 h p.t. Bands are obtained from samples with equal protein content and a single exposure time.
Mutations K77R and N81E in M affect mitochondrial function.
Construct M1.2, which is mostly targeted to the mitochondria and which produces a more dramatic reduction of CcO activity than other M mutants, contains one α-helix (α2) surrounded by two β-sheets (β1 and β2). This α-helix contains hydrophobic amino acids flanked by positively charged residues and may act as a putative mitochondrial targeting domain inhibiting the function of the CcO1. Among the eight positions differentiating MOK and THA on fragment M1.2, three are localized in this predicted α2 domain (between aa 69 and 82). In this study, we focused on mutations K to R in position 77 (K77R) and N81E, observed in THA in comparison with MOK, as most proapoptotic viral proteins acting on mitochondria share amphipathic α-helices in the N terminus or in the central portion (6). We examined the effects of these changes by generating mutants of M-MOK (K77R/N81E) and of M-THA (R77K/E81N). These mutant proteins were then expressed in HeLa cells and compared with wild-type M-MOK and M-THA. The M-MOK mutations K77R and N81E abolished the induction of TUNEL labeling (Fig. 7A). These mutations maintained mitochondrial permeability, as shown by the absence of release of cyt-c (Fig. 7B) and the restoration of CcO activity (Fig. 7C), comparable to that of wild-type M-THA. Symmetrically, the reverse mutations R77K and E81N in M-THA confer a phenotype closer to that produced by M-MOK, with a higher induction of apoptosis and release of cyt-c and a dramatic reduction of CcO activity. None of these mutations affect the expression level compared to that for wild-type M (Fig. 6D).
FIG. 7.
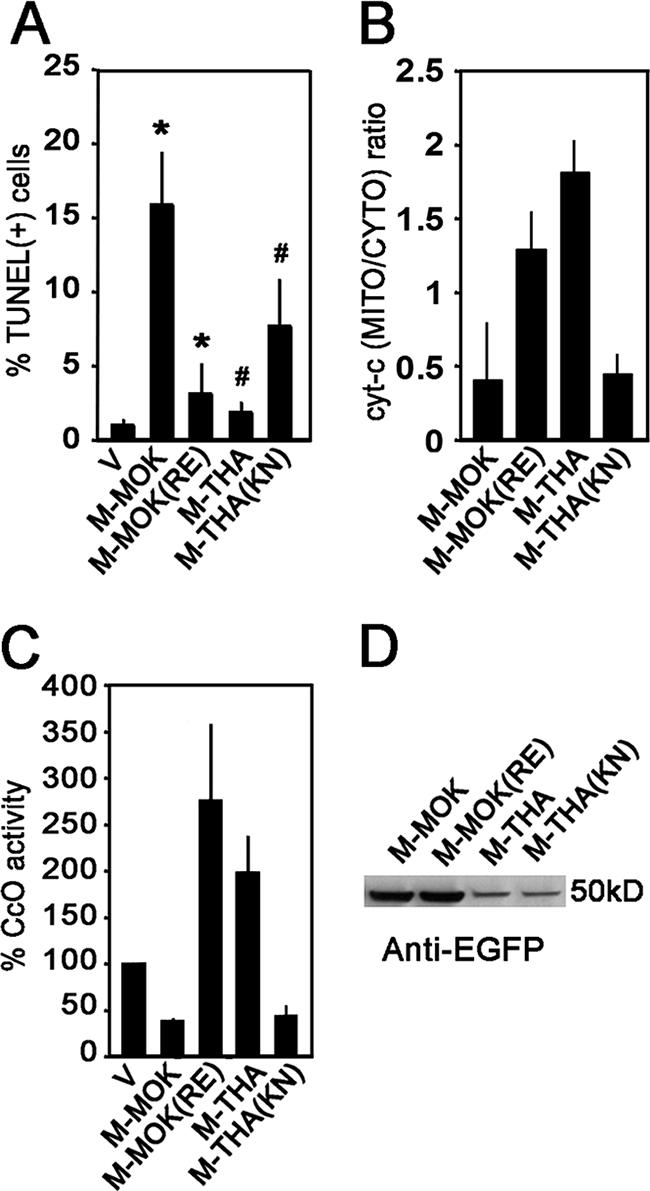
Residues at positions 77 and 81 in matrix proteins of lyssaviruses govern mitochondrial dysfunction, as shown by the release of cyt-c and Cco activity, and apoptosis 24 h posttransfection (p.t.). Single mutations at residues 77 and 81 of M-MOK and -THA molecules resulted in interchanges between their apoptotic activities. This was demonstrated (A) by TUNEL assay, (B) by cyt-c release (bars signify the ratios between mitochondrion-enriched [MITO] and cytosolic [CYTO] fractions of cyt-c, as quantified by densitometry of WB gels), and (C) CcO activity, as demonstrated by spectrometry. Activities of M-MOK, M-THA, M-MOK K77R/N81E [M-MOK(RE)], and M-THA R77K/E81N [M-THA(KN)] were compared with that for cells expressing only EGFP as a control. Average values and standard deviations in graphs are obtained from two independent experiments. Significant effects are indicated by asterisks and pound signs (P < 0.05). (D) Representative immunoblot with anti-EGFP antibody demonstrating expression levels of wild-type and mutant molecules of M-MOK and M-THA.
DISCUSSION
