

Summary
We have employed our CD4+ T cell model named HRF(+) (HIV-1 Resistance Factor) to study the inducible anti-HIV-1 responses mediated through novel soluble molecules. We found that exposure to the soluble products of HRF(+) cells activated CTCF (CCCTC binding factor) mRNA expression in HIV-1 susceptible primary and transformed CD4+ T cells and overlapped with their acquisition of transient resistance to virus. Conversely, the interference with the expression of CTCF gene in HRF(+) cells reversed the resistant phenotype and eliminated the biological potential of their cell culture supernatant to induce “HRF-like” activity in target cells. Band-shift analysis upon the nuclear fractions from HIV-1 resistant cells showed that CTCF protein bound to HIV-1 promoter and this binding prevented the formation of NF-κB/LTR complex. This evidence suggests that CTCF is an intracellular effector of HRF activity and that the acquisition of resistance to HIV-1 in CD4+ T cells is a consequence of the prior activation of CTCF gene by the soluble entity secreted by HRF(+) cells.
Keywords: CD4, T cell, HIV-1, CTCF, resistance
2. Introduction
CTCF (CCCTC binding factor) has been called a first “truly multivalent” nuclear factor performing multiple regulatory tasks in gene expression [1,2]. Through the binding of highly divergent DNA sequences and formation of various protein complexes, this multifunctional protein might suppress or activate various genes [3]. The transcription suppressive activity of CTCF protein was best described for the chicken lysozyme [4] and chicken, mouse and human c-myc genes [5]. CTCF gene is highly conserved and ubiquitously expressed throughout the Animal Kingdom, and although much has been uncovered about its structure and function, there are still some problematic issues that need to be defined. For example several mature CTCF mRNA isoforms have been detected in chicken cells [6] and CTCF gene protein products show some degree of size variability including smaller forms of unknown function [3,6,7].
In previous studies we characterized our CD4+ T cell model for studies of acquired resistance to HIV-1 infection [8–12]. In this model, HIV-1 resistant cells named HRF(+), restricted transcription of viral genome through the protective activity of a novel soluble protein factor(s) named HRF (HIV-1 Resistance Factor) acting on the level of nuclear NF-κB/DNA binding [9–11]. Thorough analysis of genes involved in transcription regulation revealed that the expression of CTCF mRNA was consistently up-regulated in HRF(+) cells [8]. As CTCF is a potent suppressor of transcription [3,5,13], a characteristic that is also the “landmark” of HRF antiviral activity [10,12], we explored the significance of CTCF mRNA up-regulation and its link to the acquired resistance to HIV-1 in CD4+ T cells.
This investigation demonstrated that the increased transcription of CTCF gene is associated with the acquired antiviral activity in CD4+ T cells. We tested the CTCF mRNA expression in several HRF(+) clones by real-time PCR, and found, that elevated activity of this gene could be correlated to the extent of the antiviral protection; the higher CTCF transcriptional activity corresponded to the stronger antiviral responses. The exposure of human macrophages and transformed CD4+ T cells to the soluble products of HRF(+) cell line also increased the expression of their endogenous CTCF mRNA and the elevated activity of CTCF gene in these cells coincided with the induction of transient “HRF-like” resistance to virus. The antiviral activity of HRF(+) cell culture supernatant was eliminated by the RNAi - mediated depletion of CTCF. To understand the link between the HRF-mediated up-regulation of CTCF transcriptional activity and the cellular resistance to virus we analyzed the affinity of nuclear CTCF to DNA sequences on HIV-1 promoter and found that CTCF binds to LTR and this CTCF/DNA association blocks the formation of NF-κB/DNA complex. Taken altogether these observations suggest that CTCF is an intracellular effector of HRF activity regulating cellular resistance to HIV-1 through the interference with HIV-1 transcription and show a novel function for this potent transcription regulator.
3. Materials and Methods
Cell cultures and viruses
HRF(+) and HRF(-) cells distinguished by HIV-1 resistance and HIV-1 susceptibility have been described previously [8–12,14,15]. 1G5 cells stably expressing luciferase gene driven by HIV-1 long terminal promoter (LTR) [16] were obtained through NIH AIDS Research and Reagent Program Division of AIDS, NIAID. Human monocytes from healthy, HIV-1 negative volunteers were obtained by elutriation from the whole blood. Monocytes were allowed to differentiate in 12-well plates (Corning Scientific) under culture conditions as described before [17]. Briefly: cells were seeded into 12-well plates at concentration of 1.5x106/well and allowed to differentiate into macrophages in DMEM (Sigma) supplemented with 10% endotoxin-free, heat inactivated human serum, 10% giant cells tumor conditioned medium (Sigma), glutamate and antibiotics.
Continuous cultures of 1G5, HRF(+), and HRF(-) cell lines were maintained in RPMI 1640 (Sigma) supplemented with 5% fetal bovine serum, antibiotics and glutamate at 37°C in a 5% CO2 95% air-humidified incubator. To prepare biologically active supernatants HRF(+) and control HRF(-) cells were cultured as described before [9,10]. Briefly; 2x106/ml HRF(+) and control HRF(-) cells were cultured overnight in protein free Hybridoma medium (Sigma) supplemented with glutamate. Subsequently, cells were removed by centrifugation and supernatants were filtered through 0.45mM membranes (Millipore) and concentrated by lyophilization from 30-ml aliquots using Labconco Freeze-Dryer. Protein powder was resuspended in 10 ml distilled water and subjected to dialysis against 10mM Tris-Cl, pH 8.0 using benzoylated cellulose tubing with MW cut-off of 1.2KDa (Sigma). Dialyzed material was concentrated again by lyophilization and stored at 40C. For cell treatments lyophilized samples were resuspended in sterile distilled water to 100x concentrate followed by second dilution in Hybridoma medium (Sigma) to 1x concentrate. This material was used to prepare conditioned culture media for macrophages.
Virus isolates used for this study was NL4-3 [18] and HIV-1 ADA [19].
Testing of HRF biological activity by Rapid Suppression Assay (RSA)
RSA was performed essentially as described before [9,10]. Briefly: 1G5 cells that are stably transfected with an inducible luciferase gene driven by HIV-1 LTR [16] were washed in PBS, resuspended in hybridoma medium to concentration of 5x106 cells/ml. For control titration curves, 100μl aliquots of 1G5 cells were supplemented with 0.01% to 50% volume of HRF(+), HRF(-) supernatants or medium alone, brought to a final volume of 200μl and incubated for three hours at 370C. Subsequently, all cells were induced with PMA at concentration of 5ng/ml. Two control tubes contained 1G5 cells were resuspended in hybridoma medium with or without PMA. Three hours later cells were collected by centrifugation and lysed in the same tubes using Reporter Lysis buffer (Promega). The expression of luciferase protein was tested based on manufacturers protocol. Cell culture supernatants for RSA testing were dialyzed for 48 hours against 10mM Tris-Cl, pH 8.0, concentrated by lyophilization and tested as 1%/vol.
RNA analyses
Total RNA was isolated using RNaeasy mini kit (Qiagen). For real-time PCR analyses total of 3μg RNA was reverse transcribed to cDNA with random hexamers using the SuperScript III First-Strand Synthesis kit (Invitrogen). The real-time detection of CTCF and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) genes expression was performed using TaqMan Gene Expression Assay on 7500 real-time PCR System (Applied Biosystems). Briefly; triplicates of total reaction mixture of 25μl were composed of 2μl of cDNA, 12.5μl of 2xABI TaqMan PCR Master Mix, and 0.25μl (90μM) of both sense and antisense primers and 0.5μl of the probe. After the initial denaturation at 950C (10min), target genes were amplified through 40 cycles of universal cycling conditions (950C/10s, 600C/1min). The GAPDH target gene was amplified using pre-designed pair of primers and a probe (Applied Biosystems) and CTCF gene was amplified by pair of primers spanning through zinc finger 4: CTCF-F 5’-TTCAAGTGTTCCATGTGCGATT-3’ and CTCF-R 5’-GTATGAGAGCGAATGTG ACGTTTTA-3’ (Invitrogen) and detected by probe CTCF-p 5’-6 FAMCAGTGTAGAAGTCAGCAA AMGBNFQ-3’ (Applied Biosystems). Standard cell dilution curves were calibrated using serial dilutions of control plasmid pTA_CTCF representing 1x106, 1x105, 1x104, 1x103, 1x102 and 10 copies of CTCF cDNA per reaction and all values are expressed as means ± standard deviation of the mean. The absolute value representing ratio target gene expression was calculated based on formula:
Investigation of the induction of CTCF expression in transformed CD4+ T cell line and in human macrophages upon exposure to soluble products of HRF(+) cell line
HRF(-) and HRF(+) cells were cultured adjacent to each other in a double chamber apparatus (Cornig). Macrophages were cultured in 7.5% conditioned HRF(+) and HRF(-) medium or hybridoma medium alone. Three days later treatments were removed and cells were cultured alone in DMEM complete growth medium for up to eight days. Expression of CTCF control GAPDH genes were tested by real-time PCR using pairs of primers described above; the biological activity of their cell culture supernatants was tested by RSA and replication of HIV-1 was tested by p24 Elisa (Perkin Elmer).
siRNA silencing of CTCF mRNA in HRF(+) cells
Homo sapiens CTCF mRNA sequence (U25435) was subjected to BLAST analysis in order to exclude the common coding sequences. Based on this analysis we selected a 190bp mRNA fragment unique to CTCF as a template for dsRNA synthesis. Two primers sense 5’-GGTCTGCTATCAGAGGTTAAT GCGG-3’ and antisense 5’-GTGACGATCCAAATTTGAACGCCGT-3’, with appended T7 promoter sequences (5’-TAATACGACTCACTATAGG-3’) were used to amplify a CTCF cDNA fragment from mRNA of HRF(+) cells. Subsequently, cDNA fragment was cloned into pCRTA vector (Invitrogen) and amplified in DH5α E. coli. CTCF cDNA fragment enlarged by T7 polymerase promoter was excised from the plasmid by EcoRI digestion, separated through agarose gel electrophoresis and purified through Ultrafree-DA columns (Millipore). 3μg of this material and control Litmus 38iluc served as templates for dsRNA synthesis using ShortCut RNAi kit based on manufacturer’s protocol (New England Biolabs Inc). 10μg of dsRNA from both samples was subjected to processing by shortcut RNase III for 20 min at 370C and purified by ethanol precipitation. The concentration of siRNA fragments was calculated based on UV absorbance and by band intensity comparison of siRNA fragments run next to serial dilutions of 21bp RNA standard (New England Biolabs Inc) on 20% TBE polyacrylamide gel (Invitrogen).
For the initial CTCF knock-out experiments 3x106 HRF(+) cells were transfected with 9nM of CTCF and control Litmus 38iluc siRNA fragments by lipofectamine 2000 (Invitrogen) in Hybridoma medium (Sigma). Complete growth medium was added after 4 hours and cells were cultured in concentration of 1x106cells/ml for another eight days. The CTCF mRNA degradation was tested every two days by real-time PCR as described above. The inhibition of protein synthesis was confirmed by Western blot analysis using α-CTCF and control α-tubulin antibodies (Santa Cruz biotechnology, Inc.).
Assay of the effect of CTCF siRNA silencing on susceptibility of HRF(+) cells to HIV-1 replication
To test the correlation of CTCF to HRF activity 2.1x107 HRF(+) and 1.2x107 HRF(-) cells were infected with HIV-1 NL4-3 [18] at concentration of 0.5pg/cell. Three days later HRF(+) culture was divided into three parts: one served as a control for restrictive infection and two others were subjected to siRNA silencing by CTCF and Litmus 38iluc fragments at 9nM concentration as described above. HIV-1 infection was monitored by Elisa (Coulter) measuring levels of extracellular antigen p24 and by RT-PCR of the single spliced vif transcript using pair of primers described before [9].