Mitochondria play a central part in cellular survival and regulation of programmed cell death. Indeed, apart from harboring several death factors that are released upon apoptotic stimuli, mitochondria also provide the cell with ATP. Thus, all mitochondrial alterations, such as mitochondrial membrane permeabilization, release of sequestered apoptotic proteins, production of reactive oxygen species, and disruption of the electron transport chain, are involved in cell death (19). Several viruses modulate apoptosis by disrupting the mitochondrial machinery (6, 33, 52).
We have previously studied apoptosis induction in cultured cells during lyssavirus infections, and we have shown that N2a and HeLa cells demonstrate common apoptotic hallmarks during the course of infection. We have also shown that M is involved in inducing apoptosis of neuronal cells by a death receptor-dependent mechanism (29). M is a small (202-aa, 25-kDa) multifunctional protein that is responsible for the assembly and budding of virus particles (21, 22, 36). It is also involved in regulating the balance between transcription and replication (14) and inhibiting translation in virus-infected cells (30). M also induces cell death in related vesiculoviruses (31).
In this study, we demonstrated that mitochondrial functionality of neuronal cells is highly disturbed after infection with MOK, a lyssavirus strain. The decrease in ATP synthesis and mitochondrial dehydrogenase activity and the increase in NO production suggested a reduction of the mitochondrial electron transfer system during MOK infection. Finally, the release of cyt-c into the cytosol, associated with caspase-9 activation, indicated the involvement of the mitochondrial pathway in MOK-induced neuronal cell death. We also report that M-MOK induced rapid aponecrosis if expressed in HeLa cells, similar to that observed during viral infection of neuronal cells. The biological relevance of M-induced mitochondrial dysfunction is of importance, as M-MOK physically interacts with CcO1, a component of the terminal enzyme of MRC, and significantly down-regulates its activity. M-MOK has been shown to colocalize with mammalian CcO, an enzymatic complex of the inner mitochondrial membrane with 13 subunits (9, 37). CcO1 and CcO2 form the catalytic center of the enzyme (25). CcO catalyzes electron transfer from cyt-c to molecular oxygen, reducing it later to water. The redox energy in this process is then converted to a proton motive force, which subsequently drives ATP synthesis. Interestingly, the positive yeast isolates obtained by the two-hybrid screen mapped to a region corresponding to the binuclear center of CcO1 (data not shown), a proton pump responsible for reducing oxygen to water (7). Thus, in our model (Fig. 8), the interaction between M-MOK and CcO1 is associated with a depletion of cellular ATP; this interaction disrupts the electron transport chain at the inner mitochondrial membrane, leading to cell death by an aponecrotic mode. Amino acid positions 77 and 81 in M proteins appeared to be critical in regulating these functions; M proteins with K77 and N81, as in wild-type M-MOK, produce CcO dysfunction and apoptosis, but M proteins with R77 and E81, similar to wild-type M-THA, are harmless. Further studies should determine if these findings obtained with MOK are related to its peculiar pathogenesis (3, 29). In particular, several studies report that neuronal apoptosis is inversely correlated with pathogenesis of lyssaviruses (39). Within this context, it is interesting that MOK and THA, representing two genotypes of lyssaviruses, exhibit low and high neurovirulence, respectively (3). Indeed, the function and integrity of the nervous system rely heavily on a large proportion of ATP generated during oxidative phosphorylation. Thus, minor disruptions of the mitochondrial respiration by M may amplify the previously described TRAIL- and caspase-8-dependent apoptotic mechanism after lyssavirus infection (29). Thus, soluble cell signals, such as nitric oxide, produced during lyssavirus infection could play a synergetic role in TRAIL-mediated neuronal apoptosis (2, 23, 29, 40, 57). Recombinant lyssaviruses with targeted mutations in the M1.2 region of M could be used to evaluate that particular question.
FIG. 8.

Diagram summarizing the link between M-MOK binding to Cco1 and aponecrotic activity reported in this study. The CcO complex, located at the inner membrane (IM), in normal conditions catalyzes electron transfer. This is converted to a proton motive force, which subsequently drives ATP synthesis (CcO complex “on”). M produced during MOK infection is transported into the mitochondrial intermembrane space (IS) by passage through the mitochondrial outer membrane. The physical interaction between M and CcO1 decreases CcO activity and mitochondrial oxidative phosphorylation (CcO complex “off”). This event leads to the necrotic component of lyssavirus-induced cell death, essentially by mitochondrial changes and ATP depletion. This response is completed by the extrinsic death receptor-dependent and caspase-dependent pathways, as shown previously (29). The death receptors are synergistically stimulated by death ligands, such as TRAIL, and NO, produced by the mitochondria during lyssavirus infection. If activated by the death-inducing signaling complex (DISC), caspase-8 (C-8) in turn triggers the downstream effector cascade either directly through caspase-3 (C-3) or through engaging the mitochondrial amplification loop. In this process, activated C-8 indirectly triggers cyt-c release and caspase-9 (C-9) activation. This event leads to the apoptotic component of lyssavirus-induced cell death. Thus, in this model, mitochondrial dysfunction plays a key role in M-MOK-mediated cell death.
Many viral proteins directly target mitochondria (35) and modulate cell death by disrupting mitochondrial morphology or regulating mitochondrial membrane permeabilization (6). However, the exact mechanisms through which viral mitochondrial apoptosis modulators exert their local action remain to be elucidated for most viral proteins (33). Thus, our data represent the first evidence of a viral protein (i) physically associating with one of the four membrane-bound electron-transporting protein complexes of the MRC, (ii) causing defects in CcO activity, and (iii) inducing programmed cell death.