Assay of the effect of CTCF siRNA silencing on the biological activity of HRF(+) cell Culture supernatant
Macrophages were cultured in 7.5% conditioned HRF(+), HRF(+) CTCF siRNA and HRF(-) medium or hybridoma medium alone. Three days later treatments were removed and cells were cultured alone in DMEM complete growth medium for up to four days. The expression of CTCF and control GAPDH genes was tested by the real-time PCR using pairs of primers described above and biological activity of their cell culture supernatants was tested by RSA.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear translocation of transcription factors in HRF(+) or control HRF(-) cells was induced with 0.1pg/cell HIV-1 NL4-3 for 30 min. at 370C. Subsequently, the induced and un-induced cells were washed in PBS and lysed in lysis buffer (20mM Hepes, pH 7.9, 10mM NaCl, 3mM MgCl2, 1mM DTT, 0.5mM EDTA, 10% glycerol, 0.1% NP-40, 1mM Na3VO4 and protease inhibitor cocktail) and nuclear fractions were collected by sedimentation at 500 x g. Nuclear proteins were extracted with extraction buffer (20mM Hepes, 400mM NaCl, 1mM DTT, 0.5mM EDTA, 20% glycerol, 1mM Na3VO4 and protease inhibitors) and tested for their ability to bind labeled nucleotides representing consensus NF-κB site provided by manufacturer (Promega) or YB-1 probe 5’-361-CTGGGGACTT TCCAGGGAGGCGTGGCCTGGGCGGGACTGGGGA-404-3’ synthesized by Invitrogen Inc. All DNA-protein binding reactions were based on manufacturer’s protocol (Promega) and described before [10]. Briefly; 0.5 ng of labeled probe was incubated for 20 min at 40C with 5μg of nuclear extracts in the presence of 1x gel shift buffer (Promega).
For the supershift band analysis 5μg of nuclear extracts were incubated for 90 min. on ice with 4μg of relevant antibody. All antibodies to p65 NF-κB, Sp1, YB-1 and CTCF were purchased from Santa Cruz, Inc. Subsequently, 1.5μl of a 10x loading buffer was added into all reactions, followed by separation by electrophoresis on 4% polyacrylamide gel until free probe was close to the bottom of the gel.
Sequencing and cloning of CTCF gene from HRF(+) cells
CTCF cDNA was amplified from HRF(+) cells by high fidelity pfu polymerase (Perkin Elmer) using pair of primers: sense CTCFF2 5’-ATGGAAGGTGATGCAGTCGAAGC-3’ and antisense CTCFR1 5’-TCACCGGTCCATCATGCTGAGGAT-3’. Subsequently the CTCF orf was cloned into TA cloning vector (Invitrogen) and subjected to sequencing at DNA sequencing Core Facility at Columbia University using consecutive forward primers: CTCFF2 (as above), CTCFF5 5’-ACATGAGAACCC ATTCAGGGGA-3’, CTCFF8 5’-TGAAGTGTCTAAAGAGGGCCT-3’ and reverse CTCFR1 (as above) . The pTA_CTCF was used to prepare standard curves for amplification of CTCF mRNA by real-time PCR.
4. Results
The level of CTCF expression correlates with the level of antiviral protection in HRF(+) cells
We reported previously that HRF(+) cells have elevated levels of CTCF mRNA [8]. The over-expression of a potent transcription inhibitor [5,20–22] in cells known for secreting a novel protein factor that inhibits transcription of HIV-1 was interesting and prompted speculation that these two activities are interrelated to the acquired resistance to virus. Since the previous data were obtained for HRF(+) cell line selected for the maximum antiviral responses, we evaluated this hypothesis measuring levels of CTCF mRNA by real-time PCR in other HRF(+) clones in hand (Table 1). Although expression of CTCF was up-regulated in all HRF(+) clones; clones differed in CTCF expression. To evaluate the relation of this diversity in CTCF mRNA level to HIV-1 resistance, we challenged all tested cell lines with HIV-1, and seven days later, measured virus replication by indirect immunofluorescence for HIV-1 antigens (IF). Again, all HRF(+) clones but not the control HRF(-) cell line restricted HIV-1 infection but they could be distinguished by the extent of their anti-viral responses, and more interestingly these variations in the antiviral protection correlated to the level of CTCF mRNA up-regulation (Table 1). For example the CTCF expression in control HRF(-) cells was at 2,28 mRNA copies/cell and at the seven-day time point 72% of the cell population was infected with HIV-1. On the other hand, the 2.4-fold increase of CTCF transcription in HRF(+) clone 5 (HRF(+)5) correlated with the 58% inhibition of HIV-1 replication and greater than 3-fold increase of CTCF mRNA overlapped with the inhibition of HIV-1 replication from 66 to 70% in clones 15, 28 and 22 respectively (Table 1). These data indicate that antiviral activity in HRF(+) cells could be associated with the expression of CTCF gene and prompted subsequent question, as to whether HRF mediated protection in HIV-1 susceptible target cells could be also linked to the elevated expression of CTCF gene.
Table 1.
Correlation between the levels of CTCF mRNA expression and the levels of antiviral protection in several HRF(+) clones.
| Analysis | HRF(-) | HRF(+)5 | HRF(+)15 | HRF(+)28 | HRF(+)22 |
|---|---|---|---|---|---|
| CTCF mRNA (copy/cell) | 2.28 | 5.56 | 7.4 | 8.1 | 12.8 |
| CTCF mRNA (fold increase) | 1 | 2.4 | 3.25 | 3.55 | 5.6 |
| HIV-1 replication (% IF+) | 72 | 14.3 | 5.8 | 2.9 | 1.6 |
The CTCF gene is up-regulated in HIV-1 susceptible target cells upon their exposure to HRF(+) cell culture supernatant and elevated expression of CTCF coincides with the induction of “HRF-like” activity
The correlation between CTCF gene up-regulation and levels of the antiviral protection in several HRF(+) clones suggested that this protein could be the intracellular effector molecule for extracellular HRF and with this in mind we investigated whether the induction of antiviral resistance in HIV-1 susceptible cells coincided with the increased activity of CTCF transcription. Based on our previous findings that HIV-1 susceptible cells cultured in adjacent chamber with HRF(+) cells were protected from virus infection [11,12] we assayed CTCF gene expression in HRF(-) cells that were cultured for three days next to HRF(+)28 clone in a double chamber apparatus. Exposed HRF(-) cells were harvested daily throughout the period of three days and the transcription levels of CTCF gene were analyzed by the real-time PCR (Table 2). HRF(+) treatment induced the expression of CTCF gene in HIV-1 susceptible target cells and two days after the initial exposure the CTCF mRNA was up-regulated by 1.54-fold and three-day co-culture further increased this gene expression, raising CTCF mRNA copy number to 7.4 copies/cell (Table 2).