Most of the proapoptotic viral proteins acting on mitochondria have sequences that direct import and contain amphipathic α-helices in the N terminus or the central portion of the protein (6), as in proteins of human immunodeficiency virus type 1 and human T-cell leukemia virus type 1 (11, 27, 50, 53). M-MOK does not seem to follow the classical protein import pathway into mitochondria, which involves cleavable targeting signals located at the N-terminal end of the preprotein (44, 48). Deletion analysis of M-MOK shows that, in addition to the Cco1 dysregulation motif, a major mitochondrial import signal remains between residues 46 and 110 and consequently is independent of the late viral budding domain, located between positions 1 and 48 of M (24). Indeed, secondary structure prediction of the corresponding M1.2 portion revealed the presence of one α-helix (position 69 to 82) surrounded by two β-sheets. This α-helix may act as a putative mitochondrial targeting signal (MTS) for the M protein. This would constitute an internal MTS in a noncleaved protein, as previously described for metabolite carriers of the inner membrane (44). For some of these molecules, several MTSs distributed across the molecule cooperate in receptor recruitment and mitochondrial membrane translocation (58). Therefore, the slight distribution of M2 (aa 106 to 202 of the M protein) in the mitochondria indicates that another weaker MTS localized in the C terminus of the M protein also contributes to its targeting. The interactions between mitochondrial receptors and MTS-containing proteins are mediated by cytoplasmic chaperones, such as heat shock protein 70 (HSP70) and HSP90, which recognize and help to maintain the mitochondrial protein in an import-competent state (54, 60). Further analyses are needed to better understand the mechanism for transport of M to the mitochondria. In this context, it has been reported that rabies virus incorporates HSP70 (49). However, no evidence has been demonstrated for the biological relevance of this phenomenon or for an eventual interaction between HSP and any lyssavirus proteins. A more precise evaluation of the M-CcO1 interaction may also yield novel opportunities for therapeutic intervention in rabies disease.
Supplementary Material
Acknowledgments
We are grateful to E. Perret and S. Shorte at the Plateforme d'Imagerie Dynamique at the Institut Pasteur for invaluable experimental help, discussions, and advice on data processing.
Raïd Kassis was supported by a predoctoral scholarship from the regional council of Guadeloupe, France. Alireza Gholami is supported by a predoctoral scholarship from Total.
Published ahead of print on 5 March 2008.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Adle-Biassette, H., H. Bourhy, M. Gisselbrecht, F. Chretien, L. Wingertsmann, M. Baudrimont, Y. Rotivel, B. Godeau, and F. Gray. 1996. Rabies encephalitis in a patient with AIDS: a clinicopathological study. Acta Neuropathol. (Berlin) 92415-420. [DOI] [PubMed] [Google Scholar]
- 2.Antunes, F., A. Boveris, and E. Cadenas. 2004. On the mechanism and biology of cytochrome oxidase inhibition by nitric oxide. Proc. Natl. Acad. Sci. USA 10116774-16779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badrane, H., C. Bahloul, P. Perrin, and N. Tordo. 2001. Evidence of two Lyssavirus phylogroups with distinct pathogenicity and immunogenicity. J. Virol. 753268-3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benedict, C. A., P. S. Norris, and C. F. Ware. 2002. To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 31013-1018. [DOI] [PubMed] [Google Scholar]
- 5.Bourhy, H., B. Kissi, and N. Tordo. 1993. Molecular diversity of the Lyssavirus genus. Virology 19470-81. [DOI] [PubMed] [Google Scholar]
- 6.Boya, P., T. Roumier, K. Andreau, R. A. Gonzalez-Polo, N. Zamzami, M. Castedo, and G. Kroemer. 2003. Mitochondrion-targeted apoptosis regulators of viral origin. Biochem. Biophys. Res. Commun. 304575-581. [DOI] [PubMed] [Google Scholar]