Table 2.
Exposure to soluble products of HRF+ cell line induces expression of CTCF mRNA in target cells.
| Analysis | HRF(-)/HRF(+) day 1 | HRF(-)/HRF(+) day2 | HRF(-)/HRF(+) day 3 | HRF(+)28 | HRF(-) |
|---|---|---|---|---|---|
| CTCF mRNA (copy/cell) | 2.1 | 3.4 | 7.4 | 9.8 | 2.2 |
| CTCF mRNA (fold increase) | 0.95 | 1.54 | 3.36 | 4.45 | 1 |
Similar studies were subsequently performed in human macrophages – a primary target cells for HIV-1 infection. Based on our previous observations three days exposure to HRF induced endogenous “HRF-like” resistance in these cells, and the induction of antiviral activity could be achieved through soluble molecules present in HRF(+) cell culture supernatant [23]. On average, five days from the initial exposure to HRF(+) cell culture supernatant or three days after the cessation of HRF treatment, macrophages acquired an “HRF-like” activity which could be detected in their culture supernatants by Rapid Suppression Assay – (RSA) [9].
We considered the possibility, that the acquisition of “HRF-like” activity in human macrophages could be linked to the activation of CTCF mRNA transcription and to test this hypothesis macrophages from two different HIV-1 negative donors were cultured alone or in the presence of 7.5% HRF(+) or control HRF(-) conditioned medium for three days, then treatments were terminated. Expression of CTCF gene was assayed by real-time PCR, and the induction of “HRF-like” activity by RSA. In this experimental setting we investigated the “early” and “late” changes of gene expression, where the “early” time-points defined the first 48 hours of continuous treatments and the “late” time-points tested both the CTCF mRNA expression and the induction of antiviral resistance after treatments were terminated (Fig. 1).
Fig. 1. Elevated expression of CTCF mRNA in primary human macrophages exposed to soluble products of HRF+ coincides with the induction of antiviral resistance.

Expression of CTCF mRNA (linear plot, left y axis) was analyzed by real-time PCR; each point of the linear plot represents a mean value of three measurements. The antiviral activity of cell culture supernatants (bars right y axis) was tested by RSA. Grey boxes show continuous exposure of target cells to treatments; RSA values show percent inhibition of HIV-1 LTR-promoted expression of luciferase gene by 1%/Vol of the sample. MQ denotes macrophages.
As observed for transformed T cells, HRF(+) but not HRF(-) treatment induced the expression of CTCF gene in human macrophages, as early as 1 hour from the initial exposure the concentration of CTCF mRNA increased by 100- fold in cells from donor 1 and 5.5-fold in cells from donor 2 (Fig. 1 left y axis). Two days later the activation of CTCF gene still exceeded controls by 100-fold (donor 1) and by 2.4-fold (donor 2) (Fig. 1 left y axis ). Most remarkably, the cessation of HRF(+) treatment did not stop the CTCF activation and high levels of its mRNA were still present for up to seven days later (Fig. 1 left y axis, 4 and 7 day-time points) which indicated that the soluble factor(s) present in HRF(+) supernatant elicited a long-term effect in target cells.
RSA evaluation of cell culture supernatants collected after treatments were terminated showed that macrophages secreted “HRF-like” activity and its induction coincided with the induction of CTCF mRNA expression (Fig. 1 right y axis). HRF treatment altered the composition of proteins secreted by macrophages and their culture supernatants now inhibited expression of the reporter gene by 29 to 41% 24 hours after the termination of treatment and by 30 to 50% four days later in donor 1 and 2 respectively (Fig. 1 right y axis; 4 and 7-day time points).
Employing the same experimental setting we assayed the efficiency of HIV-1 replication in human macrophages induced to resist virus infection. One day after the cessation of HRF treatment cells were challenged with 0.01pg/cell ADA, a well characterized macrophage tropic HIV-1 isolate [19]. The activity of CTCF mRNA was tested by Q-PCR and replication of virus was assayed by levels of the extracellular p24 core antigen. As shown in Fig. 2, replication of virus was reduced by 35 and 50% six and eight days after the initial exposure of macrophages to HRF treatment. The inhibition of HIV-1 replication correlated with the 2.2-fold increase in the activity of CTCF mRNA and provided evidence that the acquisition of antiviral activity and the induction of CTCF transcription are interrelated and suggested that CTCF protein might be the cellular partner of HRF.
Fig. 2. Replication of R5 HIV-1 ADA is inhibited in human macrophages after their exposure to soluble products of HRF(+) cells.

Expression of CTCF mRNA (linear plot, left y axis) was analyzed by real-time PCR; each point of the linear plot represents a mean value of three measurements. HRF-treated macrophages were challenged with HIV-1 after termination of treatment. Replication of HIV-1 was analyzed by p24 Elisa (bars, right y axis) at one, three and five days after infection which corresponded to four, six and eight days from the initial exposure to HRF treatment. Grey box shows continuous exposure of target cells to treatments;
Expression of CTCF is indispensable for HRF function in both intra- and extracellular compartments
To evaluate the hypothesis that CTCF protein is involved in the regulation of antiviral resistance in CD4+ T cells, we tested the consequences of transient interference with CTCF expression to the antiviral activity in HRF(+) cells. We used siRNA gene targeting [24] and established experimental conditions ensuring the viability of cell culture and significant down-regulation of CTCF expression.
As shown in Table 3, CTCF but not control Litmus siRNA fragments reduced the expression of CTCF gene in these cells by 81% two days after the delivery. CTCF mRNA levels were still reduced by 65%, 50% and 34% four, six and eight days after transfection and matched the significant reduction of CTCF protein (Fig. 3).
Table 3.
Comparison of CTCF mRNA levels in cells subjected to treatment with CTCF or control litmus siRNA fragments.
| Analysis | CTCF mRNA copy/cell | |||
|---|---|---|---|---|
| Day 2 | Day 4 | Day 6 | Day 8 | |
| HRF(+) CTCF siRNA | 2.4 | 4.5 | 6.2 | 8.6 |
| HRF(+) litmus siRNA | 8.3 | 13 | 12 | 12 |
| HRF(+) | 13 | 12.8 | 12.5 | 13 |
Fig. 3. The siRNA silencing of CTCF gene.
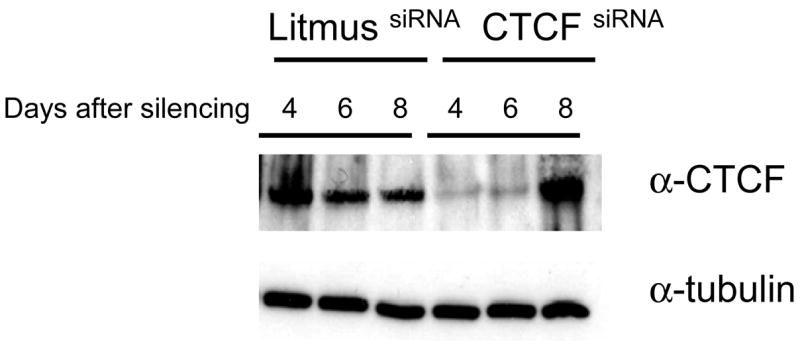
HRF(+) cells were subjected to treatments by 9 nM of CTCF or control litmus siRNA fragments and effects of this interference with CTCF gene expression were assayed over time by Western blot using antibodies to CTCF and α-tubulin. Data presented in this figure show one of the three separate experiments.
These experimental conditions were used to test the effect of CTCF gene silencing on the antiviral activity in HRF(+) cells challenged with 0.5pg/cell NL4-3 HIV-1. Replication of HIV-1 was tested by RT-PCR of the single-spliced vif transcript and detection of the p24 core antigen (Fig. 4A & B). Consistent with our previous observations [10,11], replication of HIV-1 was inhibited in HRF(+) cells and the exposure to litmus siRNA fragments did not have any affect. However, the “knock-out” of CTCF gene expression allowed virus replication and both HIV-1 vif mRNA and p24 protein were synthesized at levels surpassing those observed in control HRF(-) cells. Eight days after CTCF silencing, vif mRNA levels in HRF(+) cells were increased by 2-fold and extracellular p24 protein by 28-fold respectively (Fig. 4A and B). Subsequent measurements of HRF activity in cell culture supernatants by RSA revealed that the silencing of CTCF gene resulted also in substantial reduction of activity in the extracellular compartment (Fig. 4C) and suggested that the elevated expression of CTCF has direct effect in the maintenance of HRF-mediated activity.
Fig. 4. Transient interference with the expression of CTCF mRNA reverses the HIV-1 resistant phenotype in HRF(+) cells.

HRF(+) and HRF(-) cells were transfected by CTCF or control Litmus siRNA fragments then challenged with NL4-3 HIV-1 at concentration of 0.5pg/cell. Replication of HIV-1 was tested by RT-PCR of the single-spliced vif transcript (A); detection of the p24 core antigen (B); biological activity of cell culture supernatants was tested by RSA (C). Data presented in this figure show one of the three separate experiments.
To clarify this proposition we treated human macrophages with the extracellular material from HRF(+) cells subjected to siRNA-mediated depletion of CTFC. As shown in Fig. 5, elimination of CTCF in HRF(+) cells altered the composition of their cell culture supernatant and this reagent did not induce “HRF-like” activity in macrophages; both, the expression of CTCF mRNA (Fig. 5 left y axis) and levels of antiviral activity (Fig. 5 right y axis) were comparable to cells treated with the control HRF(-) supernatant or cultured alone thus confirming our suggestion that the induction of endogenous protection in HIV-1 susceptible cells is a consequence of the prior up-regulation of CTCF mRNA expression initiated by the exposure of target cells to the soluble entity present in HRF(+) cell culture supernatant.
Fig. 5. Transient interference with the expression of CTCF mRNA in HRF(+) cells inactivates the biological potential of their cell culture supernatant to induce the antiviral activity in target cells.

Expression of CTCF mRNA (linear plot, left y axis) was analyzed by real-time PCR, and the antiviral activity of cell culture supernatants (bars, right y axis) was tested by RSA. Grey box shows continuous exposure of target cells to treatments; RSA values show percent inhibition of HIV-1 LTR-promoted expression of luciferase gene by 1%/Vol of the sample and the each point of the linear plot represents a mean value of three measurements.
HIV-1 exposure induces CTCF/LTR binding in HRF(+) cells
The CTCF protein binds DNA targets with a remarkable plasticity through various combinations of its zinc fingers. It also forms complexes with other proteins leading to acquisition of novel functions by these CTCF/protein formations [25,26]. We reported previously that the exposure to HRF affected the formation of NF-κB/LTR complex [10,27]. As CTCF is a potent inhibitor of transcription we considered the possibility that the increased concentration of this protein alters the events on HIV-1 promoter. Although so far no single consensus sequence for CTCF has been defined, data suggest that this protein binds with high affinity to GC reach motifs [3]. Sequence analysis of HIV-1 LTR revealed possible CTCF binding sites, one of them was detected within the YB-1 binding sequences. In the absence of scientific precedence of CTCF/LTR interactions we considered two possibilities: (i) that CTCF could bind LTR directly or (ii) through its interaction with YB-1 protein [25,28,29] could affect the NF-κB sequences separated from the YB-1 cognate sequence by only six nucleotides (Fig 6). To evaluate this proposition we synthesized a 43-mer probe (called here a YB-1 probe) encompassing the binding sites for NF-κB, YB-1 and Sp1 proteins and applied band–shift analyses to compare the nuclear translocation and binding activity of NF-κB, Sp1, YB-1 and CTCF proteins from HRF(+) and HRF(-) cells to this fragment of HIV-1 promoter (Fig. 6). The nuclear translocation of transcription regulators was initiated by 30 min exposure to HIV-1 and nuclear fractions were isolated based on methods described before [10]. As shown in Fig. 6A (left panel; YB-1 probe), HIV-1 exposure induced the nuclear translocation and formation of protein/DNA complex in both tested cell lines; however, nuclear extracts derived from the induced control HRF(-) cells bound to YB-1 probe with about 50% higher efficiency as compared to band intensity in sample derived from induced HRF(+) cells. The NF-κB binding site was minimally shifted by HRF(+) sample (Fig. 6A- right panel-NF-κB probe), but proteins in HRF(-) nuclei clearly shifted the probe mobility suggesting that nuclei from HRF(-) cells have more NF-κB binding activity than nuclei from HRF(+) cells. The visible YB-1 probe shifts in HRF(+) samples (Fig. 6A- left panel -YB-1 probe) could be formed by either Sp-1 or YB-1/DNA complexes as these proteins bind LTR in the exclusive manner [30].