- 7.Bratton, M. R., L. Hiser, W. E. Antholine, C. Hoganson, and J. P. Hosler. 2000. Identification of the structural subunits required for formation of the metal centers in subunit I of cytochrome c oxidase of Rhodobacter sphaeroides. Biochemistry 3912989-12995. [DOI] [PubMed] [Google Scholar]
- 8.Brenner, C., and G. Kroemer. 2000. Apoptosis. Mitochondria—the death signal integrators. Science 2891150-1151. [DOI] [PubMed] [Google Scholar]
- 9.Bruno, C., A. Martinuzzi, Y. Tang, A. L. Andreu, F. Pallotti, E. Bonilla, S. Shanske, J. Fu, C. M. Sue, C. Angelini, S. DiMauro, and G. Manfredi. 1999. A stop-codon mutation in the human mtDNA cytochrome c oxidase I gene disrupts the functional structure of complex IV. Am. J. Hum. Genet. 65611-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coleman, P. G., E. M. Fevre, and S. Cleaveland. 2004. Estimating the public health impact of rabies. Emerg. Infect. Dis. 10140-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Agostino, D. M., L. Ranzato, G. Arrigoni, I. Cavallari, F. Belleudi, M. R. Torrisi, M. Silic-Benussi, T. Ferro, V. Petronilli, O. Marin, L. Chieco-Bianchi, P. Bernardi, and V. Ciminale. 2002. Mitochondrial alterations induced by the p13II protein of human T-cell leukemia virus type 1. Critical role of arginine residues. J. Biol. Chem. 27734424-34433. [DOI] [PubMed] [Google Scholar]
- 12.Everett, H., and G. McFadden. 1999. Apoptosis: an innate immune response to virus infection. Trends Microbiol. 7160-165. [DOI] [PubMed] [Google Scholar]
- 13.Faber, M., R. Pulmanausahakul, S. S. Hodawadekar, S. Spitsin, J. P. McGettigan, M. J. Schnell, and B. Dietzschold. 2002. Overexpression of the rabies virus glycoprotein results in enhancement of apoptosis and antiviral immune response. J. Virol. 763374-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finke, S., R. Mueller-Waldeck, and K. K. Conzelmann. 2003. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 841613-1621. [DOI] [PubMed] [Google Scholar]
- 15.Fromont-Racine, M., J. C. Rain, and P. Legrain. 1997. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet. 16277-282. [DOI] [PubMed] [Google Scholar]
- 16.Fu, Z. F., and A. C. Jackson. 2005. Neuronal dysfunction and death in rabies virus infection. J. Neurovirol. 11101-106. [DOI] [PubMed] [Google Scholar]
- 17.Gietz, R. D., R. H. Schiestl, A. R. Willems, and R. A. Woods. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11355-360. [DOI] [PubMed] [Google Scholar]
- 18.Gizatullina, Z. Z., Y. Chen, S. Zierz, and F. N. Gellerich. 2005. Effects of extramitochondrial ADP on permeability transition of mouse liver mitochondria. Biochim. Biophys. Acta 170698-104. [DOI] [PubMed] [Google Scholar]
- 19.Green, D. R., and G. Kroemer. 2004. The pathophysiology of mitochondrial cell death. Science 305626-629. [DOI] [PubMed] [Google Scholar]
- 20.Green, D. R., and J. C. Reed. 1998. Mitochondria and apoptosis. Science 2811309-1312. [DOI] [PubMed] [Google Scholar]
- 21.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 7510623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harty, R. N., J. Paragas, M. Sudol, and P. Palese. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 732921-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huerta-Yepez, S., M. Vega, A. Jazirehi, H. Garban, F. Hongo, G. Cheng, and B. Bonavida. 2004. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-kappa B and inhibition of Bcl-xl expression. Oncogene 234993-5003. [DOI] [PubMed] [Google Scholar]
- 24.Irie, T., J. M. Licata, H. R. Jayakar, M. A. Whitt, P. Bell, and R. N. Harty. 2004. Functional analysis of late-budding domain activity associated with the PSAP motif within the vesicular stomatitis virus M protein. J. Virol. 787823-7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwata, S., C. Ostermeier, B. Ludwig, and H. Michel. 1995. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 376660-669. [DOI] [PubMed] [Google Scholar]
- 26.Jackson, A. C., and J. P. Rossiter. 1997. Apoptosis plays an important role in experimental rabies virus infection. J. Virol. 715603-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacotot, E., K. F. Ferri, C. El Hamel, C. Brenner, S. Druillennec, J. Hoebeke, P. Rustin, D. Metivier, C. Lenoir, M. Geuskens, H. L. Vieira, M. Loeffler, A. S. Belzacq, J. P. Briand, N. Zamzami, L. Edelman, Z. H. Xie, J. C. Reed, B. P. Roques, and G. Kroemer. 2001. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J. Exp. Med. 193509-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karplus, K., C. Barrett, and R. Hughey. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14846-856. [DOI] [PubMed] [Google Scholar]
- 29.Kassis, R., F. Larrous, J. Estaquier, and H. Bourhy. 2004. Lyssavirus matrix protein induces apoptosis by a TRAIL-dependent mechanism involving caspase-8 activation. J. Virol. 786543-6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komarova, A. V., E. Real, A. M. Borman, M. Brocard, P. England, N. Tordo, J. W. Hershey, K. M. Kean, and Y. Jacob. 2007. Rabies virus matrix protein interplay with eIF3, new insights into rabies virus pathogenesis. Nucleic Acids Res. 351522-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopecky, S. A., and D. S. Lyles. 2003. Contrasting effects of matrix protein on apoptosis in HeLa and BHK cells infected with vesicular stomatitis virus are due to inhibition of host gene expression. J. Virol. 774658-4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroemer, G. 2003. Mitochondrial control of apoptosis: an introduction. Biochem. Biophys. Res. Commun. 304433-435. [DOI] [PubMed] [Google Scholar]
- 33.Li, D., X. Z. Wang, J. P. Yu, Z. X. Chen, Y. H. Huang, and Q. M. Tao. 2004. Cytochrome C oxidase III interacts with hepatitis B virus X protein in vivo by yeast two-hybrid system. World J. Gastroenterol. 102805-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li, X. Q., L. Sarmento, and Z. F. Fu. 2005. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J. Virol. 7910063-10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lichty, B. D., H. McBride, S. Hanson, and J. C. Bell. 2006. Matrix protein of vesicular stomatitis virus harbours a cryptic mitochondrial-targeting motif. J. Gen. Virol. 873379-3384. [DOI] [PubMed] [Google Scholar]
- 36.Mebatsion, T., F. Weiland, and K. K. Conzelmann. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73242-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michel, H., J. Behr, A. Harrenga, and A. Kannt. 1998. Cytochrome c oxidase: structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 27329-356. [DOI] [PubMed] [Google Scholar]
- 38.Morimoto, K., H. D. Foley, J. P. McGettigan, M. J. Schnell, and B. Dietzschold. 2000. Reinvestigation of the role of the rabies virus glycoprotein in viral pathogenesis using a reverse genetics approach. J. Neurovirol. 6373-381. [DOI] [PubMed] [Google Scholar]
- 39.Morimoto, K., D. C. Hooper, S. Spitsin, H. Koprowski, and B. Dietzschold. 1999. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J. Virol. 73510-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamichi, K., S. Inoue, T. Takasaki, K. Morimoto, and I. Kurane. 2004. Rabies virus stimulates nitric oxide production and CXC chemokine ligand 10 expression in macrophages through activation of extracellular signal-regulated kinases 1 and 2. J. Virol. 789376-9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouali, M., and R. D. King. 2000. Cascaded multiple classifiers for secondary structure prediction. Protein Sci. 91162-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park, J., K. Karplus, C. Barrett, R. Hughey, D. Haussler, T. Hubbard, and C. Chothia. 1998. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 2841201-1210. [DOI] [PubMed] [Google Scholar]