Fig. 6. EMSA analysis of the nuclear fractions from HRF(+) and HRF(-) cells upon HIV-1 NL4-3 induction.

The nuclear translocation of transcription factors was initiated by 30 min exposure to HIV-1 at concentration of 0.1pg p24/cell. (A) The nuclear translocation of proteins was tested by formation of protein/DNA complexes to YB-1 or to consensus NF-κB probes; (B through E) Protein complexes bound to the YB-1 probe were detected by antibodies raised to p65-NF-κB, Sp-1, YB-1 and CTCF proteins; [C] annotation in the gels indicates that competing unlabeled nucleotide was added into the reaction]; (F) The YB-1 probe enclosing cognate sequences for NF-κB (363–373); Sp1-III (376–385); Sp1-II (387–396); Sp1-I (398–402); YB-1 (381–402) was designed based on the HIV-1 HXB2 promoter sequence; Data presented in this figure show one of the three separate experiments.
To resolve this question the composition of protein complexes bound to YB-1 probe was analyzed by supershift EMSA using antibodies to p65 NF-κB, Sp-1 and YB-1 proteins (Fig. 6B-D). The NF-κB dimer bound to its cognate sequence on YB-1 probe only in the control HRF(-) but not in HRF(+) sample, thus confirming our conclusions of shift analysis shown in Fig. 6A. HIV-1 exposure induced also the Sp-1/LTR binding in HRF(-) cells (Fig. 6C). Binding of Sp1 protein was less efficient in HRF(+) sample, which suggested that these sequences could be occupied by YB-1 protein. This proposition was confirmed by reaction with antibody to YB-1 which detected more YB-1 protein from HRF(+) nuclei bound to the probe under both the “constitutive” and “virus-induced” culture conditions. We also tested nuclear extracts from HRF(+) and HRF(-) cells for the presence of CTCF/LTR binding and found that antibody to CTCF formed a supershift band in samples from HRF(+) but not from HRF(-) cells (Fig. 6E). This data showed for the first time that CTCF binds to HIV-1 LTR sequences and this protein/DNA association interfered with the NF-κB/LTR complex formation. It is noteworthy that the nuclear CTCF and YB-1 proteins isolated from HRF(+) cells formed complexes with the LTR probe with similar affinity in both “constitutive” and “inducible” culture conditions thus suggesting that the cellular exposure to HRF rather than virus induction enables CTCF affinity to regulate this new target.
5. Discussion
Novel functions of CTCF protein in regulation of cellular latency of EBV and HSV-1 have been reported recently by other groups [27,31]. In one system cellular exposure to EBV activated transcription of CTCF followed by accumulation of its protein product. Consequently, CTCF protein bound to EBV sequences and inhibited transcription of virus encoded transactivator-EBNA2. In other studies a short autonomous repression motif located within N’terminal domain of CTCF was found to be important for the suppression of basal activity of SV40- and CMV promoters [13]. These newly discovered functions of CTCF protein demonstrated its remarkable regulatory diversity. We found that the exposure to active entity secreted by HIV-1 resistant cells also induced the activity of CTCF gene. To understand this phenomenon we set our research objective to verify two hypotheses; (1) that the up-regulation of CTCF gene is linked directly to the HRF-mediated protection in HIV-1 susceptible target cells, and (2) that the CTCF gene product could be in fact an intracellular co-factor of HRF activity.
The first theory was particularly interesting in conjunction with our recent observations, suggesting that HRF treatment induced the production of endogenous “HRF-like” activity in human macrophages [23]. The acquisition of antiviral protection in HIV-1 target cells resulted from their initial exposure to the soluble products of HRF(+) cell line and persevered after treatment was terminated. Newly acquired “HRF-like” resistance was maintained through soluble molecule(s) secreted into the extracellular compartment and like “native” HRF, restricted replication of HIV-1 in virus susceptible, primary cells.
We explored the possibility that the acquisition of “HRF-like” activity in target cells could be correlated to the increased transcription of CTCF and tested this gene’s activity in transformed and primary HIV-1 susceptible CD4+ T cells upon their exposure to soluble products of HRF(+) cells. Probing over time the CTCF mRNA expression in target cells cultured next to HRF(+) clone confirmed our hypothesis. HRF treatment elicited rapid activation of CTCF expression coinciding with the induction of endogenous resistance. This effect was particularly strong in human macrophages thus excluding the concern that the source of CTCF observed in transformed T cell lines could be induced by a transforming virus rather than by the HRF.
Since the acquisition of “HRF-like” antiviral activity coincided with the dramatic increase of CTCF mRNA, we postulated that CTCF is in fact an intracellular effector of HRF activity and the rapid accumulation of this gene product (unpublished data) contributes directly to the “switch” from “HIV-1 susceptible” to “HIV-1 resistant” phenotype. This hypothesis was confirmed by two observations; (i) the resistant phenotype in HRF(+) cells after siRNA directed silencing of CTCF expression was reversed, and (ii) the soluble products derived from HRF(+) cells subjected to CTCF siRNA silencing did not induce the antiviral responses in human macrophages. These observations suggested that that the increased activity of CTCF gene was indispensable for the maintenance of antiviral resistance and the increased expression of CTCF protein is required to regulate the expression or export of HRF itself.
The “landmark” of HRF mediated antiviral activity in CD4+ T cells is the inhibition of virus transcription through the interference with the formation of NF-κB/DNA complex [10,23]. We reported previously that HRF treatments did not restrict the NF-κB nuclear translocation and the active entity in HRF(+) supernatants did not compete with NF-κB dimer for its cognate site on HIV-1 promoter. We hypothesized that the intracellular HRF effector molecule could bind to LTR through its own unique DNA site, or through its association with other LTR regulatory proteins and disable the formation of NF-κB/LTR complex. The evidence of CTCF binding to HIV-1 promoter supported this proposition and enabled construction of a hypothetical model of the HRF-mediated antiviral activity involving CTCF (Fig. 6). Based on our data we suggest that HRF induces the activation of CTCF gene through the ligation of cell surface receptor or its cellular uptake. The activation of CTCF gene expression might be induced by the cellular up-take of HRF or through the activation of other cellular partners. The enhanced activity of CTCF gene is followed by the increase of the protein concentration (data not shown). Detection of CTCF/LTR complex in un-induced cells indicates that the nuclear translocation of CTCF is independent of virus stimuli; however, upon the virus invasion the cytosolic CTCF protein is relocated to the nucleus where it binds to HIV-1 LTR with a higher efficiency shown by the increase of CTCF/DNA band density. Subsequently the CTCF/LTR complex or CTCF-YB-1/LTR complex disables the capacity of NF-κB to bind its DNA target - possibly through changing the conformation of DNA as it was found for κB proteins [32].
Although we are closer to the understanding of HRF-mediated responses to virus invasion; there are still many questions waiting for an answer; for example the identity of HRF itself. This elusive molecule is yet to be defined and we believe that the identification of its intracellular co-factor will facilitate its discovery. It is also puzzling why CTCF protein bound HIV-1 promoter only in HRF(+) cells? Analysis of mRNA did not show any diversity between CTCF transcripts in HRF(+) and control HRF(-) systems (data not shown) which suggests, that HRF treatment along with the induction of transcription enables also a new function upon CTCF gene product. This new function can be achieved through the formation of CTCF/protein complexes or through the posttranslational modification. Comparable kinetics of YB-1 and CTCF binding suggested that HRF exposure enables the YB-1/CTCF binding and this protein complex targets HIV-1 LTR. Despite of these questions one fact remains solid: the CTCF is an intracellular co-factor of a novel inducible anti-HIV-1 activity in CD4+ T cells; This research along with reports from other research groups [13,27,31] shows that in addition to well-defined activities of CTCF, this protein might be involved in regulation of cellular responses to the viral invasion.
Fig. 7. The hypothetical scenario for HRF-mediated antiviral activity in HIV-1 susceptible CD4+ T cells.

Through the ligation of cell surface receptor and activation of the intracellular effector or through its intracellular uptake HRF induces transcription of CTCF mRNA followed by the increased protein levels (1–4). Upon virus invasion (5) CTCF protein is relocated to the nucleus where it binds to HIV-1 LTR possibly through its interaction with YB-1 protein (6–7) and inhibits transcription of HIV-1 through direct interaction with LTR; this CTCF/LTR complex blocks binding of NF-κB to its cognate sequence on HIV-1 promoter (8).
Acknowledgments
This work was supported by MH070282 and AI061286 from the National Institutes of Health. We thank Dr. David J. Volsky for giving us access to Q-PCR facility. 1G5 cells were obtained through the NIH AIDS Research and Reagent Program, Division of AIDS, NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klenova EM, Morse HC, 3rd, Ohlsson R, Lobanenkov VV. The novel BORIS + CTCF gene family is uniquely involved in the epigenetics of normal biology and cancer. Semin Cancer Biol. 2002;12:399–414. doi: 10.1016/s1044-579x(02)00060-3. [DOI] [PubMed] [Google Scholar]
- 2.Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520–7. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 3.Dunn KL, Davie JR. The many roles of the transcriptional regulator CTCF. Biochem Cell Biol. 2003;81:161–7. doi: 10.1139/o03-052. [DOI] [PubMed] [Google Scholar]
- 4.Lutz M, Burke LJ, LeFevre P, Myers FA, Thorne AW, Crane-Robinson C, Bonifer C, Filippova GN, Lobanenkov V, Renkawitz R. Thyroid hormone-regulated enhancer blocking: cooperation of CTCF and thyroid hormone receptor. Embo J. 2003;22:1579–87. doi: 10.1093/emboj/cdg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16:2802–13. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol. 1993;13:7612–24. doi: 10.1128/mcb.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klenova EM, Nicolas RH, U S, Carne AF, Lee RE, Lobanenkov VV, Goodwin GH. Molecular weight abnormalities of the CTCF transcription factor: CTCF migrates aberrantly in SDS-PAGE and the size of the expressed protein is affected by the UTRs and sequences within the coding region of the CTCF gene. Nucleic Acids Res. 1997;25:466–74. doi: 10.1093/nar/25.3.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kartvelishvili A, Lesner A, Szponar M, Simm M. Microarray analysis of differentially expressed genes in cells resistant to HIV-1. Immunol Lett. 2004;93:79–86. doi: 10.1016/j.imlet.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Lesner A, Kartvelishvili A, Lesniak J, Nikolov D, Kartvelishvili M, Trillo-Pazos G, Zablotna E, Simm M. Monoubiquitinated histone H1B is required for antiviral protection in CD4(+)T cells resistant to HIV-1. Biochemistry. 2004;43:16203–11. doi: 10.1021/bi0492758. [DOI] [PubMed] [Google Scholar]
- 10.Lesner A, Li Y, Nitkiewicz J, Li G, Kartvelishvili A, Kartvelishvili M, Simm M. A soluble factor secreted by an HIV-1-resistant cell line blocks transcription through inactivating the DNA-binding capacity of the NF-kappa B p65/p50 dimer. J Immunol. 2005;175:2548–54. doi: 10.4049/jimmunol.175.4.2548. [DOI] [PubMed] [Google Scholar]