- 43.Penninger, J. M., and G. Kroemer. 2003. Mitochondria, AIF and caspases—rivaling for cell death execution. Nat. Cell Biol. 597-99. [DOI] [PubMed] [Google Scholar]
- 44.Pfanner, N., and A. Geissler. 2001. Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol. 2339-349. [DOI] [PubMed] [Google Scholar]
- 45.Prehaud, C., S. Lay, B. Dietzschold, and M. Lafon. 2003. Glycoprotein of nonpathogenic rabies viruses is a key determinant of human cell apoptosis. J. Virol. 7710537-10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pulmanausahakul, R., M. Faber, K. Morimoto, S. Spitsin, E. Weihe, D. C. Hooper, M. J. Schnell, and B. Dietzschold. 2001. Overexpression of cytochrome c by a recombinant rabies virus attenuates pathogenicity and enhances antiviral immunity. J. Virol. 7510800-10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raposo, G., M. J. Kleijmeer, G. Posthuma, J. W. Slot, and H. J. Geuze. 1996. Immunogold labeling of ultrathin cryosections: application in immunology, p. 1-11. In D. M. Weir, L. A. Herzenberg, and C. Blackwell (ed.), Weir's handbook of experimental immunology in four volumes, vol. 4. Blackwell Science, Cambridge, MA. [Google Scholar]
- 48.Rehling, P., N. Wiedemann, N. Pfanner, and K. N. Truscott. 2001. The mitochondrial import machinery for preproteins. Crit. Rev. Biochem. Mol. Biol. 36291-336. [DOI] [PubMed] [Google Scholar]
- 49.Sagara, J., and A. Kawai. 1992. Identification of heat shock protein 70 in the rabies virion. Virology 190845-848. [DOI] [PubMed] [Google Scholar]
- 50.Schuler, W., K. Wecker, H. de Rocquigny, Y. Baudat, J. Sire, and B. P. Roques. 1999. NMR structure of the (52-96) C-terminal domain of the HIV-1 regulatory protein Vpr: molecular insights into its biological functions. J. Mol. Biol. 2852105-2117. [DOI] [PubMed] [Google Scholar]
- 51.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21377-423. [DOI] [PubMed] [Google Scholar]
- 52.Shirakata, Y., and K. Koike. 2003. Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J. Biol. Chem. 27822071-22078. [DOI] [PubMed] [Google Scholar]
- 53.Somasundaran, M., M. Sharkey, B. Brichacek, K. Luzuriaga, M. Emerman, J. L. Sullivan, and M. Stevenson. 2002. Evidence for a cytopathogenicity determinant in HIV-1 Vpr. Proc. Natl. Acad. Sci. USA 999503-9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stojanovski, D., A. J. Johnston, I. Streimann, N. J. Hoogenraad, and M. T. Ryan. 2003. Import of nuclear-encoded proteins into mitochondria. Exp. Physiol. 8857-64. [DOI] [PubMed] [Google Scholar]
- 55.Thoulouze, M. I., M. Lafage, V. J. Yuste, L. Baloul, L. Edelman, G. Kroemer, N. Israel, S. A. Susin, and M. Lafon. 2003. High level of Bcl-2 counteracts apoptosis mediated by a live rabies virus vaccine strain and induces long-term infection. Virology 314549-561. [DOI] [PubMed] [Google Scholar]
- 56.Thoulouze, M. I., M. Lafage, V. J. Yuste, G. Kroemer, S. A. Susin, N. Israel, and M. Lafon. 2003. Apoptosis inversely correlates with rabies virus neurotropism. Ann. N. Y. Acad. Sci. 1010598-603. [DOI] [PubMed] [Google Scholar]
- 57.Ubol, S., C. Sukwattanapan, and Y. Maneerat. 2001. Inducible nitric oxide synthase inhibition delays death of rabies virus-infected mice. J. Med. Microbiol. 50238-242. [DOI] [PubMed] [Google Scholar]
- 58.Wiedemann, N., N. Pfanner, and M. T. Ryan. 2001. The three modules of ADP/ATP carrier cooperate in receptor recruitment and translocation into mitochondria. EMBO J. 20951-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida, M. 2001. Multiple viral strategies of HTLV-1 for dysregulation of cell growth control. Annu. Rev. Immunol. 19475-496. [DOI] [PubMed] [Google Scholar]
- 60.Young, J. C., N. J. Hoogenraad, and F. U. Hartl. 2003. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 11241-50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.