- 11.Simm M, Miller LS, Durkin HG, Allen M, Chao W, Lesner A, Potash MJ, Volsky DJ. Induction of secreted human immunodeficiency virus type 1 (HIV-1) resistance factors in CD4-positive T lymphocytes by attenuated HIV-1 infection. Virology. 2002;294:1–12. doi: 10.1006/viro.2001.1300. [DOI] [PubMed] [Google Scholar]
- 12.Simm M, Pekarskaya O, Potash MJ, Volsky DJ. Prolonged infection of peripheral blood lymphocytes by Vif-negative HIV type 1 induces resistance to productive HIV type 1 infection through soluble factors. AIDS research and human retroviruses. 2000;16:943–52. doi: 10.1089/08892220050058353. [DOI] [PubMed] [Google Scholar]
- 13.Drueppel L, Pfleiderer K, Schmidt A, Hillen W, Berens C. A short autonomous repression motif is located within the N-terminal domain of CTCF. FEBS Lett. 2004;572:154–8. doi: 10.1016/j.febslet.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 14.Simm M, Su Z, Huang EY, Chen Y, Jiang H, Volsky DJ, Fisher PB. Cloning of differentially expressed genes in an HIV-1 resistant T cell clone by rapid subtraction hybridization, RaSH. Gene. 2001;269:93–101. doi: 10.1016/s0378-1119(01)00456-5. [DOI] [PubMed] [Google Scholar]
- 15.Su ZZ, Chen Y, Kang DC, Chao W, Simm M, Volsky DJ, Fisher PB. Customized rapid subtraction hybridization (RaSH) gene microarrays identify overlapping expression changes in human fetal astrocytes resulting from human immunodeficiency virus-1 infection or tumor necrosis factor-alpha treatment. Gene. 2003;306:67–78. doi: 10.1016/s0378-1119(03)00404-9. [DOI] [PubMed] [Google Scholar]
- 16.Aguilar-Cordova E, Chinen J, Donehower L, Lewis DE, Belmont JW. A sensitive reporter cell line for HIV-1 tat activity, HIV-1 inhibitors, and T cell activation effects. AIDS research and human retroviruses. 1994;10:295–301. doi: 10.1089/aid.1994.10.295. [DOI] [PubMed] [Google Scholar]
- 17.Choe W, Volsky DJ, Potash MJ. Induction of rapid and extensive beta-chemokine synthesis in macrophages by human immunodeficiency virus type 1 and gp120, independently of their coreceptor phenotype. J Virol. 2001;75:10738–45. doi: 10.1128/JVI.75.22.10738-10745.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–91. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–41. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnold R, Maueler W, Bassili G, Lutz M, Burke L, Epplen TJ, Renkawitz R. The insulator protein CTCF represses transcription on binding to the (gt)(22)(ga)(15) microsatellite in intron 2 of the HLA-DRB1(*)0401 gene. Gene. 2000;253:209–14. doi: 10.1016/s0378-1119(00)00271-7. [DOI] [PubMed] [Google Scholar]
- 21.Awad TA, Bigler J, Ulmer JE, Hu YJ, Moore JM, Lutz M, Neiman PE, Collins SJ, Renkawitz R, Lobanenkov VV, Filippova GN. Negative transcriptional regulation mediated by thyroid hormone response element 144 requires binding of the multivalent factor CTCF to a novel target DNA sequence. J Biol Chem. 1999;274:27092–8. doi: 10.1074/jbc.274.38.27092. [DOI] [PubMed] [Google Scholar]
- 22.Burcin M, Arnold R, Lutz M, Kaiser B, Runge D, Lottspeich F, Filippova GN, Lobanenkov VV, Renkawitz R. Negative protein 1, which is required for function of the chicken lysozyme gene silencer in conjunction with hormone receptors, is identical to the multivalent zinc finger repressor CTCF. Mol Cell Biol. 1997;17:1281–8. doi: 10.1128/mcb.17.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li G, Aaron S, Kazmierczak K, Lesner A, Li Y, Ivanova A, Bentsman G, Potash MJ, Simm M. Inhibition of HIV-1 or bacterial activation of macrophages by products of HIV-1-resistant human cells. Immunol Cell Biol. 2007:1–7. doi: 10.1038/sj.icb.7100092. [DOI] [PubMed] [Google Scholar]
- 24.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 25.Chernukhin IV, Shamsuddin S, Robinson AF, Carne AF, Paul A, El-Kady AI, Lobanenkov VV, Klenova EM. Physical and functional interaction between two pluripotent proteins, the Y-box DNA/RNA-binding factor, YB-1, and the multivalent zinc finger factor, CTCF. J Biol Chem. 2000;275:29915–21. doi: 10.1074/jbc.M001538200. [DOI] [PubMed] [Google Scholar]
- 26.Lutz M, Burke LJ, Barreto G, Goeman F, Greb H, Arnold R, Schultheiss H, Brehm A, Kouzarides T, Lobanenkov V, Renkawitz R. Transcriptional repression by the insulator protein CTCF involves histone deacetylases. Nucleic Acids Res. 2000;28:1707–13. doi: 10.1093/nar/28.8.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amelio AL, McAnany PK, Bloom DC. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol. 2006;80:2358–68. doi: 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ansari SA, Safak M, Gallia GL, Sawaya BE, Amini S, Khalili K. Interaction of YB-1 with human immunodeficiency virus type 1 Tat and TAR RNA modulates viral promoter activity. J Gen Virol. 1999;80 ( Pt 10):2629–38. doi: 10.1099/0022-1317-80-10-2629. [DOI] [PubMed] [Google Scholar]
- 29.Klenova E, Scott AC, Roberts J, Shamsuddin S, Lovejoy EA, Bergmann S, Bubb VJ, Royer HD, Quinn JP. YB-1 and CTCF differentially regulate the 5-HTT polymorphic intron 2 enhancer which predisposes to a variety of neurological disorders. J Neurosci. 2004;24:5966–73. doi: 10.1523/JNEUROSCI.1150-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawaya BE, Khalili K, Amini S. Transcription of the human immunodeficiency virus type 1 (HIV-1) promoter in central nervous system cells: effect of YB-1 on expression of the HIV-1 long terminal repeat. J Gen Virol. 1998;79 ( Pt 2):239–46. doi: 10.1099/0022-1317-79-2-239. [DOI] [PubMed] [Google Scholar]
- 31.Chau CM, Zhang XY, McMahon SB, Lieberman PM. Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J Virol. 2006;80:5723–32. doi: 10.1128/JVI.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schreck R, Zorbas H, Winnacker EL, Baeuerle PA. The NF-kappa B transcription factor induces DNA bending which is modulated by its 65-kD subunit. Nucleic Acids Res. 1990;18:6497–502. doi: 10.1093/nar/18.22.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]